T T C G G T G A T A T CC A G G C G G C G G G C A A T C A T C T T G T T C G G C A A A C C C T G GG C A A T C A G C T T G A G A A T A T C G C G C T C G C G T G G G G T T AA C T G G T T A A C A T C T C A G A A A A T G C G C T C C T G A T G C A C C C A T A C C G C T G C T T C C A C G C G A G A C T T G A G C T T C A T T T T C T T C A G C A T G T G C T T G A C G T G C A C T T T T A C T G T G C T T T C G G T G A T A T C C A G G C G G C G G G C A A T C A T C T T
MCCMB ’09
P R O C E E D I N G S N
OF THE I TERNATIONAL
O O
M SCOW C NFERENCE O
O N C M P U T A T I O N A L
O I
M L E C U L A R B O L O G Y
July 20-23, 2009
Department of Bioengineering and Bioinformatics of M.V. Lomonosov Moscow State University
INRIA, France The
,
Scientific Council on Biophysics RAS Institute for Information Trasnsmission Problems, RAS State Scientific Centre GosNIIGenetika
Biological Department
of M.V. Lomonosov Moscow State University
1930
У
Engelhardt Institute of Molecular Biology Russian Academy of Sciences
Russian Fund of Basic Research
Organizers
Sponsored by
А Ф УК
ЛЬТЕТ
БИОИНЖЕ НЕ Р И И И БИ
ОИ Н Ф
ОРМАТ
КИ И
Р И
INRIA, France the French National Institute for Research in Computer Science and Control
( )
TheScientific Council on Biophysics,RussianAcademy of ciencesS Institute for Information Trasnsmission Problems, RussianAcademy of ciencesS State Scientific Centre GosNIIGenetika Biological Department of M.V. Lomonosov Moscow State University
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences Russian Fund of Basic Research with financial support of
MCCMB ’09
Moscow, Russia July 20-23, 2009
P R O C E E D I N G S
July 20–23, 2009
NEW METHOD TO IMPROVE ERROR PROBABILITY ESTIMATION APPLIED TO ILLUMINA SEQUENCING
IRINA ABNIZOVA1, TOM SKELLY1, YUMI YAN1, TONY COX1
The new short read sequencing technique introduced new technological and computational challenges. It requires reconsideration of well-known error estimation algorithms, taking into account different sequencing platforms.
DETECTION OF GENES THAT UNDERWENT POSITIVE SELECTION IN DEEP-SEA ARCHAEBACTERIA OF
PYROCOCCUS GENUS
K.V. GUNBIN1, D.A. AFONNIKOV2, N.A.KOLCHANOV2
Pressure is an environmental parameter of crucial importance for organisms. Archaeal species of the Pyrococcus genus live under both normal (~0,1MPa) and high pressures (>10MPa). To date, the genomes of three Pyrococcus species have been completely sequenced: P. furiosus bacteria live under normal pressure, whereas P. horikoshii and P. аbyssi are piezophilic (live in deep sea environment under high pressure at 14MPa and 20MPa, respectively). In this work we analyze the rate of nucleotide substitution in search for genes underwent positive selection in deep-sea species of Pyrococcus genus.
A phylogenetic analysis was performed to determine the evolutionary relatedness of the piezophilic species of the Pyrococcus genus and T.
kodekaraensis as outgroup. The analysis of phylogenetic tree demonstrates that piezophilic species have a common origin and the ancestor of piezophilic species emerged from archaebacteria phylogenetically close to the extant species of Pyrococcus genus inhabiting in normal pressure environments.
Events of positive selection (PS) for adaptation of life under high pressure were searched for the set of 508 homologous genes which protein sequences are close homologs (amino acid sequence identity greater than 40%) and have no paralogs in genomes. We reconstructed genes and proteins of the most recent ancestor of piezophilic species of the Pyrococcus genus and the common ancestor of P. furiosus, P. horikoshii and P. аbyssi species.
Reconstructed ancestral sequence of genes and proteins were compared with extant sequences using nonsynonymous to synonymous substitution rate ratio, radical to conservative amino acid replacement rate ratio, also amino acid dissimilarity measures. We use ArCOG functional classification of analyzed genes and demonstrated that positive selection events occurred in genes and proteins of ‘Coenzyme transport and metabolism’ and ‘Energy production and conversion’ functional groups (Table 1). The results suggest
1 Institute of Cytology and genetics SB RAS, [email protected]
2 Institute of Cytology and genetics SB RAS, Novosibirsk State University [email protected]; [email protected]
July 20–23, 2009
that genes of these functional classes may be important for adaptation of piezophilic Pyrococcus species to deep-sea environment.
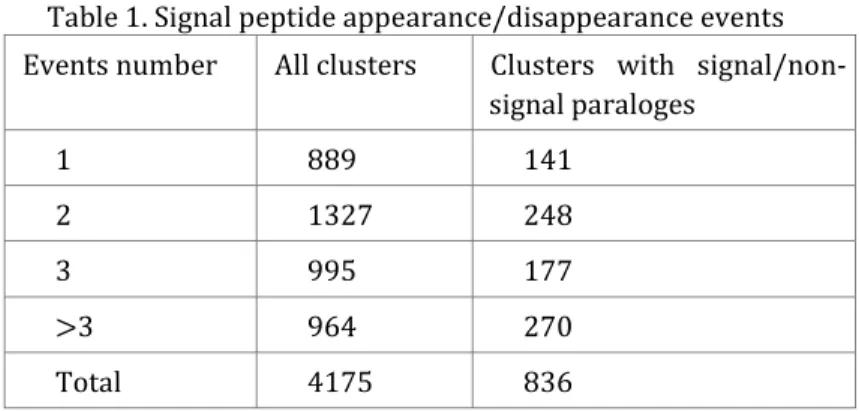
Table 1. ArCOG group enrichment in the full set of analyzed genes and in genes with identified positive selection events. Last column represents the probablilty of difference in number of genes in full and PS sets observed by chance according to Monte Carlo shuffling test with 105 replicas. ArCOG groups with statistical significant difference (p<0.05) shown in bold.
ArCOG group ArCOG group ArCOG group
ArCOG group Number Number Number Number in full in full in full in full dataset datasetdataset dataset
Number Number Number Number in PS in PS in PS in PS group groupgroup group
ppp
p----value of observing value of observing value of observing value of observing by random chance by random chance by random chance by random chance Amino acid transport and metabolism 34 8 0.18225
Carbohydrate transport and metabolism 22 2 0.90471 Cell cycle control; cell division;
chromosome partitioning 8 0 *
Cell motility 7 2 0.32479
Cell wall/membrane/envelope biogenesis 13 1 0.90748
Coenzyme transport and metabolism 15 6 0.02416
Defense mechanisms 3 0 *
Energy production and conversion 33 11 0.01072
Inorganic ion transport and metabolism 16 0 * Intracellular trafficking; secretion; and
vesicular transport 6 0 *
Lipid transport and metabolism 5 0 *
Nucleotide transport and metabolism 24 5 0.36181 Posttranslational modification; protein
turnover; chaperones 18 4 0.34395
Replication; recombination and repair 24 4 0.58199 Secondary metabolites biosynthesis;
transport and catabolism 5 1 0.59636
Signal transduction mechanisms 3 1 0.42142
Transcription 28 4 0.70909
Translation; ribosomal structure and
biogenesis 76 14 0.36997
Function unknown 87 6 0.99903
General function prediction only 75 14 0.34782
Not annotated 6 1 0.66633
Total 508 84
The work was supported by SB RAS integration project №109, Scientific School НШ-2447.2008.4, RAS program “Origin and evolution of Biosphere”
and CRDF REC-008 grant.
