Universität für Bodenkultur Wien
University of Natural Resources and Life Sciences, Vienna
UNIVERSITY OF NATURAL RESOURCES AND APPLIED LIFE SCIENCES, VIENNA
Department of Food Sciences and Technologies Institute of Food Biotechnology
Master Thesis
Production and Characterization of Several Oxidases for Pentosan- Crosslinking
(as a part of the PentoGlu Project)
To acquire the academic degree
Master of Science (MSc.)
Eva Vojta, BSc
This part of the PentoGlu Project was carried out at the Institute of Food Biotechnology and supervised by:
Univ. Prof. Dipl.-Ing. Dr. techn. Dietmar Haltrich Dipl.-Ing. Dr. Roman Kittl
April 2015 – November 2015
"Broad, wholesome, charitable views of men and things cannot be acquired by vegetating in one little corner of the earth all one's lifetime."
Mark Twain
Acknowledgement
This work would have been impossible to accomplish without quite a number of people...
Therefore, I would like to thank first and foremost my main supervisor Roman Kittl, who overviewed the whole project, checked on the fermenter with me even late at night and had an answer to any of my questions. Next, I would like to give credit to Regina Paukner, who kept an eye on my practical work, explained every procedural step to me in detail, with patience and kindness, and who literally wracked her brain to solve some of the problems with me.
Furthermore I want to thank my supervisors from the Working Group Food Technology; Stefano, whose door was always open, allowing me to stop by for a brief discussion at any moment, and Denisse, who still had a smile on her face even during the most stressful time and strongly supported me regarding the rheological measurements. I would also like to thank Cindy, the good spirit of the Food Biotechnology laboratory, who closely watches over the labs tidiness and without whom nothing would work out as it should. I had fantastic lab mates from Austria, Croatia (at this point special thanks to my hilariously sarcastic fellow graduate students Nenad and Anita), Vietnam, Thailand, and Ireland. Thanks for the highly amusing and constructive lunch breaks to Roman, Wolfgang and Elena. I wish all of them a successful scientific career and a full life, and I am very happy that our time at BOKU overlapped. I truly appreciate that the Institute of Food Technology and its head Dietmar Haltrich provided me with all necessary resources and material, and that they allowed me to work with such great equipment and in a relaxed working atmosphere. Above all, I thank my parents for financial and emotional support, for not frequently pressuring me about completion dates, and for their sincere interest in my work. Last, but not least, my boyfriend deserves a medal of honor for providing me with delicious sushi-lunchboxes and for having an open ear for anything and everything.
Abbreviations
2,6-DMP 2,6-dimethoxyphenol
ABTS 2,2’-Azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) AnGOx Aspergillus niger glucose oxidase
AX Arabinoxylan
BaLac Botrytis aclada laccase BliLac Bacillus licheniformis laccase
Cgl1 Corynebacterium glutamicum laccase FA ferulic acid
HIC Hydrophobic interaction chromatography IMAC Immobilized metal ion affinity chromatography IEX Ion exchange chromatography
LAB lactic acid bacteria P2O pyranose 2-oxidase
PcPOx Phanerochaete chrysosporium pyranose 2-oxidase PCR polymerase chain reaction
PnGOx Penicillium notatum glucose oxidase PMSF phenylmethylsulfonyl fluoride
RO H2O reverse osmosis filtered water
TmP2O Trametes multicolor pyranose 2-oxidase UHQ H2O ultra high quality water
WEAX water-extractable arabinoxylan WUAX water-unextractable arabinoxylan
Abstract
The challenge with baking gluten-free bread is to deal with the absence of gluten as the most important structure-building protein in dough and to find an appropriate alternative to it.
According to various publications laccase, glucose 1-oxidase and pyranose 2-oxidase are able to cross-link the polysaccharide arabinoxylan, resulting in a network in gluten-free bread that has functional properties similar to gluten protein. Several suitable enzymes were selected to study their crosslinking ability. Bacterial laccase from Corynebacterium glutamicum (Cgl1) and glucose 1-oxidase from the fungus Penicillium notatum (PnGOx) were recombinantly expressed. The expression of Cgl1 in E. coli was not successful, the fermentation of the yeast Pichia pastoris transformed with a vector carrying the PnGOx gene for overexpression resulted in 2985 ml of supernatant with 2.0 U/mL PnGOx activity. Additionally, the following enzymes were chosen for rheological characterization: Botrytis aclada laccase (BaLac), Phanerochaete chrysosporium pyranose 2-oxidase (PcPOx), OxyPlus cellobiose dehydrogenase (CDH Oxyplus) and bacterial laccase from Bacillus licheniformis (BliLac double mutant).
The rheological measurements of the crosslinking of arabinoxylan by the enzymes were carried out by recording the increase in viscosity and the characteristic curves of storage modulus and loss modulus by means of a rheometer. PcPOx, BaLac and BliLac showed a positive crosslinking effect. Based on the results of the preliminary rheological experiments, 11742 U of PcPOx were produced in a 30 L fermentation and 71722 U of BaLac in a 10 L fermentation for subsequent baking tests. Purified BaLac has a specific activity of 69.3 U / mg, purified PcPOx one of 3.7 U / mg.
In addition, recombinant PcPOx was biochemically characterized; its temperature optimum is 60°C at pH 5.5, also PcPOx still has more than 50% of the original activity after 24 hours of incubation at this temperature. Even after 26 days of incubation at 28°C no loss of activity was found.
Abstract
Die Herausforderung beim Backen von glutenfreiem Brot ist das Fehlen von Gluten, dem wichtigsten strukturgebenden Protein in Teig, und daher das Auffinden einer geeigneten Alternative zu Gluten. Laccase, Glucose 1-Oxidase und Pyranose 2-Oxidase sind laut diversen Publikationen in der Lage das Polysaccharid Arabinoxylan so querzuvernetzen, dass das resultierende Netzwerk funktionelle Eigenschaften ähnlich denen des Glutenproteins hat.
Mehrere geeignete Enzyme wurden gewählt, um ihre Fähigkeit zur Quervernetzung zu ermitteln.
Bakterielle Laccase von C. glutamicum (Cgl1) und Glucose 1-Oxidase aus dem Pilz P. notatum (PnGOx) wurden rekombinant exprimiert. Die Expression von Cgl1 in E. coli war nicht erfolgreich. Die Fermentation von PnGOx in P. pastoris ergab 2985 mL Überstand mit 2.0 U/mL PnGOx-Aktivität. Zusätzlich wurden folgende Enzyme zur rheologischen Charakterisierung ausgewählt: P. chrysosporium Pyranose 2-oxidase (PcPOx), B. aclada Laccase (BaLac), OxyPlus Cellobiose-Dehydrogenase und bakterielle B. licheniformis Laccase (BliLac, Doppelmutante).
Die rheologische Messung der enzymatischen Arabinoxylan-Quervernetzung erfolgte durch Aufzeichnung des Anstiegs der Viskosität und des Verlaufs von Speichermodul und Verlustmodul mittels Rheometer. Einen positiven Quervernetzungseffekt zeigten dabei PcPOx, BaLac und BliLac. Auf Basis der Ergebnisse dieses Vorversuchs wurden anschließend 11742 U PcPOx in einer 30L-Fermentation und 71722 U BaLac in einer 10L-Fermentation für nachfolgende Backversuche produziert. Gereinigte BaLac weist eine spezifische Aktivität von 69.3 U/mg auf, gereinigte PcPOx eine von 3.7 U/mg.
Zusätzlich wurde rekombinante PcPOx biochemisch charakterisiert; ihr Temperaturoptimum liegt bei pH 5.5 bei 60°C, auch ist nach 24-stündiger Inkubation bei 60°C noch mehr als 50% der ursprünglichen Aktivität vorhanden. Sogar nach 26-tägiger Inkubation bei 28°C wurde kein Aktivitätsverlust festgestellt.