MATHEMATICAL MODELING OF THE MOLECULAR GENETIC SYSTEMS REGULATING A PLANT
DEVELOPMENT
ILYA AKBERDIN1, FEDOR KAZANTSEV1, STANISLAV FADEEV2, IRINA GAINOVA2, VITALY LIKHOSHVAI1
Keywords: auxin metabolism, gene network, automatic generation, mathematical model, plant development
Indole-3-acetic acid (IAA) is physiologically active in the form of the free acid, but can also be found in conjugated forms in plant tissues. IAA can be degraded and redundant pathways lead to its synthesis. Auxin participates in regulation of cell differentiation in development of embryo, leaves, vascular tissue, fruit, primary and lateral root and in controlling apical dominance and tropisms. The regulation of the IAA metabolism (synthesis, conjugation and degradations) is enough complex and may explain in some aspects how this simple substance is able to influence such diverse processes. Mathematical modeling of IAA metabolic gene network can help reveal the main factors governing this complex process. To reach this aim, we first reconstructed a gene network of auxin biosynthesis, conjugation degradation by annotating experimental data from 107 published papers into GeneNet computer system.
This gene network after reduction was input into converter to generate the mathematical model of auxin metabolism. We have reconstructed the gene network and develop the mathematical model of auxin metabolism in arabidopsis shoots. The model allows to reproduce some phenomenological and molecular-genetic aspects of the auxin role in the plant development. The obtained results confirm adequacy of the developed model. In silico experiments testify to qualitatively rapid processes of the molecular genetic regulation of the systems homeostasis. The cumulative experimental data allowed starting construction of spatial distributed hierarchical model that describe both molecular genetic processes and processes on the level of cell- cell interactions simultaneously. So earlier we’ve developed the cellular automaton model that imitates morphodynamics of embryo development by means of regulation of signals produced by different embryonic cells is a first
1 The Institute of Cytology and Genetics SB RAS, Russian Federation, [email protected]
2 The Institute of Mathematics SB RAS, Russian Federation
July 20–23, 2009
step in modelling the process of development in general and in modelling the gene network for morphogenesis in particular [1]. The next step in mathematical modeling application to studying of the plant development rules is integration of the spatial distributed hierarchical model with model of the intracellular auxin metabolism.
Akberdin I.R., Ozonov E.A., Mironova V.V., Gorpinchenko D.N., Omelyanchuk N.A., Likhoshvai V.A., Kolchanov N.A. (2007). “A cellular automaton to model the development of shoot meristems of Arabidopsis thaliana”, Journal of Bioinformatics and Computational Biology Vol. 5, pp. 641-650.
WATER-MEDIATED HYDROGEN BONDS ARE ESSENTIAL FOR LOOP STABILIZATION IN PROTEIN STRUCTURES
EVGENIY AKSIANOV1, SERGEI SPIRIN1,2, ANNA KARYAGINA1,3,4, ANDREI ALEXEEVSKI1,2
Keywords: protein structure, water, hydrogen bond, water-mediated bond
Protein structures are mostly composed of secondary structural elements (SSE): alpha-helices and beta-strands. SSEs are connected by unstructured regions (loops). Loops resolved in Х-ray experiments are not flexible; they are stable, at least in a crystal. Regular nets of hydrogen bonds (H-bonds) stabilize both helices and sheets and are important for SSE's stability. No regular hydrogen bond networks are known to stabilize loop conformations. Based on a number of examples we hypothesized that intradomain hydrogen bonds mediated by water molecules significantly contribute to the stabilization of loops. To test our hypothesis, we analyzed intradomain direct hydrogen bonds and water-mediated hydrogen bonds in a non-redundant set of protein domain X-ray structures with high resolution.
Methods. 995 protein domains were obtained from the SCOP 1.73 database; sequence identity between each pair of domains was ≤90 %, all structures are X-ray with resolution better than 1.5 Å. Secondary structural elements (β-strands and α-helices) were detected using DSSP algorithm. An H- bond was defined as a pair of atoms such that (1) one of atoms may be proton donor and other proton acceptor, (2) the distance between atoms is 2.3–3.7 Å and (3) the angles between the direction of the H-bond and the optimal direction of H-bond is ≤ 40° for both atoms.
Results. The number (per 20 residues of the corresponding SSEs) of H- bonds and water-mediated bonds between helices, strands and loops are shown in Table 1. The numbers of backbone-backbone H-bonds per 20 residues in helices and sheets are less than the maximal possible 20 (11.5 for strands and 12.1 for helices) mainly due to large number of short helices and
1 Belozersky Institute, Moscow State University, Moscow, Russia, [email protected]
2 Scientific Research Institute for System Studies (NIISI RAN), Moscow
3 Gamaleya Institute of Epidemiology and Microbiology, 18 Gamaleya st., Moscow, 123098, Russia
4 Institute of Agricultural Biotechnology, 42 Timiryazevskaya st., Moscow, 127550, Russia
July 20–23, 2009
hairpins (where the number of regular H-bonds is twice smaller) and irregularities in SSE's H-bond networks.
Table 1. Number of direct/water-mediated hydrogen bonds between helices, strands, and loops.
Strands Helixes Loops
Side chain Backbone Side chain Backbone Side chain Backbone
Length (1) 40897 40897 47681 47681 37062 37062
Atoms (2) 14.13 40.00 17.93 40.00 89.75 40.00
0.51 / 1.23
(3) 0.00 / 0.02 1.49 / 2.45 12.14(5) / 0.05 3.18 / 4.57 1.78 / 0.83 BONDS WITHIN
THE SAME SSE 0.10 / 0.07
(backbone to side chain) 0.14 / 0.07 0.25 / 0.13 BONDS BETWEEN DIFFERENT SSEs
Strands (s.c.) Strands (bb.)