Table of contents
1 Introduction ... 1
2 Aims of the project ... 2
3 Background ... 4
3.1 Gluten free bread – The challenge ... 4
3.2 Sourdough – The enzymes working environment ... 4
3.3 Enzymes as additive in industrial bread-making ... 5
3.4 Pentosans (Arabinoxylans) ... 6
3.5 Enzymatic cross-linking of pentosans ... 6
3.6 Selection of enzymes from Literature ... 8
3.7 Enzymes – Biochemical characterization ... 10
3.8 Oxidases ... 10
3.8.1 Pyranose oxidases ... 10
3.8.2 Laccase ... 13
3.8.3 Cellobiose Dehydrogenase ... 17
3.9 Protein production and expression systems ... 19
3.9.1 Molecular cloning process ... 19
3.9.2 Expression hosts ... 20
3.9.3 Cloning vectors ... 21
3.10 Rheometry and characterization of viscoelastic behavior of a solution ... 24
4 Material and Methods ... 26
4.1 Material ... 26
4.1.1 Biological material ... 26
4.1.2 Chemicals ... 26
4.1.3 Media ... 26
4.1.4 Antibiotics ... 26
4.1.5 Media for E. coli cultivation ... 27
4.1.6 Media for P. pastoris cultivation ... 28
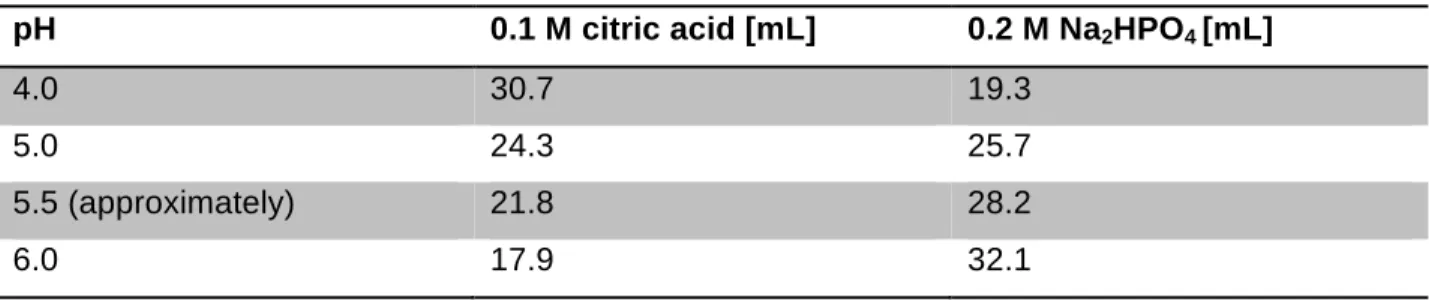
4.1.7 Buffers ... 29
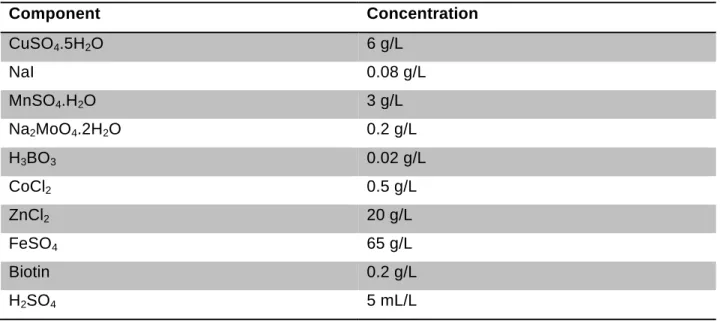
4.1.8 Other Solutions ... 31
4.1.9 Vectors ... 33
4.1.10 Primers ... 33
4.1.11 Genes ... 34
4.1.12 Enzymes ... 35
4.1.13 Kits ... 35
4.1.14 Equipment ... 36
4.2 Methods ... 37
4.2.1 Cultivation methods ... 37
4.2.2 Cloning methods ... 38
4.2.3 Protein expression and purification ... 43
4.2.4 Characterization of Enzymes ... 49
5 Results and Discussion ... 53
5.1 Expression of Cgl1 ... 53
5.1.1 Preliminary activity screening tests ... 53
5.2 Expression and purification of PnGOx ... 53
5.2.1 Preliminary activity screening tests ... 53
5.2.2 Expression on a medium scale for preliminary rheological tests ... 55
5.3 Expression and purification of PcPOx ... 59
5.3.1 Expression on a medium scale for preliminary rheological tests ... 59
5.3.2 Expression on a large scale for constructive rheological tests and baking trials ... 60
5.4 Expression and purification of BaLac ... 62
5.4.1 Expression on a large scale for constructive rheological tests and baking trials ... 62
5.5 Biochemical Characterization of PcPOx ... 67
5.5.1 Temperature optimum of recombinant PcPOx at pH 5.5 ... 67
5.6 Rheological Characterization ... 70
5.6.1 PnGOx ... 71
5.6.2 PcPOx ... 73
5.6.3 BaLac ... 74
5.6.4 BliLac ... 75
5.6.5 CDH OxyPlus ... 76
6 Conclusions and Outlook ... 78
7 Appendix ... 81
7.1 Sequences ordered by Invitrogen, GeneArt® Gene Synthesis service ... 81
7.2 Activity correction factors ... 83
8 Literature ... 86
9 List of Figures ... 93
10 List of Tables ... 94
1 Introduction
Coeliac disease is a genetically determined chronic inflammatory intestinal disease induced by gluten, the storage protein of wheat. Its treatment is based on a strict gluten-free diet for life, which implies total avoidance of wheat, barley and rye. Approximately one out of 250 people is affected by coeliac disease (Green et al, 2003), which points out the necessity of innovative approaches for the production of gluten-free products.
Pseudocereals like amaranth, quinoa and buckwheat may serve as alternative raw materials in the production of gluten-free products. Apart from dietary treatment of coeliac patients, pseudocereals are known to improve the nutritional balance of gluten-free food as they contain high quality protein and significant amounts of fibre, minerals such as calcium and iron and polyphenols. (Alvarez-Jubete et al., 2009; Alvarez-Jubete et al., 2010)
The challenge with gluten-free bread based on pseudocereals is now to deal with the absence of gluten as the most important structure-building protein in dough. The covalent structure of a gluten network is mainly formed by bonds in and between gluten proteins and is crucial for the baking quality of wheat and its unique rheological dough properties, water absorption capacity, viscosity, elasticity and the gas retention during fermentation. (Wieser, 2007). A cereal biopolymer that can be treated in a way to serve as an alternative to gluten is the polysaccharide pentosan. It consists of an arabinoxylan-backbone, to which ferulic acid is linked by ester linkage (Izydorczyk et al., 1995). Prior to its addition to dough, pentosan has to be extracted from raw material, for instance from rye. In dough, pentosan chains can be oxidatively cross-linked to each other by dimerization of ferulic acid residues in order to form a network. This kind of cross- linkage can be performed by oxidizing enzymes like laccase (Figueroa-Espinoza et al, 1998), glucose oxidase und pyranose oxidase (Decamps et al, 2013).
In the course of the present study several oxidases are produced for cross-linking of ferulic acid residues of rye pentosans in gluten-free dough. Similar research has so far only been conducted with a focus on gluten containing wheat flour, and gluten free oat flour respectively (Flander et al, 2011; Renzetti et al, 2010).
The pentosan-cross-linking effect of enzymes has so far only been studied for commercially available enzymes from a few organisms; therefore studies like the following on more efficient
enzymes are necessary to subsequently optimize the process and to reduce the costs of enzyme application for this purpose.
2 Aims of the project
This work is performed as a part of the PentoGlu Project at the Institute of Food Biotechnology in cooperation with the Institute of Food Technology. The overall aim of the PentoGlu project is to imitate the dough formation of rye bread to optimize the texture and quality of gluten-free bread;
for this purpose, bread dough made from pseudocereals and millet/sorghum is supplemented with pentosans whose isolation from rye bran has been optimized before. To improve dough properties, such as higher viscosity of the dough, sourdough technology is applied at the same time as certain enzymes are added. The latter are supposed to cross-link pentosans and thus to build a stable hemicellulose-network.
The aim of this part of the PentoGlu project in particular is first of all to identify enzymes from previous research whose optimal operating conditions correspond to the reaction conditions in sourdough – low pH and temperature - and whose analysis and production is realizable with the available resources. Using molecular biological and microbiological methods, the selected enzymes are then produced in smaller amounts that are sufficient for characterization. In the next step, the selected enzymes are characterized on the one hand biochemically –if relevant properties have not been published yet- while considering their proposed working conditions. On the other hand they are characterized rheologically in terms of their ability to cross-link water- extractable arabinoxylan and therefore to increase the viscosity of an arabinoxylan-standard- solution. Rheological characterization is supposed to give an idea of each enzyme’s effectiveness in dough improvement and also of the enzyme quantity needed for this same effect. Finally, the effective enzymes are produced in larger quantities that are required for subsequent baking trials at the Institute of Food Technology in Vienna.
Figure 1 Chronological course of this project
3 Background
3.1 Gluten free bread – The challenge
According to the COMMISSION REGULATION (EC) No 41/2009, the term ‘gluten-free’ on the labeling is reserved to products whose content of gluten does not exceed 20 mg/kg. ‘Gluten’ is defined as a protein fraction from wheat, rye, barley, oats or their crossbred varieties and derivatives thereof, to which some persons are intolerant. It is stated that oats can be included in gluten-free diet by most intolerant people, but its role still has to be further investigated.
However, the major concern is the contamination of oats with wheat, rye or barley that can occur during grain harvesting, transport, storage and processing. Gluten is composed of the protein fractions gliadin and glutenin, which give in combination cohesive, elastic and viscous properties to the gluten network. If the gluten network is missing, doughs are rather liquid and lack in appealing texture, colour and other quality characteristics of bread. An approach for the improvement of gluten-free product formulations is the use of starch and hydrocolloids like xanthan or guar gum. (Gallagher et al., 2004)
3.2 Sourdough – The enzymes working environment
Sourdough technology can be applied to compensate textural and sensoric deficiencies of gluten-free dough based on pseudocereals. Usually, for this purpose flour and water are fermented with lactic acid bacteria and yeasts. The positive effects of this process on gluten-free bread include e.g. improved gas retention, dough softening and improved mineral bioavailability.
Furthermore, the production of prebiotic exopolysaccharides from LAB is supposed to replace more expensive hydrocolloids that are usually added as bread improvers. Also, the drop in pH - due to the production of organic acid - leads to an increase in protease and amylase activity and therefore to a reduction of bread staling. The pH of sourdough varies with the nature of the process and starter culture. Usually 20 % sourdough is added to bread dough; the pH of this sourdough-supplemented bread dough has been reported to range from 4.7 to 5.4. (Arendt et al., 2008; Moroni et al., 2009) The use of sourdough technology thus defines the reaction conditions of the enzymes that are used for pentosan cross-linking: since the enzymes are added to the sourdough preparation and are supposed to work during sourdough fermentation, the enzymes have to be sufficiently active at a pH varying from 5 to 6 and 30°C for at least 2 hours (intended conditions of sourdough fermentation in the further course of the project).
3.3 Enzymes as additive in industrial bread-making
Almost one third of the enzymes that are applied in food are used in the production of bread.
The following enzymes are currently most commonly used in the baking industry:
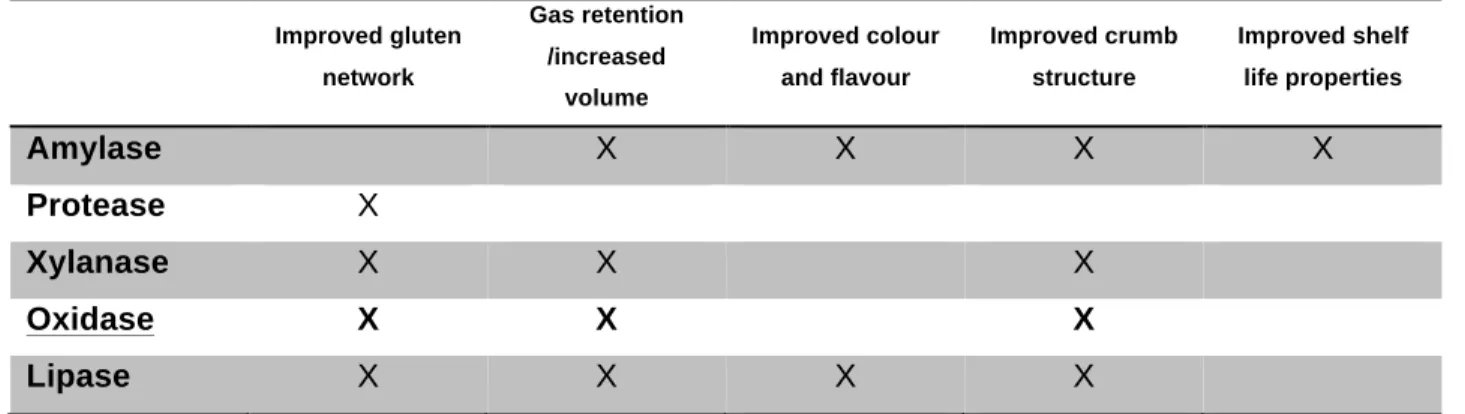
Table 1 General effect of enzymes in bread making (Whitehurst & van Oort, 2010)
Improved gluten network
Gas retention /increased
volume
Improved colour and flavour
Improved crumb structure
Improved shelf life properties
Amylase X X X X
Protease X
Xylanase X X X
Oxidase X X X
Lipase X X X X
Among them, amylase is the most commonly used enzyme in bread making. Amylases, also endoglucanases, hydrolyze starch to create low molecular weight sugars (small dextrins, maltose and glucose). Glucose and maltose reinforce the browning effect during the baking process. The addition of amylases also leads to a reduction in dough viscosity during starch gelatinization. The mechanism of xylanases is not yet fully understood, but the effect is based on reducing the size and thus water binding of water-unextractable and water-extractable arabinoxylan. Phospholipases basically replace emulsifiers in dough, but the precise effect remains unclear. Proteases contribute among others to flavour development by releasing amino acids from protein. (Whitehurst & Van Oort, 2010) Oxidases are relevant for the present thesis.
From the group of oxidases especially glucose oxidase has so far been commercially used; one of its purposes in dough is the production of H2O2 in order to oxidize the sulfhydryl groups of the gluten protein to disulfide bonds. The latter leads to an increase of the gluten networks strength.
Therefore, glucose-oxidase is a safe alternative to the previously used oxidizing agent potassium bromide (KBrO3), which was classified as a carcinogen and banned in most countries. Moreover, glucose-oxidase is the reason for gel formation by H2O2-induced crosslinking of water-soluble pentosans. The closely related pyranose 2-oxidase was found to have the same effects as glucose-oxidase, but its application in food is still rare. (Decamps et al., 2013) Laccases also belong to the group of oxidases and are commercially available. Still, they are not designated to be used in the baking industry, but rather for the production of juice and beer.
3.4 Pentosans (Arabinoxylans)
Pentosans are hemicelluloses and main components of the endosperm cell wall of various cereal grains. Relating to the whole grain, wheat contains 4-9% pentosan and rye 8-12.1%, however, wheat and rye cell walls consist of 60-70% pentosan (and those of barley of 25%).
Chemically, they are defined as arabinoxylans because they have a linear backbone of (1à4) - linked beta-D-xylopyranose with alpha-L-arabinofuranose residues at positions O2 and/or O3.
Figure 2 Structure of arabinoxylan (Cui, 2005)
The arabinose content of arabinoxylans varies and has strong influence on its physicochemical properties. Furthermore, ferulic acid is covalently attached to the C5 position of arabinose by ester linkage, but only at a low amount: WEAX contain 0.2 to 0.4% ferulic acid, WUAX 0.6 to 0.9% (i.e. there are 2 to 4 or 6 to 9 molecules ferulic acid per 1000 xylose molecules). Ferulic acid is able to form dehydrodimers, which leads to crosslinking of arabinoxylan chains. Since ferulic acid has antioxidant activity, this property is transferred to arabinoxylan and arabinoxylan can therefore be considered as functional food. Arabinoxylans are divided into water-soluble and water-insoluble arabinoxylans, whose solubility depends on the ratio of arabinose to xylose, the degree of substitution and their molecular weight. WUAX is bound to other cell wall components (proteins, lignin), whereas WEAX is only loosely associated with the cell wall. It is known that viscosity and water absorption of wheat-based dough decreases if water-soluble arabinoxylans are added to dough. This leads to lower hardness and better plasticity of the finished bread. In addition, a reduced starch retrogradation was observed in wheat bread supplemented with arabinoxylan. (Rosicka-Kaczmarek et al., 2016)
3.5 Enzymatic cross-linking of pentosans
As an alternative to gluten network in bread, a pentosan network using various enzymes can be formed. The potential of laccase, glucose-oxidase and pyranose-oxidase to crosslink pentosans via dehydrodimers of ferulic acid residues has been reported repeatedly. Carvajal-Millan et al.