Helixes
(s.c.) Helixes (bb.) Loops (s.c.) Loops (bb.) Strands (s.c.(4)) 2.07 / 8.45 0.23 / 1.27 0.36 / 0.76 0.03 / 0.14 1.40 / 3.95 0.85 / 2.05 Strands (bb. (4)) 0.23 / 1.27 11.48 / 0.5 0.06 / 0.34 0.03 / 0.05 0.32 / 0.93 1.31 / 0.82 Helixes (s.c.) 0.36 / 0.76 0.06 / 0.34 1.22 /
10.13 0.49 / 0.99 1.76 / 4.10 0.80 / 1.77 Helixes (bb.) 0.03 / 0.14 0.03 / 0.05 0.49 / 0.99 0.07 / 0.38 0.74 / 1.02 2.00 / 0.52 Loops (s.c.) 1.40 / 3.95 0.32 / 0.93 1.76 / 4.10 0.74 / 1.02 2.65 /
20.75 3.48 / 9.04 Loops (bb.) 0.85 / 2.05 1.31 / 0.82 0.80 / 1.77 2.00 / 0.52 3.48 / 9.04 1.52 / 4.54 (1) The total length of all elements in the investigated structures (in amino acids).
(2) Number of hydrogen donors and acceptors per 20 residues
(3) 0.51 direct bonds and 1.23 water-mediated bonds per 20 amino acids. The same notations are used in all other cells of the table.
(4) s.c. means side chains, bb. means backbone atoms.
(5) Numbers greater than 4 bonds per 20 residues are shown bold and large.
From table 1 it follows that water-mediated bonds between side chains of two helices or two strands were detected for approximately a half of residues.
In the case of loop-to-loop interactions water-mediated bonds on average were detected for each residue, and their contribution to loop – loop interactions exceeds the contribution of direct H-bonds.
We conclude that intra-domain water-mediated bonds are common feature in protein structures. Such bonds may be especially important for loop stabilization.
The work is partly supported by the Russian Foundation for Basic Research, grants 07-04-91560 and 08-04-91975.
July 20–23, 2009
GENOMIC INSIGHTS INTO THE ORIGINS OF METAZOAN CELL DIFFERENTIATION
KIRILL V. MIKHAILOV1, A.V. KONSTANTINOVA1, M.A. NIKITIN1, V.V. ALEOSHIN1, L.YU. RUSIN2, YURI V. PANCHIN2
Keywords: Mesomycetozoea; molecular phylogenetics; origin of Metazoa;
Choanoflagellates and mesomycetozoeans are two groups of unicellular organisms that are the closest relatives of animals [1]. The ongoing genome sequencing effort aimed at their members is an attempt to understand the origin of animals and multicellularity in the context of evolution of genes and genomes [2]. These studies have brought about a notion of “Metazoa-specific”
genes, genes found exclusively in metazoans, which are thus considered likely to be novelties specifically associated with the development multicellularity.
The “Metazoa-specific” genes code a large number of cell signalling and adhesion proteins such as cadherins and TGFb pathway components, to name a few. However the list of “Metazoa-specific” genes is rapidly contracting as the number of sequenced genomes of unicellular relatives of metazoans increases. The genomes of choanoflagellates were found to contain a multitude of tyrosine kinases – proteins involved in the regulation of cell proliferation and motility that were originally considered to be a metazoan novelty [3]. Another example is a mesomycetozoean that possesses components involved in cell-matrix adhesion, such as focal adhesion kinase and integrin beta [4]. Here we present evidence for the exclusion of yet another set of genes from the “Metazoa-specific” list by demonstrating their presence in another mesomycetozoean and showing that they are actively expressed. The premetazoan ancestry of metazoan transcription factor families and signal transduction pathways is poorly accommodated by the traditional view of the metazoan ancestors as blastula-like colonies, which had subsequently undergone cell differentiation. The new data suggests that the elements of the genetic toolkit for the development of multicellular animals were possibly already in use by their unicellular relatives. Mapping of major gene families and ecological traits onto the phylogeny indicates that presence of different cell types at different stages of life cycle and appearance of
1 Belozersky Institute for Physicochemical Biology, Lomonosov Moscow State University, Moscow, Russian Federation, [email protected]
2 Institute for Information Transmission Problems, Russian Academy of Sciences,
multicellular aggregates is not an intrinsic property of metazoans, but of a much wider group of organisms – Opisthokonta [5]. The emerging scenario regards the last common ancestor of multicellular animals as an integration of different stages of the unicellular ancestor’s life cycle.
1. E.T.Steenkamp, J.Wright, S.L.Baldauf (2006) The protistan origins of animals and fungi, Molecular Biology and Evolution, 23: 93–106.
2. I.Ruiz-Trillo, G.Burger , P.W.Holland, N.King, B.F.Lang, et al. (2007) The origins of multicellularity: a multi-taxon genome initiative, Trends in Genetics, 23:113–118.
3. N.King, M.J.Westbrook, S.L.Young, A.Kuo, M.Abedin, et al. (2008) The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans, Nature, 451: 783–788.
4. K.Shalchian-Tabrizi, M.A.Minge, M.Espelund, R.Orr, T.Ruden, et al.
(2008) Multigene phylogeny of choanozoa and the origin of animals, PLoS ONE, 3: 2098.
5. K.V.Mikhailov, A.V.Konstantinova, M.A.Nikitin, P.V. Troshin, L.Yu. Rusin, V.A. Lyubetsky, Y.V. Panchin, et al. (2009) The origin of Metazoa: a transition from temporal to spatial cell differentiation, Bioessays, 31: (in press).
July 20–23, 2009
INHERENT POTENTIALITIES OF VORONOI-DELAUNEY TESSELLATION AS APPLIED TO BIOLOGY PROBLEMS
ANASTASYA ANASHKINA1, NATALIA ESIPOVA1, VLADIMIR TUMANYAN1
Researchers of different areas of interest effectively used Voronoi- Delaunay tessellation to solve various problems for a long time. During last years the interest to this method arises due to its possibilities in complex biological studies along with crystallography and chemistry. By definition, Voronoi polyhedron or Voronoi region is a part of space which points locate closer to this center than to any other center of the system. Tetrahedron (based on four centers of the system) is a Delaunay simplex whether inside the circumsphere there are no other centers of the system. The set of all Delaunay simplexes of a system as well as the set of Voronoi polyhedrons fills space without slits and overlaps. These tessellations are dual and topologically equivalent.
Single-valued character of Voronoi-Delaunay tessellation make this method extremely attractive for researchers as well as it’s independence of any parameters. Mathematical rigorousness and exactness of exploration are very rare occur in biological sciences. The method is developed both for two- dimensional and three-dimensional cases. Modifications of the basic method provide additional capabilities and allow analyzing not only systems of points but systems of spheres of similar radii, systems of spheres of different radii, systems of bodies of arbitrary shapes [1].
Voronoi-Delaunay tessellation encounters some problems in practical use.
In particular, boundary conditions should be set. Another problem consists in time-consuming during computations for multi-atomic systems.