(2005) demonstrated the central role of ferulic acid in the gelation of WEAX and for structural and rheological properties of an arabinoxylan network. Decamps et al. proved in 2013 that cross-linking of arabinoxylans with glucose-oxidase and pyranose 2-oxidase is due the production of H2O2. In addition, they showed that the extent of cross-linking depends on the concentration or rate of production of H2O2. Irrespective of pyranose oxidases, it has been previously shown that the oxidative gelation of arabinoxylans is attributed to H2O2, which serves as acceptor of hydrogen atoms while ferulic acid works as their donor. (Whitaker, 1993) Figueiroa-Espinoza stated the potential of laccase to cross-link feruloylated WEAX and to form an arabinoxylan gel in 1998. The reaction mechanism is based on laccase catalyzing the dehydrogenation of the hydroxyl group at C4 of ferulic acid into a highly reactive phenoxy radical. It is possible that the phenoxy radical isomerizes into a radical with a trivalent carbon with an unpaired electron at C5 or C8. The phenoxy radical is finally able to dimerize and react with another radical to form C-C or C-O linkages. Table 2 gives an overview of the cross-linking mechanisms:
Table 2 Pentosan-crosslinking mechanism of relevant oxidases
Enzyme Reaction mechanism of enzyme Non-enzymatic dimerization Laccase
(EC 1.10.3.2)
Oxidation of phenols (here: ferulic acid) to radicals
Dimerization of ferulic acid radicals
Glucose oxidase (EC 1.1.3.4.) Pyranose oxidase
(EC 1.1.3.10.)
Oxidation of glucose or aldopyranose in conjunction with production of H2O2
H2O2 acts as hydrogen acceptor, ferulic acid as
hydrogen donor
In any case cross-linked arabinoxylan in baking implicates the advantage that it can bind much more water per gram of polymer than arabinoxylan that has not been cross-linked. (Vinkx &
Delcour, 1996)
3.6 Selection of enzymes from Literature
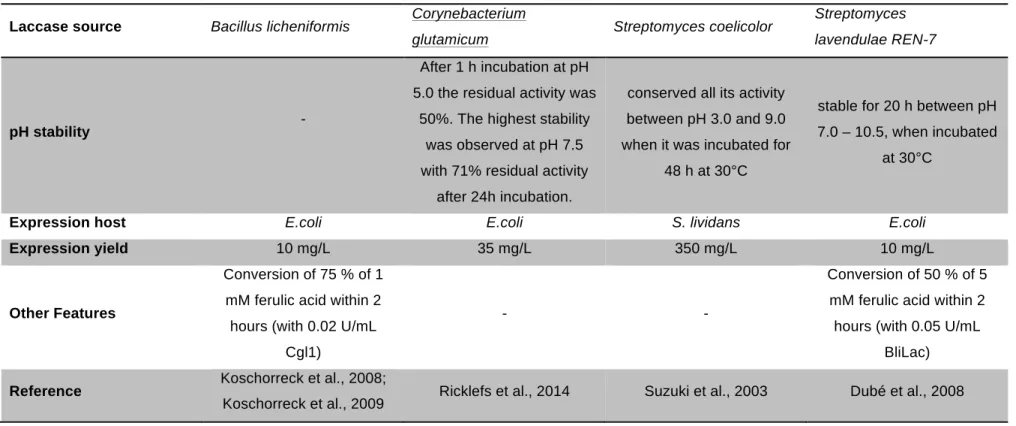
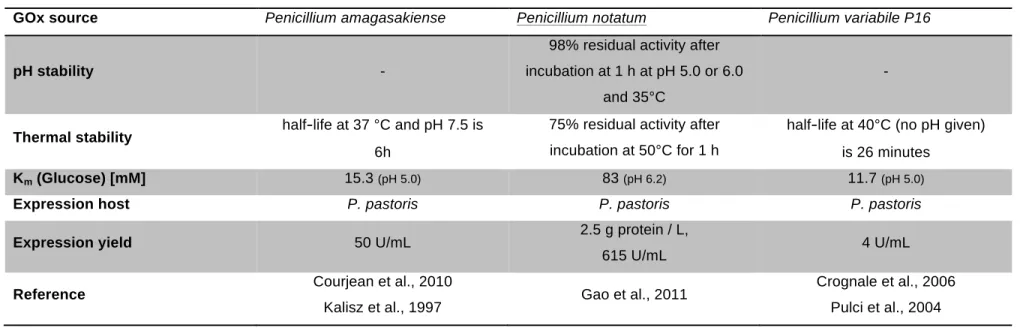
Prior to the practical work, two oxidases (laccase and glucose 1-oxidase) from relevant publications were selected, which seemed to be particularly suitable for cross-linking of rye pentosan besides the already fixed enzymes BaLac, PcPOx, and CDH Oxyplus. The focus was put on finding a suitable bacterial laccase and a glucose oxidase different from the commercially available and most investigated Aspergillus niger glucose oxidase. The enzymes were expected to be optimally active at pH 5 to 6 and 30°C, and to be producible in easy-to-handle host strains at high yields. Underlining in the table below highlights the finally selected enzymes.
Table 3 Comparison of Bacterial Laccases from Literature
Laccase source Bacillus licheniformis Corynebacterium
glutamicum Streptomyces coelicolor Streptomyces lavendulae REN-7
pH stability -
After 1 h incubation at pH 5.0 the residual activity was
50%. The highest stability was observed at pH 7.5 with 71% residual activity
after 24h incubation.
conserved all its activity between pH 3.0 and 9.0 when it was incubated for
48 h at 30°C
stable for 20 h between pH 7.0 – 10.5, when incubated
at 30°C
Expression host E.coli E.coli S. lividans E.coli
Expression yield 10 mg/L 35 mg/L 350 mg/L 10 mg/L
Other Features
Conversion of 75 % of 1 mM ferulic acid within 2 hours (with 0.02 U/mL
Cgl1)
- -
Conversion of 50 % of 5 mM ferulic acid within 2 hours (with 0.05 U/mL
BliLac)
Reference Koschorreck et al., 2008;
Koschorreck et al., 2009 Ricklefs et al., 2014 Suzuki et al., 2003 Dubé et al., 2008
Table 4 Comparison of Glucose Oxidases from Literature
GOx source Penicillium amagasakiense Penicillium notatum Penicillium variabile P16
pH stability -
98% residual activity after incubation at 1 h at pH 5.0 or 6.0
and 35°C
-
Thermal stability half-life at 37 °C and pH 7.5 is 6h
75% residual activity after incubation at 50°C for 1 h
half-life at 40°C (no pH given) is 26 minutes
Km (Glucose) [mM] 15.3 (pH 5.0) 83 (pH 6.2) 11.7 (pH 5.0)
Expression host P. pastoris P. pastoris P. pastoris
Expression yield 50 U/mL 2.5 g protein / L,
615 U/mL 4 U/mL
Reference Courjean et al., 2010
Kalisz et al., 1997 Gao et al., 2011 Crognale et al., 2006
Pulci et al., 2004
3.7 Enzymes – Biochemical characterization
The activity of an enzyme is heavily dependent on external conditions, such as temperature, pH and ionic strength of the surrounding medium. pH affects enzyme activity in two different ways:
Reactive groups such as carboxyl, amino and thiol groups in the catalytic center suffer from protonation or deprotonation as pH changes and hence vary between active and inactive state.
In addition, large changes in pH damage the three-dimensional protein structure irreversibly. For enzymatic assays usually the optimum pH is set as the standard pH-value, which is for most enzymes situated in the physiological range around 7.5. Regarding influence of temperature, more and more protein is denatured with increasing temperature. The highest levels of activity may be measured at the temperature optimum, but at the same time denaturation is well advanced. The enzyme molecule, which is normally in its native active state N, usually is in equilibrium with the partially denatured, enzymatically inactive U state. The temperature at which N = U is called the melting temperature Tm of the enzyme. The Tm of enzymes lie in a range from 30°C to more than 100°C, but in aqueous solution is usually somewhere between 60 and 80°C.
(Bisswanger, 2015; Misset, O., & van Dijk, 1998)
3.8 Oxidases
3.8.1 Pyranose oxidases
Pyranose oxidases are flavoproteins as they contain flavin adenine dinucleotide (FAD) as a cofactor. Glucose 1-oxidase and pyranose 2-oxidase belong to this group of enzymes, and simultaneously to the superfamily of glucose-methanol-choline oxidases (GMC). They catalyze the oxidation of aldopyranoses to the corresponding keto-aldoses and hydrogen peroxide, with molecular oxygen serving as the electron acceptor. The first half reaction involves the reduction of the flavin cofactor, with a hydride equivalent being transferred from the pyranose to FAD. In the second half-reaction an oxidation of molecular oxygen to hydrogen peroxide takes place, in which two electrons are transferred from the reduced FAD to the molecular oxygen. (Hille et al., 2013) Homogeneous pyranose oxidase solutions are of bright yellow color and exhibit a typical flavoprotein spectrum with absorption maxima at 276, 373 and 456 nm. (Artolozaga et al., 1997) 3.8.1.1 Pyranose 2-oxidase
Pyranose 2-oxidase (EC 1.1.3.10., pyranose:oxygen 2-oxidoreductase) catalyses the oxidation of several aldopyranoses at the C2 position, using O2 as electron acceptor, to give the
corresponding 2-keto-aldoses and hydrogen peroxide. At first two electrons are transferred as a hydride equivalent from sugar molecule on FAD. In doing so, reduced FADH2 and the corresponding 2-keto-sugar are generated. Thereafter, two electrons are transferred from the reduced cofactor to O2, resulting in H2O2 formation. The overall reaction mechanism of this enzyme – as for most flavoprotein oxidoreductases – is classified as a ping-pong bi-bi mechanism.
Figure 3 Reaction catalyzed by P2O with D-glucose as a substrate (Hille et al., 2013)
Figure 4 Reductive (1) and oxidative (2) half-reactions of P2O (Hille et al., 2013)
P2O occurs in white-rot fungi, such as in the species Trametes versicolor or Phanerochaete chrysosporium, in which their natural role is supposed to be the production of H2O2. H2O2 is then serving as a substrate necessary for the lignin-degrading enzyme of the particular fungus. Some species need P2O to produce antibiotics like β-pyrone-cortalcerone from D-glucose via 2-keto- D-glucose, which protect the fungus from bacterial infestation. In biotechnology P2O is used for the biotransformation of D-glucose and D-galactose to the corresponding 2-keto-sugars, which are then further reduced to D-fructose and D-tagatose. Bio-fuel cells are in discussion as another potential area of application for P2O. (Hille et al., 2013) Most pyranose oxidases have a molecular weight of about 280 to 310 kDa and are composed of four identical subunits of 70 kDa each. (Artolozaga et al., 1997)
3.8.1.1.1 PcPOx
The native enzyme is derived from white rot fungus Phanerochaete chrysosporium, where its main function is to provide H2O2 from D-glucose for the ligninolytic system. Phylogenetic analysis of de Koker et al. (2004) revealed rather low genetic identity of PcPOx with POx from other organisms. PcPOx has the highest identity of 53% with Lyophyllum shimeji (LsPOx), followed by 41% genetic identity with POx from Peniophora subspecies (PsPOx) and Trametes ochracea (ToPOx). Unlike other pyranose 2-oxidases, PcPOx is not glycosylated. The substrate specificity is as broad as common for pyranose 2-oxidases, with the highest relative activity of
100% for D(+)-glucose (Km =1.09 ± 0.02 mM) (Artolozaga et al., 1997). Moreover, the relative activity amounts to 65% for D-glucono-delta-lactone and, which is quite unusual, to 17% for D- cellobiose. Artolozaga et al. (1997) state that both α- and β-anomers of D-Glucose can be utilized at almost identical rates. According to Artolozaga et al. (1997), native PcPOx is active within a range from pH 5.0 to pH 9.0, with an activity maximum at pH 8.0 to 8.5 and 55°C. The wide optimum pH range and the comparatively high temperature optimum for the oxidation of glucose are rather unusual features when compared to other pyranose 2-oxidase, as they use to have a relatively sharp pH optimum. Apart from that, Artolozaga. reported a size of 65 kDa per subunit of this homotetrameric enzyme, whereas de Koker et al. (2004) found it to be slightly larger with 70 kDa. The latter is also in compliance with the size prediction by the Expasy translation tool based on the cDNA sequence that was found by De Koker et al. in 2004. There is supposed to be only one single P2O gene in the genome. Their cDNA sequence (GenBank accession no. AY522922) was finally used by Pisanelli et al. (2009) to successfully express the protein in E. coli BL21 (DE3).
3.8.1.2 Glucose 1-oxidase
Glucose 1-oxidase (EC 1.1.3.4, β-D-glucose:oxygen 1-oxido-reductase) catalyzes the oxidation of D-glucose specifically at the C1-position to gluconolactone. At the same time the enzyme bound flavin cofactor is reduced. Subsequently, hydride equivalents from the reduced cofactor are transferred to molecular oxygen, and H2O2 is formed. The overall reaction mechanism is referred to as ping-pong mechanism.
Figure 5 Reaction catalyzed by glucose 1-oxidase (Hille et al., 2013)
Glucose oxidase can also use derivatives of D-glucose and alpha-hydroxycarbonyl compounds as substrates, such as dihydroxyacetone or glyceraldehyde. However, the oxidation of D- glucose is by far the most effective. Glucose oxidase can also use derivatives of D-glucose and alpha-hydroxycarbonyl compounds such as dihydroxyacetone, glyceraldehyde, etc., as substrates. However, the oxidation of D-glucose is by far the most effective. Particularly well characterized is glucose oxidase derived from the species Aspergillus niger and Penicillium amagasakiense, in fact as a glycosylated homodimer with one molecule of FAD non-covalently
bound per subunit. The molecular mass of the native enzyme varies from 130 to 325 kDa depending on the degree of glycosylation. In terms of application in industry, glucose oxidase is used in biosensors, such as in glucose-sensing electrodes. (Hille et al., 2013)
3.8.1.2.1 PnGOx
In the 1940ies, glucose oxidase from Penicillium notatum was originally referred to as thermolabile antimicrobial "notatin", or "penicillin B" due to its bactericidal effect. Keiling and Hartree first described the biochemical properties of PnGOx in 1948. They noted a pH optimum of 5.6 and a Km for glucose of 4.2 mM, with glucose being the clearly preferred substrate. Apart from that only mannose and xylose showed 1% of the rate of glucose oxidation. Since then, there were only few further studies on this enzyme. As, according to Houbraken et al. (2011), Penicillium notatum is a synonym of Penicillium chrysogenum, the work of Leiter et al. (2004) can be counted among them. To be specific, Leiter et al. (2004) investigated glucose oxidase from the fungus Penicillium chrysogenum, which was proven to be a good producer of beta- lactam and basic antifungal protein. The cytotoxic activity of the fungus during its exponential phase of growth was attributed to PcGOx, which releases the oxidative agent H2O2. Again, D- Glucose was described as the preferred substrate (Km=9.5 mM). Xylose is also biochemically implemented, but in a less efficient way (Km=690 mM). The determined kinetic properties of PcGOx coincided well with those of glucose oxidases from other Penicillium species. A value of 155 kDa was found for the molecular weight of PcGOx, and accordingly a value of 75 kDa for its subunit. Finally, Gao et al. (2012) successfully expressed the wildtype gene (Gen-Bank accession number JN809249) and the codon-optimized gene (GenBank accession number JN809250) of PnGOx from strain Penicillium notatum F4 in Pichia pastoris in 2011. From its gene sequence they concluded a molecular mass of 64.5 kDa per subunit for wild-type PnGOx.
The recombinant wild-type enzyme was found to have an optimum pH of 6.2 and optimum activity at 35-40°C. Its Km-value for D-glucose was determined to be 83 mM. Gao et al. claim to have expressed the codon-optimized PnGOx gene at a yield of 2.5 gprotein/L (and 615 U/mL respectively) in a 3 L fermenter.
3.8.2 Laccase
Laccases (EC 1.10.3.2, benzenediol:oxygen oxidoreductases) belong to the superfamily of multi- copper proteins, i.e. they contain copper atoms in their catalytic center. Laccases oxidize various phenolic substances to their radicals while reducing molecular oxygen to water. In the beginning laccases were found mainly in eukaryotes: laccases from plants such as peach, sycamore and tobacco were not further characterized or are rarely used as their purification from crude plant
extract is difficult due to the presence of many other oxidative enzymes. Laccase-producing fungi are most notably white rot fungi. The latter produce laccase primarily extracellularly (95%
of the enzyme are located outside of the cell), but also intracellularly. In plants approximately 45% of the enzyme are glycosylated based on galactose, mannose, N-acetylglucosamine, in fungi the glycosylation adds up to about 10-20%. Fungal laccases are usually monomeric proteins, but homodimers do also exist. Their molecular weight is usually in the range from 50 to 130 kDa. Fungal laccases work optimally within an acidic pH range - more specifically about pH 3.5 to 5, provided that organic hydrogen donors serve as substrates. The temperature optimum of most laccases is between 50 and 70°C. But recently genes for laccases were also found in prokaryotes, in both gram-positive and gram-negative bacteria. The exact biological function of laccases in bacteria remains unknown, but the best-studied bacterial laccase CotA is, for example, part of the outer coat of Bacillus subtilis spores and involved in the biosynthesis of a brown spores pigment. This pigment is similar to melanin and probably responsible for the protective effect of the spore from UV light and H2O2. (Driks, 2004) In comparison to fungal laccases, bacterial laccases work at higher pH values and temperatures, exhibit lower redox potentials and are easier to handle. Their size varies in a range from 28 to 180 kDa and they occur both intra- and extracellularly depending on the species .For instance SLAC from S.
coelicolor is present extracellularly (Machczynski et al., 2004), whereas laccase from B.
licheniformis is located intracellularly (Koschorrek et al., 2008).