Two-dimensional Voronoi-Delaunay tessellation is used even for cell cultures architecture analysis. Voronoi facet as well as Delaunay edge is a natural unambiguous non-parametric way to reveal the nearest neighbors in tridimensional space. This procedure is equivalent to revelation of contacts between atoms. Consequently Voronoi-Delaunay tessellation allows calculating of local atomic density and contacts between biopolymer molecules. A contact between two atoms, in this case, is a common facet of Voronoi polyhedron. As a result the contact between two residues is defined as a set of common facets of Voronoi polyhedrons of appropriate atoms. So it
1 Engelghardt Institute of Molecular Biology RAS, Russian Federation, [email protected]
is possible to explore the statistics of contacts between atoms or residues/nucleotides in protein-protein [2] and protein-nucleic [3] interfaces.
Knowledge of rules which control interactions in protein-protein interfaces is necessary for correct prediction of interaction sites on the surface of protein or protein complexes. Also, it may well be that application of this powerful method will decide the question of existence of kind of code of nucleic acid- protein recognition.
Voronoi network (more specifically, Voronoi S-network) is the main tool for empty interatomic space analysis. This network penetrates through interatomic space of the system and represents locus located outermost from atoms [4].
1. N.N. Medvedev (2000) Metod Voronogo-Delone v issledovanii struktury nekristallicheskih sistem, Novosibirsk: NIC OIGGM SO RAN.
2. A. Anashkina et al. (2007) Comprehensive statistical analysis of residues interaction specificity at protein-protein interfaces, Proteins, 67(4):
1060-77.
3. A.A. Anashkina et al. (2008) Geometricheskij analiz DNK-belkovyh vzaimodejstvij na osnove metoda Voronogo-Delone, Biofizika, 53(3):
402-6.
4. N.N Medvedev, V.P. Voloshin (2003) Issledovanie mezhatomnyx pustot v molekulyarnyh sistemah, Struktura i dinamika molekulyarnyh sistem, X (1): 299-304.
This research was supported (funded) by Russian Foundation for Basic Research Grants 07-04-01765а and 08-04-01770а.
July 20–23, 2009
COMPUTATIONAL ANTI-AIDS DRUG DESIGN RESULTING FROM THE STUDY ON SPECIFIC INTERACTIONS OF
IMMUNOPHILINS WITH THE HIV-1 GP120 V3 LOOP
ALEXANDER ANDRIANOV1
Keywords: HIV-1, V3 Loop, 3D Structure, Computer Modeling, Molecular Docking Currently, special emphasis of the research teams involved in the anti-AIDS drug studies is attracted to the HIV-1 V3 loop (reviewed in [1]). The higher interest in V3 is caused by numerous experimental data testifying to the fact that exactly this gp120 site gives rise to the principal target for neutralizing antibodies and accounts for the choice of co-receptor determining the preference of the virus in respect with T-lymphocytes or primary macrophages. Since the V3 loop governs the cell tropism and cell fusion (see, e.g., [1], one of the strategic ways in developing the anti-HIV-1 drugs may be based on the approach anticipating the search for the chemicals capable of the efficacious blockading this functionally significant stretch of gp120.
Comprehensive analysis of the data of study [2] allows one to suppose that immunophilins exhibiting specific high-affinity interactions with the HIV-1 V3 loop may be utilized as a basic substance to set out of the search for the potential anti-AIDS therapeutic agents.
This work proceeds with my previous study [3] where the virtual molecule presenting the promising anti-HIV-1 pharmacological substance was designed by means of the computer modeling based on the analysis of specific interactions between the FK506-binding protein and synthetic peptide imitating the immunogenic crown of the V3 loop.
The object of the present study was to generate the model describing the structural complex of cyclophilin A with the HIV-MN V3 loop followed by the computer-aided design of the immunophilin-derived peptide able to mask the biologically important V3 segments.
To this end, the following problems were solved: (i) the NMR-based conformational analysis of the HIV-MN V3 loop was put into effect, and its low energy structure fitting the input experimental observations was determined;
(ii) molecular docking of this V3 structure with the X-ray conformation of CycA was carried out, and the energy refining the simulated structural
1 Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, Kuprevich Street., 5/2, 220141 Minsk, Republic of Belarus,
complex was implemented; (iii) the matrix of inter-atomic distances for the amino acids of the molecules forming part of the built over-molecular ensemble was computed, the types of interactions responsible for its stabilization were analyzed, and the CycA stretch which accounts for the binding to V3 was identified; (iv) the most probable 3D structure of this stretch in the unbound state was predicted, and its collation with the X-ray structure for the corresponding site of CycA was performed; (v) the potential energy function and its constituents were studied for the structural complex generated by molecular docking of the V3 loop with the CycA peptide offering the virtual molecule which imitates the CycA segment making a key contribution to the interactions of the native protein with the HIV-1 principal neutralizing determinant; (vi) as a result, the designed molecule was shown to be capable of the effictive blocking the functionally crucial V3 sites; and (vii) starting from the joint analysis of the results derived here and in study [3], the composition of the peptide cocktail presenting the promising anti-AIDS pharmacological substance was developed.
The molecules simulated here by molecular modeling methods may become the first representatives of a new class of chemicals (immunophilin- derived peptides) offering the forward -looking basic structures for the design of efficacious and safe antiviral agents.
The author appreciates the Belarusian Republican Foundation for Basic Research for financial support (project No X08-003).
1. S.Sirois, T.Sing, K.C.Chou (2005) HIV-1 gp120 V3 loop for structure- based drug design, Curr. Protein Pept. Sci., 6: 413-422.
2. M.M.Endrich, H.Gehring (1998) The V3 loop of human immunodeficiency virus type-1 envelope protein is a high-affinity ligand for immunophilins present in human blood, Eur. J. Biochem., 252: 441- 446.
3. A.M.Andrianov (2008) Computational anti-AIDS drug design based on the analysis of the specific interactions between immunophilins and the HIV-1 gp120 V3 loop. Application to the FK506-binding protein, J.
Biomol. Struct. Dynam., 26: 49-56.
July 20–23, 2009
HOMOLOGY MODELING AND MOLECULAR DYNAMICS IN STRUCTURAL STUDIES ON THE HIV-1 GP120 V3
LOOPS: INSIGHT INTO THE VIRUS SUBTYPE A
IVAN ANISHCHENKO1, ALEXANDER ANDRIANOV2
Keywords: HIV-1, V3 Loop, 3D Structure, Computer Modeling, Molecular Docking The V3 loop of the HIV-1gp120 glycoprotein presenting 35-residue-long, frequently glycosylated, highly variable, and disulfide bonded structure plays the central role in the virus biology and forms the principal target for neutralizing antibodies and the major viral determinant for co-receptor binding. Here we present the computer-aided studies on the 3D structure of the HIV-1 subtype A V3 loop (SA-V3 loop) in which its structurally inflexible regions and individual amino acids were identified and the structure-function analysis of V3 aimed at the informational support for anti-AIDS drug researches was put into practice.