The active site of all laccases contains in general 3 types of copper: there is one "mononuclear"
type 1 copper atom per active site, and additionally 3 copper ions of type 2 and 3, which are arranged as a trinuclear cluster. As a conclusion four electrons are required to render laccase fully oxidized. Due to its high redox potential the substrate oxidation takes place at the type 1 copper atom, which is bound by conserved ligands (two histidine and one cysteine residues), whereas the reduction of molecular oxygen and the release of water takes place at the trinuclear cluster. Inhibition of laccases is often effected by small inorganic anions such as chloride, azide, fluoride and hydroxide anions, which may bind to the trinuclear cluster of type 2 and type 3 copper ions, and thus prevent the electron transfer from the type 1 copper
Figure 6 Reaction catalyzed by laccase (Baldrian, 2006)
to the trinuclear cluster. The greater their molecular radius, the less efficient is the inhibition of molecules (fluoride > chloride > bromide).
Type 1 copper is also responsible for the characteristic blue color of laccase solutions. Similar enzymes that lack type 1 copper are called "yellow" or "white" laccases, or are not considered as laccases at all by some authors. Phenols, polyphenols, aromatic amines and various non- phenolic substances are suited as substrates; of course laccases from different organisms differ in their substrate specificity. Generally, the substrate specificity is rather low. In order to measure the enzymatic activity, especially syringaldazine, guaiacol and the non-phenolic ABTS are in use. The radicals formed during the oxidation of the monomeric substances react further in the following non-enzymatic reactions:
• Cross-linking of monomers
This leads to the formation of dimers, oligomers or polymers coupled by C-C, C-O, and C-N bonds. Depending on the pH of the surrounding medium and the reactivity of the intermediate radicals different final products are formed. This property of laccases leads to ligninification in higher plants, and is of particular interest in this thesis.
• Degradation of polymers
Enzymes are unable to interact directly with substrates due to steric hindrance, but only via mediators. These mediators are small organic molecules or metals, such as manganese, which are oxidized by laccase, and then in turn react with the polymer.
• Ring cleavage of aromatics
Biotechnological applications include their employment as amperometric biosensors to analyze various phenolic compounds like catecholamines and tea tannins, but also ascorbic acid.
Another possible application of laccase is the oxidation of toxic substances in industrial wastewater in a way that enables the subsequent separation of the resulting insoluble precipitates. This is relevant for the food industry too as, for example, phenolic compounds in wastewater from olive mills cause environmental problems in the Mediterranean region. Food technological applications of enzymes also include the stabilization of wine, beer and fruit juices, where laccases are supposed to target specific polyphenols in order to prevent haze and sediment formation that is stimulated by protein-polyphenol interactions. Laccases also show promise for obtaining thermo-irreversible gels from the new functional ingredient sugar beet pectin, as it is able to oxidatively cross-link ferulic acid residues of beet pectin. Sugar beet pulp is an interesting raw material as it is an alternative pectin source to apple pomace and citrus peel. Laccase-mediated gelation enables in consequence the heating of food products while maintaining their gel structure. Generally, laccases are beneficial for their environment as they produce water as the only by-product, and as they do not require cofactors. (Baldrian et al.,
2006; Claus, 2003; Claus, 2004; Johannes et al., 2000; Minussi et al., 2002; Morozova et al., 2007; Santhanam et al., 2011; Sharma et al., 2007)
3.8.2.1 BaLac
Oliver Mann cultured, purified and characterized laccase from the phytopathogenic fungus Botrytis aclada in 2009. After a two-stage cleaning a specific activity of 120 U / mg was obtained.
Unfortunately, Botrytis aclada produces large amounts of an extracellular polysaccharide, which results in high viscosity and thus causes problems with processing of the supernatant.
Heterologous expression of BaLac in Pichia pastoris is a solution to this problem and was successfully carried out by Kittl et al. (2012) with a high fermentation yield (495 mg/L laccase with the inducible AOX1 system, 517 mg/L with the constitutive GAP system). Regarding the difference between the expression systems, production is faster with the GAP system and higher specific laccase activity is measured in the supernatant (41.3 U/mg with the GAP system, 14.2 U/mg with the AOX1 system). Both the wild-type enzyme and the recombinant enzyme have a molecular weight of 75 kDa, or 65 kDa in the deglycosylated state. Both have a rather low temperature optimum of 45°C and higher activity in the acidic range as their pH optimum is pH 3.0 for ABTS or 6.0 for syringaldazine. The thermal stability of BaLac is rather poor; the half-life of the enzyme is only 23.4 minutes at 45°C. In contrast to most other laccases BaLac is not inhibited by chloride ions, and its stability even increases in their presence. This property of BaLac is interesting for its application in dough, as NaCl is usually added as an ingredient. The laccase that is most closely related to BaLac is derived from Botrytis cinerea. (Kittl et al., 2012) 3.8.2.2 BliLac
In 2008, Koschorreck et al. identified a laccase from of the soil bacterium Bacillus licheniformis DSM 13, of which the physiological role is still unknown. In the course of their work, the DNA of BliLac (in the respective publication referred to as cotA) was cloned and expressed in E. coli at a yield of 26 mg/L protein. A relatively high sequence identity of 63% of BliLac to bacterial laccase CotA from B. subtilis was found. Wild-type BliLac is a monomeric protein with a molecular weight of approximately 65 kDa and a Km value of 6.5 µM for ABTS. The enzyme shows good tolerance to high temperatures. To be more specific, activity is at its highest at 85°C, and after incubation at 70°C and 80°C wild-type BliLac has a residual activity of 43% and 8%, respectively. This property allows, for example, the facilitation of purification by heating the crude extract briefly to 70°C in order to precipitate other E.coli proteins. The optimum pH for the oxidation of ABTS is 4.2, for the oxidation of syringaldazine and 2,6-DMP it is pH 7.0. When it comes to the preferred substrates of wild-type BliLac, a dimerization of sinapic acid, caffeic acid
and ferulic acid was observed. The highest relative conversion within two hours was of sinapic acid with 66% (at a substrate concentration of 5 mM), however, only 14% of the provided ferulic acid were converted. In 2009, Koschorreck et al. constructed the double mutant K316N / D500G by combining random and site-directed mutagenesis. With this double mutant, a protein yield of 291 mg/L could be obtained. The double mutant decolorizes industrial dyes more efficiently and also converts ferulic acid and caffeic acid (21% in two hours, compared to 14% of the wild-type enzyme) much faster, which is specifically interesting for arabinoxylan crosslinking. The Km value of ABTS is 11.7 μM, which is slightly lower when compared to 16.0 μM of wild-type enzyme. Thus, it was discovered that the amino acid at position 500 has a strong influence on the expression of laccase, and that the amino acid at position 316 has a strong influence on the enzyme activity. (Koschorreck et al., 2008; Koschorreck et al., 2009)
3.8.2.3 Cgl1
Ricklefs et al. identified Cgl1 as a bacterial laccase encoded by Corynebacterium glutamicum in 2014. The Cgl1 gene ((Gen-Bank NC 003450.3 (3169415.3170896)) was amplified from the genomic DNA of C. glutamicum ATCC 13032, but without the sequence of the C. glutamicum- specific TAT-secretion signal, and heterologously expressed in E.coli. The presence of the mentioned secretion signal in C. glutamicum indicates that the enzyme is originally located outside the cell. The temperature optimum of the recombinant enzyme is 60°C, so Cgl1 is stable at higher temperatures, but also at higher pH values in the neutral to alkaline range and in organic solvents. For example, incubation of the enzyme in 40% acetone even seems to increase its activity. The stability of the enzyme at high pH values is promising for its use to decolorize wastewater from the textile industry; that kind of wastewater usually exhibits pH values of 7.0 to 11.0. As the redox potential of its copper T1 site is 260 mV, Cgl1 is classified as low potential laccase. With respect to substrate specificity Cgl1 catalyzes the oxidation of several phenolic substrates, in which the main oxidation products of all substrates except syringic acid are dimers. 75% of 1 mM dissolved ferulic acid was implemented within two hours, which indicated that ferulic acid is a suitable substrate for Cgl1. When compared to other prokaryotic laccases, Cgl1 only shows little sequence identity with them, e.g. 15.5% with B.
licheniformis laccase and 15.9% with B. subtilis laccase. (Ricklefs et al., 2014)
3.8.3 Cellobiose Dehydrogenase
Cellobiose dehydrogenase, an extracellular flavocytochrome, is produced by wood-degrading and plant pathogenic fungi. According to Kracher et al. (2016), one physiological function of
CDH in wood-degrading fungi is that of an extracellular source of electrons for lytic polysaccharide monooxygenases (LPMOs); LPMOs require extracellular electron sources to hydroxylate and consequently break the polysaccharide chain. The molecular weight of CDH is typically 90-100 kDa, of which approximately 10% are attributable to glycosylation. It is a monomeric protein consisting of two domains, which are connected by a protease-sensitive linker. The larger dehydrogenase domain contains flavin and belongs to the superfamily of glucose-methanol-choline oxidases, just like pyranose oxidases. The smaller cytochrome domain binds a heme B as prosthetic group. Kracher et al. (2016) found the cytochrome domain of CDH to be responsible for the electron transfer. The catalytic mechanism involves the transfer of electrons from electron donors such as cellobiose via its two redox centers to both two- electron acceptors (e.g. benzoquinone) and one-electron acceptors (e.g., cytochrome C, ferric ion). Mainly soluble cellodextrins, mannodextrins and lactose are oxidized by CDH to the corresponding lactones in a ping-pong mechanism. However, monosaccharides, maltose and molecular oxygen are poor substrates. Generally, CDH is a very stable enzyme regarding temperature and pH, and it can be used in biosensors, biofuel cells and bioelectrosynthesis.
(Cameron & Aust, 2001; Ludwig et al., 2010; Zamocky et al., 2006)
3.9 Protein production and expression systems
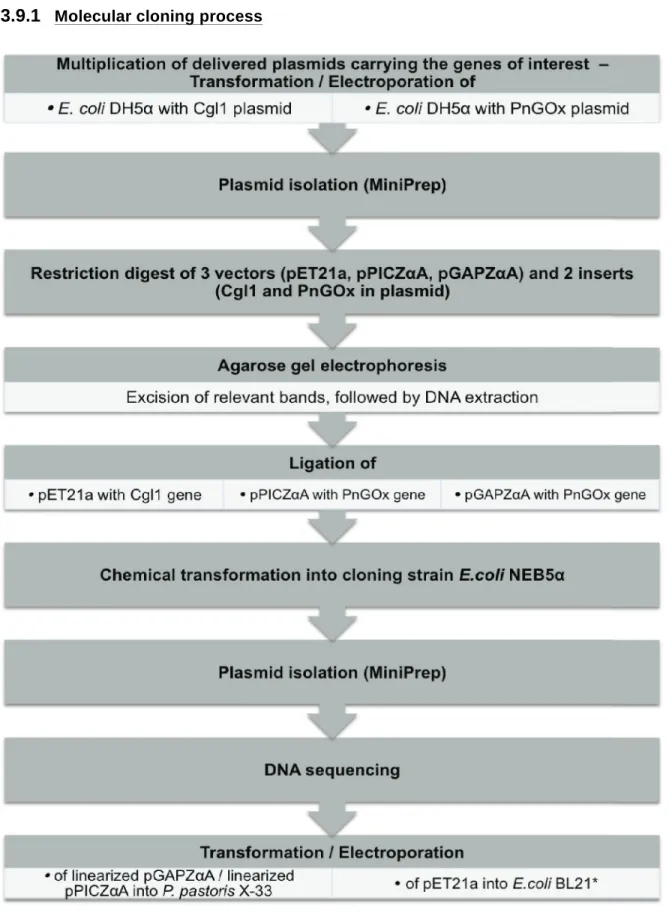
3.9.1 Molecular cloning processFigure 7 Chronology of molecular cloning of Cgl1 and PnGOx gene into expression host
3.9.2 Expression hosts
To achieve the highest expression yield within the shortest time possible in enzyme production, the choice of a suitable host organism is crucial. Suitable host organisms include bacteria, yeast, filamentous fungi as well as unicellular algae and should be easy to cultivate and to handle.
3.9.2.1 Escherichia coli
Among the advantages of the gram-negative bacterium Escherichia coli as a host organism counts its rapid growth; under optimal growth conditions the cell number doubles within 20 minutes. E. coli is easy to cultivate in inexpensive media, achieving high cell densities of up to 100 grams of dry cell mass per liter. Its genetics are very well characterized and there is a variety of cloning vectors and mutant host strains available. A prominent example is the commonly used strain BL21*(DE3), whose cells do not encode for the genes of Lon protease, that degrades foreign protein, and for the protease OmpT of the outer cell membrane, that would normally be able to degrade extracellular protein after cell lysis. The transformation with exogenous DNA can occur quickly and easily - even within only five minutes. Heterologous DNA is usually cloned into plasmids that replicate so that fifteen to a few hundred copies per cell are present. Expression of proteins of eukaryotic origin in E.coli is problematic because E. coli is not able to make post-translational modifications. Hence, it is not suitable as a host organism for the expression of proteins that contain disulfide bridges and require post-translational modifications like glycosylation, lipidation, sulfation, disulphide isomerization etc. for their function. Anyway, E.coli has so far allowed expression, characterization and structural resolution of many proteins of prokaryotic origin like Bacillus subtilis CotA, and even of a few of eukaryotic origin such as the fungal laccase from Cyathus bulleri. A major disadvantage is that excess of inducer and high- level overexpression of recombinant protein in E. coli lead to formation of misfolded and inactive proteins, also called inclusion bodies, in the cytoplasm. In general, E.coli is not able to secrete recombinant proteins beyond the periplasm into the medium; hence soluble and insoluble proteins accumulate in the cytoplasm. Inclusion bodies are aggregates of overexpressed protein, small foreign proteins and nucleic acids. Although the production of recombinant proteins as inclusion bodies may have certain advantages, the recovery of active protein from inclusion bodies is too expensive for large-scale production. Strategies to avoid inclusion body formation during fermentation include lowered temperature in combination with oxygen limitation (ergo:
measures to reduce cell growth), and also limitation of the inducing substance. Another drawback with expression in E.coli is that its codon usage is biased and therefore may lead to inefficient expression of heterologous genes that contain several codons that are rarely used by E.coli. However, this can be avoided by genetically altering target gene codons in a way that the
encoded protein product is not modified. (Hannig & Makrides, 1998; Baneyx, 1999; Piscitelli et al., 2010; Spadiut et al., 2010; Spohner et al., 2014; Rosano & Ceccarelli, 2014)
3.9.2.2 Pichia pastoris
Since 2006, Pichia pastoris has GRAS status and recombinant proteins produced with Pichia pastoris have been approved by the Food and Drug Administration (FDA), which can be considered as a breakthrough for food technological use of this expression host organism. More than 500 pharmaceutical substances and recombinant proteins today are produced using Pichia pastoris. The choice of a Pichia pastoris strain depends on its intended application; common strains include the wild-type strain X-33, or modified strains with special features as auxotrophy or protease deficiency. In comparison to E.coli, Pichia has equal possibilities of genetic manipulation and a lower speed of growth, but very high cell densities can be obtained. The standard vectors for expression in Pichia, which are described in detail below, are routinely replicated in E. coli and finally maintained in Pichia cells. In addition, yeasts can modify recombinant proteins post-translationally. Proteins can be either expressed intracellularly, or secreted into the surrounding medium in soluble form. When compared to Saccharomyces cerevisiae, Pichia pastoris secretes significantly less host protein. The latter reduces the purification costs. For this secretion an amino-terminal signal sequence attached to the protein is necessary, followed by 6 to 15 hydrophobic amino acid residues and a cleavage site. The most commonly used signal sequence is the α-factor from Saccharomyces cerevisiae. Again, codon optimization is possible in order to adapt to the codon usage of P. pastoris, and it may lead to a dramatic increase in protein expression levels. (Macauley-Patrick et al., 2005; Piscitelli et al., 2010; Spohner et al., 2014)
3.9.3 Cloning vectors
Apart from a promoter and terminator region, plasmid vectors also contain a selective marker and a multiple cloning site as essential elements. The purpose of a selective marker, i.e. an antibiotic resistance gene, is to select the cells that took up a plasmid; the cells that incorporated the plasmid are thus not harmed if the respective antibiotic is added to the growth medium.