To this effect, the following successive steps were carried out: (i) using the methods of homology modeling and simulated annealing, the ensemble of the low-energy structures was generated for the consensus amino acid sequence of the SA-V3 loop and its most probable conformation was defined basing on the general criteria widely adopted as a measure of the quality of protein structures in terms of their 3D folds and local geometry; (ii) the elements of secondary V3 structures in the built conformations were characterized and careful analysis of the corresponding data arising from experimental observations for the V3 loops in various HIV-1 strains was made; (iii) to reveal common structural motifs in the HIV-1 V3 loops regardless of their sequence variability and medium inconstancy, the simulated structures were collated with each other as well as with those of V3 deciphered by NMR spectroscopy and X-ray studies for diverse virus isolates in different environments; (iv) with the object of delving into the conformational features of the SA-V3 loop, molecular dynamics trajectory was computed from its static 3D structure followed by determining the structurally rigid V3 segments and comparing the findings obtained with the ones derived hereinbefore; and (v) to evaluate the
1 United Institute of Informatics Problems, National Academy of Sciences of Belarus, Surganov Street 6, 220012 Minsk, Republic of Belarus, [email protected]
2 Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, Kuprevich Street, 5/2, 220141 Minsk, Republic of Belarus, [email protected]
masking effect that can occur due to interaction of the SA-V3 loop with the two virtual molecules constructed previously [1, 2] by tools of computational modeling and named FKBP and CycA peptides, molecular docking of V3 with these molecules was implemented and inter-atomic contacts appearing in the simulated complexes were analyzed to specify the V3 stretches keeping in touch with the ligands.
As a matter of record, V3 segments 3-7, 15-20, and 28-32 containing the highly conserved and biologically meaningful residues of gp120 were shown to retain their 3D main chain shapes in all the cases of interest presenting the forward-looking targets for anti-AIDS drug researches. From the data on molecular docking, synthetic analogs of the CycA and FKBP peptides were suggested being suitable frameworks for making a reality of the V3-based anti-HIV-1 drug projects.
In addition, the computational V3 model proposed above provides a productive basis to gain a better insight into the principles of virus functioning, and, therefore, can be used in subsequent studies for investigating the structure-functional relationship as well as for examining the structural effects of mutations or distinguishing between various forms of the V3 loop under different conditions.
1. A.M.Andrianov (2008) Computational anti-AIDS drug design based on the analysis of the specific interactions between immunophilins and the HIV-1 gp120 V3 loop. Application to the FK506-binding protein, J.
Biomol. Struct. Dynam., 26: 49-56.
2. A.M.Andrianov (2009) Immunophilins and HIV-1 V3 loop for structure- based anti-AIDS drug design, J. Biomol. Struct. Dynam., 26: 445-454.
This study was supported by grants from the Union State of Russia and Belarus (scientific program SKIF-GRID; № 4U-S/07-111) as well as from the Belarusian Foundation for Basic Research (project X08-003).
July 20–23, 2009
3D STRUCTURE MODELING AND POSTERIOR COLLATION OF THE HIV-1 V3 VARIABLE LOOPS FOR
DISCOVERY OF THEIR STRUCTURALLY INVARIANT SITES EXPOSING THE ACHILLES' HEEL IN THE HIV-1
“REDOUBTS”
A. M. ANDRIANOV1, I.V. ANISHCHENKO2
Keywords: HIV-1, V3 Loop, 3D Structure, Computer Modeling, Molecular Docking The HIV-1 gp120 V3 loop forming the virus principal neutralizing determinant and determinants of cell tropism and cell fusion is considered as one of the promising targets for anti-AIDS drug studies (reviewed in [1]). The V3 loops derived from different HIV-1 isolates contain highly variable amino acid sequences, which prevents antibodies bound to a V3 loop of one isolate from having effect on the V3 loops of other isolates. However, the analysis of various HIV-1 V3 loop sequences makes it clear that, despite their high variability which complicates fundamentally the studies on the V3 loop structure, some of the amino acid positions located in the N- and C-terminals and especially those residing in its immunogenic tip, are highly conserved.
Conserving these V3 stands allows one to suggest that the residues occupying them may preserve their conformational states in diverse HIV-1 strains and, therefore, may present the promising targets for developing the new therapeutic agents. Therefore, one is in need of the information on the 3D structure of V3 and its inflexible regions, which is of particular importance to successful implementation of the anti-AIDS drug studies [1].
In the light of the above, the computational approaches combining the NMR-based protein structure modeling with the mathematical statistics methods were used here to define the locally accurate 3D structures of the HIV-1 gp120 V3 loops from Minnesota, Haiti, RF, and Thailand isolates in water solution as well as from Minnesota and Haiti isolates in a water/trifluoroethanol mixed solvent. To specify the structural motifs of V3 giving rise to the close spatial folds regardless of the sequence and environment variability, the simulated structures and their individual
1 Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, Kuprevich Street, 5/2, 220141 Minsk, Republic of Belarus,
2 United Institute of Informatics Problems, National Academy of Sciences of Belarus,
segments of different length were collated between themselves and with those derived previously from homology modeling [2] and X-ray crystallography [3].
As a result, the sequence and environment changes were found to trigger the considerable structural rearrangements of the V3 loop, but, at the same time, some of the functionally crucial V3 stretches were shown to keep the 3D shapes in all the cases in question. In the first place, it concerns core V3 sequence 15-20 as well as its N- and C-terminal sites 3-7 and 28-32 comprising the residues, which contribute significantly to the virus immunogenicity and cell tropism. In addition, structurally rigid V3 stretch 3-7 includes the highly conservative glycolysation site of gp120 utilized by the virus for defense against neutralizing antibodies and elevation of its infectivity. In the context of these findings, the inflexible V3 motifs identified in this study may present the weak units in the HIV-1 protection system and, therefore, their detection is of great importance to successful design of the V3- based anti-AIDS drugs being able to stop the HIV's spread.
1. S.Sirois, T.Sing, K.C.Chou (2005) HIV-1 gp120 V3 loop for structure- based drug design, Curr. Protein Pept. Sci., 6: 413-422.
2. I.V.Anishchenko, A.M. Andrianov (2008) Computer-aided modeling of the 3D structure for the HIV-1 gp120 V3 loop: exploring the virus subtype A, Proceedings of II International Conference “Advanced Information and Telemedicine Technologies for Health” (Minsk, 2008):
12-16.
3. C.C. Huang, M. Tang, M.Y. Zhang, S. Majeed, E. Montabana, R.L. Stanfield, D.S. Dimitrov, B. Korber, J. Sodroski, I.A. Wilson, R. Wyatt, P.D. Kwong (2005) Structure of a V3-containing HIV-1 gp120 core, Science, 310:
1025 – 1028.
This study was supported by grants from the Union State of Russia and Belarus (scientific program SKIF-GRID; № 4U-S/07-111) as well as from the Belarusian Foundation for Basic Research (project X08-003).
July 20–23, 2009
POLYCTLDESIGNER – THE SOFTWARE FOR CONSTRUCTING POLYEPITOPE IMMUNOGENS.
DENIS ANTONETS1, AMIR MAKSYUTOV2, SERGEY BAZHAN3
Keywords: Immunity, cytotoxic T-lymphocyte, T-cell epitope, polyepitope antigen Design of the artificial polyepitope immunogens capable of eliciting high levels of the CD8+ CTL responses to is a promising approach in creation of an efficient vaccines. When designing such immunogens, it is necessary to optimize the processing and presentation of contained epitopes. DNA vaccine constructs encoding poly-CTL-epitope immunogens containing N-terminal ubiquitin and spacer sequences ensuring correct processing and presentation of selected epitopes were shown to be highly efficient in stimulating CD8+
CTL responses. These results inspired us to create PolyCTLDesigner software, intended for designing optimal polyepitope antigens. To optimize polytope sequence for inducing high level of CTL response one should take into account major steps of MHC class I-dependent antigen processing:
proteasomal/immunoproteasomal cleavage of antigen and TAP-dependent transport of generated peptidic fragments into endoplasmic reticulum where they bind to MHC class I molecules. To prognose proteasomal/immunoproteasomal processing PolyCTLDesigner utilizes predictive models developed by Toes et al. [1]. The site of proteasomal cleavage should be located at the С-terminus of the epitope. Thus to optimize proteasomal cleavage (if necessary) C-terminus of the epitope should be extended with spacer motif with up to six aminoacid residues in length. To predict peptide binding to TAP our program uses models developed by Peters et al. [2]. Since, according to a widely accepted hypothesis, the major contributions to TAP-binding are provided by the first three N-terminal amino acid residues of the peptide and the last one (C-terminal), and given the fact, that C-terminus of the epitope must stay unchanged, only N-terminus of the antigenic peptide could be extended to optimize its interaction with TAP1/TAP2 heterodimer. According to the chosen models and algorithms for
1 Research Center of Virology and Biotechnology Vector, Russian Federation, [email protected]
2 Research Center of Virology and Biotechnology Vector, Russian Federation, [email protected]
3 Research Center of Virology and Biotechnology Vector, Russian Federation,
TAP-binding prediction the maximal length of N-terminal spacer sequence will make three residues: ARY. PolyCTLDesigner is integrated with TEpredict program (http://tepredict.sourceforge.net), created earlier. TEpredict is used by PolyCTLDesigner to predict T-cell epitopes. PolyCTLDesigner allows the user to select the minimal set of epitopes with known (or predicted) specificity towards various allelic variants of MHC class I molecules covering the selected MHC-repertoire with a specified redundancy. Currently PolyCTLDesigner utilizes two algorithms to design polyepitope immunogens.
The first one utilizes an optimal spacer motif derived from the selected predictive models (e.g., ADLVKV). And the second algorithm utilizes redundant spacer motif and minimizes formation of «non target» epitopes in the sequence of the desired polyepitope immunogen. The developed software realizes the rational approach to designing highly immunogenic poly-CTL- epitope vaccine constructs and can be used for designing new candidate polyepitope vaccines capable of eliciting high levels of the T-cell–mediated immune responses. More detailed description of the program and its source code are available at http://tepredict.sourceforge.net/PolyCTLDesigner.html.
The program is written in Python programming language (http://python.org).
1. Toes R.E. et al. (2001). Discrete Cleavage Motifs of Constitutive and Immunoproteasomes Revealed by Quantitative Analysis of Cleavage Products. J. Exp. Med., 194:1-12.
2. Peters B. et al. (2003) Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol., 171:1741–
1749.
July 20–23, 2009
GENOME-WIDE SEARCH FOR 5’-UTR OF
SACCHAROMYCES CEREVISIAE GENES AND THEIR ORTHOLOGS
KIRILL ANTONEZ1, ALSU SAIFITDINOVA2 Keywords: yeast,5'-UTR
Motivation and Aims: Prokaryotic and eukaryotic mRNAs are the important step of protein biosynthesis and consists of coding sequence and untranslated regions (UTRs). UTR’s play essential role in posttranscriptional life of mRNA and may harbor regulatory elements in addition to translation initiation sequences. Also 5’-UTRs of both prokaryotic and eukaryotic mRNA may form stable secondary structures, which influence the efficiency of translation initiation. Certain 5’-UTRs contain riboswitches that regulate protein synthesis by ligand binding and decrease or enhance translation efficiency [1].
Realization of genetic information in eukaryotes includes processing of RNA, its transport from nucleus to cytoplasm, translation and decay [2, 3]. There are regulatory elements in 5’- and 3’-UTRs that hasten decay of mRNA. Also UTR’s may contain stems which special proteins interact with leading to inhibition or initiation of translation [4]. Besides main ORF, mRNA may contain upstream ORF located in 5’-UTR that decrease efficiency of translation [5]. All these elements can regulate tissue-specific production of protein, fast response to stress or influence on development and progress of disease [6].
Therefore, it is important to identify regulatory sequences in mRNA. The frequent way to find regulatory elements is to compare the set of sequences, which harbor putative elements. Currently there is no useful tool for analysis of Saccharomyces cerevisiae 5’-UTRs. Our aim was to write program in order to get the set of 5’-UTRs of yeast genes and their orthologs.
Methods and Algorithms: We used Microsoft Visual Studio 2008 for writing program. The program was written in C# language for .NET Frameworker 3.5 with usage of Windows Workflow Foundation. To get the data about yeast genes we used Saccharomyces Genome Database (www.yeastgenome.org) and published data about length of UTR [7, 8]. The information about yeast gene orthologs was obtained from Princeton Protein Orthology Database
1 Saint-Petersburg State University, Russian Federation, [email protected]
2 Saint-Petersburg branch of Vavilov Institute of General Genetics RAS, Russian
(ppod.princeton.edu). To get the detailed data for other organisms we used WormBase (www.wormbase.org), FlyBase – A Database of Drosophila Genes
& Genomes (www.flybase.org), TAIR (www.arabidopsis.org), Mouse Genome Informatics (www.informatics.jax.org), Protein Knowledgebase (www.uniprot.org) and Homo sapiens genes (NCBI36). BioMart tool (www.ensembl.org/biomart/) was used for downloading human 5’-UTR sequences.
Results: We have designed the program UTRdbMaker for getting a set of 5’- UTRs. It obtains information corresponding to the gene names containing ORFs and 5’-UTRs sequences of yeast genes and their orthologs. UTRdbMaker analyses nucleotide composition of 5’-UTRs. Results of search are written in text files as tables and contain general descriptions of yeast genes. These results may be used for exploration of conservation of 5’-UTRs and for searching of regulatory elements in them. Code of UTRdbMaker can be extended for similar work with other regions or other databases.
1. W.C.Winkler et al. (2004) Control of gene expression by a natural metabolite-responsive ribozyme, Nature, 428: 281-286.
2. J.E.G.McCarthy (1998) Posttranscriptional Control of Gene Expression in Yeast, Microbiol. Mol. Biol. Reviews, 62: 1492-1553.
3. Ch.Dimaano et al. (2004) Nucleocytoplasmic Transport: Integrating mRNA Production and Turnover with Export through the Nuclear Pore, Mol. Cell. Biol, 24: 3069-3076.
4. A.M.Thomson et al. (1999) Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation, Int. J. Biochem. Cell Biol, 31:
1139-1152.
5. A.M.Resch et al. (2009) Evolution of alternative and constitutive regions of mammalian 5’UTRs, BMC Genomics, 10: 162.
6. J.T.Rogers et al. (2002) An iron-responsive element type II in the 5’- untranslated region of the Alzheimer’s amyloid precursor protein transcript, J.Biol.Chem. 277: 45518-45528.
7. F.Miura et al. (2006) A large-scale full-length cDNA analysis to explore the budding yeast transcriptome, PNAS, 103:17846-17851.
8. Z.Xu et al. (2009) Bidirectional promoters generate pervasive transcription in yeast, Nature, 457: 1033-1037.
July 20–23, 2009
A TRUSTY KNOWLEDGE-BASED POTENTIAL ENERGY BASED ON PAIRWISE RESIDUE CONTACT AREA
SEYED SHAHRIAR ARAB1, ARMITA SHEARI1, MEHDI SADEGHI2, CHANGIZ ESLAHCHI3, HAMID PEZESHK4
Keywords: Knowledge-based potential, decoy sets, protein structure prediction, protein folding
We develop a new approach to calculate a knowledge-based mean-force based on pairwise residue contact area. To test its effectiveness, we elaborate it on several decoy sets to measure its ability to discriminate native structure from decoys. In all cases this potential has been able to distinguish native structures from the decoys with about 100% accuracy. Also calculated Z-score shows high value for all protein datasets. This knowledge-based mean force can discriminate native structures from the decoys effectively, so it will be useful for protein structure prediction and model refinement.
Considering energy function to detect a correct protein fold from incorrect ones is very important for protein structure prediction and protein folding.
Mainly, two different types of potential energy function are currently in use either on the identification of native protein models from a large set of decoys or protein fold recognition and threading studies. The first class of potentials, the so-called physical-based potential, is based on the fundamental analysis of the forces between the particles referred to as physical energy function. The second type is knowledge-based energy function based on information from known protein structures. In physical energy function, a molecular mechanics force field is used. Molecular mechanics force fields are parameterized from ab initio calculation and small molecule structural data. They are essentially the sum of pairwise electrostatic and Van der Waals interaction energies, bonds, angles and dihedral angle terms. In addition, terms that are not included such as entropy and solvent effect are implicitly considered. Although, physical
1 Department of Bioinformatics, Institute of Biochemistry and Biophysics, University of Tehran, Iran, [email protected], [email protected]
2 National Institute of Genetic Engineering and Biotechnology, Tehran-Karaj Highway, Tehran, Iran, [email protected]
3 Department of Mathematical Sciences, Shahid Beheshti University, Tehran Iran, [email protected]
4 School of Computer Science, Institute for Studies in Theoretical Physics and
energy function is widely used in molecular dynamic simulation of proteins in their native and denatured states and can be used to distinguish the decoy and native structures, but these functions have not been efficient in protein structure prediction because of their greater computational cost. To reduce computational complexity of the protein folding problem, knowledge-based or empirical mean-force potential is widely used. Since the structure of folded proteins reflects the free energy of the interaction of all their components, including all enthalpic and entropic contribution, as well as solvent effects, such potentials provide an excellent shortcut towards a powerful objective function. It can be used to force the system to obtain potential between groups of atoms by use of experimentally determined structures. In this approach, statistical thermodynamics is used in an analysis of the frequency of observed states to estimate the underlying free energy. Most often, the distribution of pairwise distances are used to extract a set of effective potential between residues or atoms. The distribution of pairwise distances can be compiled from the protein structure database and by defining a reference state, Boltzman's Law is used to calculate the interaction energy of a particular pair.
The total potential energy of a protein is simply taken as a sum over all pairwise interactions. In most cases, one or two points for each residue are used to represent a protein. These points are usually C(alpha), C(beta) or the center of mass of each side chain. Each interaction can be distance – dependent. A large variety of knowledge-based potential of mean-force have been developed by introducing additional interactions such as surface area terms, the main chain and side chain dihedral angles, three and four body terms and heavy atoms. In the contact potential, either distance – dependent or only dependent on contact, the distance between the centers of two C(alpha), C(beta) or center of mass of two residues or the all heavy atoms of two residues are calculated and the observed frequency of contacts between residues converts to free energy using Boltzman’s equation. In this way, there is some problems that distance between two C(alfa) Atoms of two residues may be equal to the distance of two atoms of these residues in another position, but the orientation of two residue side chains may be quite different and they are considered as the pairs with equal pairwise distance. In other words, the side chains of two atoms may not have direct contact with each other and some atoms may be located in internal of the space. In this study, we develop a new approach to calculate a knowledge-based potential energy based on pairwise residue contact area. We calculated the parts of each pairwise residue surface area that are in contact in Å2 by rolling a probe ball
July 20–23, 2009
of different sizes around the atoms of a residue to determine the direct contacts surface area of each pair. This pairwise direct contact area, was used to determine statistical contact area preference between each residue pairs, when a contact area preference estimates a sum of energetic interaction and a structural constraint. A good energy function at its minimum should discriminate native structures from decoys. So, to test the effectiveness of this new potential, we elaborated it on several decoy sets to measure its ability to discriminate native structure from decoys. Several decoy sets that contain one to hundreds of decoy proteins generated in different ways were used and in all cases this potential has been able to distinguish native structures from the decoys with about 100% accuracy. Calculated Z-score, which is a useful measure of the validity of the computed potential, shows high value for all protein datasets. The knowledge-based mean force pairwise direct contact area can discriminate effectively, so it will be useful for protein structure prediction and model refinement.
EVOLUTIONARY DYNAMICS OF CRISPR-CASSETTES
VALERY SOROKIN1, IRENA ARTAMONOVA2
Keywords: prokaryotic immunity, CRISPR-cassettes, metagenome, evolution
CRISPRs, Regularly Interspaced Short Palindromic Repeats, are a new type of prokaryotic anti-phage immunity systems. A typical CRISPR system consists of a CRISPR-cassette that is a chain of almost identical repeats separated by unique spacers, a leader region, and CRISPR-associated genes [1].
Analysis of the CRISPR-systems was performed in metagenomic sequence data. There are no efficient tools for CRISPR-cassette search, since, when applied to metagenomes, all three publicly available programs, CRT [2], PILER-CR [3], and CRISPRFinder [4], produce high levels of false positive noise. Thus, to search for CRISPR-cassettes in metagenomes we developed a filtering procedure based on a combination of these three programs.
This procedure was applied to the Sorcerer II [5] metagenome data, resulting in 192 reliable cassettes. All cassettes found by at least one of the three tools were collected in a database called MeCRISPR (http://iitp.bioinf.fbb.msu.ru/vsorokin/crispr). The database interface allows browsing and analyzing pre-calculated CRISPR-cassettes and their flanking sequences; in particular, to search against spacers, repeats and metagenomic contigs containing at least one CRISPR cassette.
We clustered CRISPR-cassettes based on similarity between repeat units.
Additional analysis of flanking regions allowed us to distinguish between the lateral transfer and the parallel evolution of cassettes in related strains. For every group of homologous cassettes, we reconstructed the evolutionary history.
We observed that similarities representing phage-related spacers or lateral transfers of cassettes were significantly enriched in metagenome contigs from same geographical locations. This shows that on-going phage-host encounters of specific ocean locations involve the CRISPR-mediated response and imprint the host genome.
1 M.V. Lomonosov Moscow State University, Russian Federation, [email protected]
2 Vavilov Institute of General genetics RAS; Kharkevich Institute of Information Transmission Problems RAS, Russian Federation, [email protected]
July 20–23, 2009
We also investigated CRISPR-cassettes in close strains of Xanthomonas oryzae. The attempt to construct an experimental system for studying CRISPR systems failed because of the unresolved paradox in two strains Xo604 and Xo21. A shared spacer of homologous CRISPR-cassettes of these strains is identical to the Xp10 phage and should, theoretically, prevent the phage infection in both cases. However while Xo21 is indeed resistant for this phage, the Xo604 strain is sensitive. We explained it by identifying a mutation in the phage regulatory motif, discovered for the Xanthomonas cassettes. The comparative analyses of all known CRISPR-cassettes of Xanthomonas oryzae (five strains) will be presented.
This is joint work with Mikhail S. Gelfand, Konstantin V. Severinov, Mikhail A. Pyatnitskiy, Ekaterina Semenova and Maxin Nagronykh. This work was partially supported by the Russian Foundation of Basic Research (09-04- 01098-a) and the Russian Academy of Sciences (programs “Molecular and Cellular Biology” and “Fundamental problems of Oceanology”).
1. R. Sorek et al. (2008) CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea, Nat Rev Microbiol., 6:181-186.
2. C. Bland et al. (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats, BMC Bioinformatics. 8: 209.
3. R.C. Edgar (2007) PILER-CR: fast and accurate identification of CRISPR repeats, BMC Bioinformatics. 8: 18.
4. I. Grissa et al. (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats, Nucleic Acids Res., 35:
W52-7.
5. D.B. Rusch et al. (2007) The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific, PLoS Biol., 5: e77.
INVESTIGATING BRANCH POINT SITE CONSENSUS OF HUMAN
FEDOR GONCHAROV1, VLADIMIR BABENKO2
Splicing is commonly recognized as one of the ultimate regulation stages of gene expression. In particular, alternative splicing (AS) is a widespread mechanism with an important role in generating appropriate tissue and/or stage specific product from the same gene. On the other hand, one of the key binding sites in the course of spliceosome assembly, namely branch point site (BPS) is drastically degenerate in mammals in contrast to intron poor organisms, e.g. yeast (Gao et al., 2008).
We explored the 30bp branch point region sequences [-50, -20] relative to 3’ splice site from 28156 human introns. For analysis we built up the maximum parsimony tree for 7-mers taking into account the pairwise correlation values of the positions in the7-mers occurrence distribution. We got several resulting points after analysis:
There are several major branch point site consensi in human that supposes BPs heterogeneity.
1. The most abundant human BPS is represented by ACTGACG oligonucleotide which is consistent with (Irimia, Roy, 2008) and differs from , e.g. yeast (TACTAAC)
2. The human U2 RNP can bind to mRNA BPS not by canonical GTAGTA site, but in significant number of cases by IIa loop (Pomeratz et al., 2009), which is confirmed with extensive ATTAAAC representation as BPS in human (Henscheid, Voelker, Berglund, 2008).
3. The BPs sequence depends on the intron length, so it is closer to canonical in small to moderate introns.
4. Cassette exon –related BPS 3’ downstream possess significantly lower BPS strength (more mismatches from major consensi) than obligatory exons (p<1e-8).
In metazoan cells the increasing tissue specific complexity leads to multistage gene regulation in the course of replication, transcription and posttranscriptional phases. It was shown (IrFimia, Roy) that intron rich organisms usually belong to the top hierarchical clade of the organization complexity tree. We believe that branch point redundancy comes as the part of
1Institute of Cytology and Genetics, Russian Federation, [email protected]
2 Institute of Cytology and Genetics, Russian Federation, [email protected]
July 20–23, 2009
AS regulation evolution. In particular, strong BPS don’t allow for cis- regulatory element to affect splicing, so BPS of the canonical type could be referred to as Intronic Splicing Enchancer (ISE). On the contrary, regulated exons lack strong BPs signal apparently due to regulation.
1. Irimia M, Roy SW. Evolutionary convergence on highly-conserved 3' intron structures in intron-poor eukaryotes and insights into the ancestral eukaryotic genome. PLoS Genet. 2008. 4(8):e100014
2. Gao K, Masuda A, Matsuura T, Ohno K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 2008.36(7):2257-6
3. Henscheid KL, Voelker RB, Berglund JA. Alternative modes of binding by U2AF65 at the polypyrimidine tract. Biochemistry. 2008. 47(1):449-59.
4. Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature.
2009. 458(7237):475-80.
GLAUCOMA AND MYOPIA WHOLE GENOME ASSOCIATION STUDY
VLADIMIR BABENKO1, MARINA GUBINA1, IGOR KULIKOV1, RUSLAN AITNASAROV1
Keywords: Illumina 550, SNP analysis, glaucoma, myopia,
40 individuals were genotyped with the Illumina 550 snp array (Illumina, Inc., http://illumina.com) at the “Bioingineering” Center, RAS, Russia. The data comprises 27 healthy individuals, 5 patients with glaucoma and 8 ones with myopia diagnosis. All individuals are Caucasians from Novosibirsk urban region, Russia. The total SNP volume comprises more than 340 thousand SNPs We implemented sql database schema designed by us for maintenance of the sample and a software suite to analyze it.
Results.
We identified 44 target SNPs while analyzing 11 normal and 13 disorder cases where discrepancy between control and affected samples set was more than empirically chosen significant threshold of 9 genotypes. Using haploview software suite (www.hapmap.org) we selected 28 non-redundant unlinked SNPs. Next we scanned OMIM database (www.ncbi.nlm.nih.gov/omim) for the genes comprising the target SNP set. There we identified 5 genes with
‘glaucoma’ and ‘myopia’ as keywords, namely: myocilin (MYOC), optineurin (OPTN), cytochrome P450 family 1 subfamily B (CYP1B1), optic atrophy 1 isoform 8 (OPA1), WD repeat domain 36 (WDR36).
The gene OPA1 (optical atrophy, chrom 3) significantly associated with target SNPs is located within recently identified cluster of genes (MFN1, SOX2OT and PSARL, Andrew T et al., Plos Genetics, 2008), and proved to be associated with myopia. We thus reconfirm the impact of this gene on myopia in ethnic population considered.
1 Institute of Cytology and Genetics, Russian Federation, [email protected]