Multiple cloning sites can be described as an artificial stretch of DNA containing a high amount of restriction endonuclease sites that do not occur elsewhere in the plasmid. (Casali N., 2003) 3.9.3.1 pET-21a(+)
The expression of a gene inserted in pET-21a(+) is controlled by T7 bacteriophage transcription signals. pET-21a(+) encodes an ampicillin resistance gene and it enables a C-terminal HisTag®
fusion as well as an N-terminal T7Tag® fusion. In general, the initial cloning step is about the plasmid being transferred to a bacterial host without a T7 RNA polymerase gene, e.g. E.coli DH5α, for reasons of plasmid propagation. During this step background expression is avoided.
The plasmid is then transferred to an appropriate bacterial expression host that is a λDE3 lysogen and therefore contains amongst others a chromosomal copy of T7 RNA polymerase, a T7 lac promoter et cetera. The target gene is expressed by induction with either lactose or IPTG.
(Novagen (2005). pET system manual, 11th edition) IPTG is not ideal for large-scale expression of recombinant protein, especially because of its high costs. Expression regulated by the lac promoter in the simultaneous presence of glucose and lactose is triggered only after all glucose is metabolized by the microorganism.
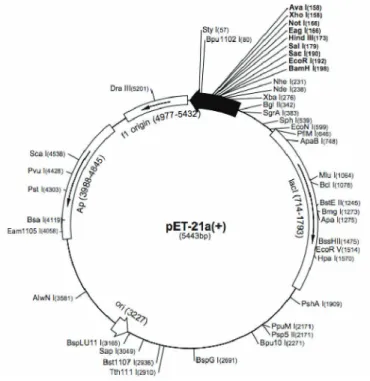
Figure 8 pET-21a(+) map (Novagen)
3.9.3.2 pPICZαA
pPICZαA is a vector for protein expression and secretion in Pichia pastoris. The fusion of a recombinant protein to an N-terminal peptide encoding the S. cerevisiae α-secretion signal during expression enables secretion. Recombinant strains carrying a pPICZαA plasmid can be selected on Zeocin™ containing media as pPICZαA contains a Zeocin™ resistance gene.
Furthermore, the plasmid contains the AOX1 promoter, the alcohol oxidase I gene that regulates the expression of the first enzyme in the pathway of methanol utilization and therefore allows a methanol-induced expression of the target gene, and an AOX1 transcription termination region.
By initial transformation to an E.coli cloning strain, e.g. DH5α, the target gene-containing
plasmid is produced in sufficient amounts for later transformation to Pichia. Before transformation to Pichia, the pPICZαA construct has to be linearized, ideally by cutting the plasmid within the 5’-AOX1 region, as linearized DNA is integrated into the Pichia genome during transformation. (Invitrogen (2010) pPICZα A, B, and C. User manual.)
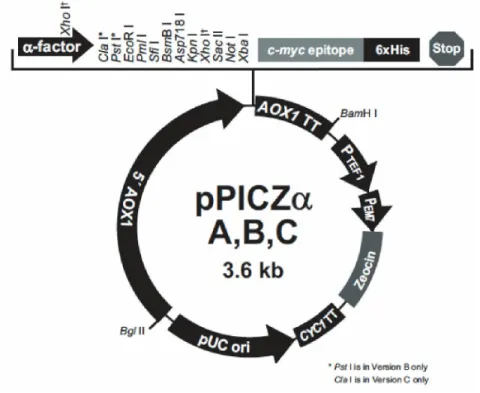
Figure 9 pPICZα A map (Invitrogen)
3.9.3.3 pGAPZαA
The enzyme GAPDH (glyceraldehyde-3-phosphate dehydrogenase) is constitutively expressed in Pichia pastoris and as pGAPZαA contains the GAP promoter, a recombinant protein inserted in this vector is also constitutively expressed. Apart from that, pGAPZαA contains a Zeocin™
resistance gene, an AOX1 transcription termination region and similar to pPICZαA the S.
cerevisiae α-factor secretion signal is fused to the N-terminal end of the target protein for secretion. As for pPICZαA, pGAPZαA is initially transformed to a DH5α E.coli cloning strain for plasmid multiplication purposes; afterwards those plasmids are linearized and used to transform competent Pichia cells. The linearized DNA is finally integrated into the host genome. (Invitrogen (2010) pGAPZα A, B, and C. User manual.) In contrast to pPICZα A, pGAPZα A – transformed Pichia cells can be fermented on glucose and glycerol throughout the whole fermentation process and are therefore of interest for large-scale protein production without the need of induction.
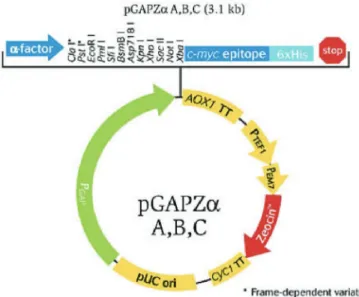
Figure 10 pGAPZαA map (www.thermofisher.com)
3.10 Rheometry and characterization of viscoelastic behavior of a solution
The instrument used for the rheological measurements, a Kinexus rheometer, is a rotational rheometer. Rotational systems are in general used to follow time-dependent behavior, as through tube systems material can only be passed once. Viscoelastic properties of matter are mostly determined from oscillatory testing, where samples are subjected to a harmonically varying strain while being held in containment systems, such as a cone and plate system. Such dynamic testing techniques may fix the deformation (i.e. the strain) and measure the resulting stress like during this work, or fix the stress amplitude and measure the resulting deformation.
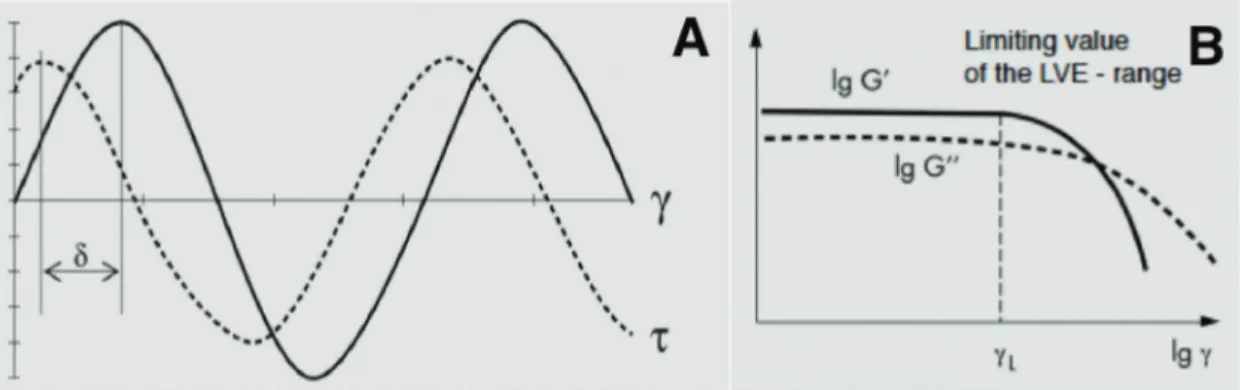
Both methods produce similar results. In the first case, the viscoelasticity of a material is evaluated according to the retardation of the measurement response (stress) to the preset oscillation. This retardation is referred to as a phase shift δ between the sine waves of preset deformation γ and resultant stress τ (see Fehler! Verweisquelle konnte nicht gefunden werden. A); if δ lies between 0° and 90°, the sample is showing viscoelastic behavior.
Figure 11 Sine curves of preset deformation and resulting stress in rheometry (A), linear viscoelastic range (B) (S. Goodyer, Anton Paar Ltd.)
The specific type of dynamic rheological test that is used to evaluate the cross-linking ability of enzymes is a so-called isothermal time sweep, where frequency and amplitude are kept constant over time. More precisely, the time sweep test is carried out at small amplitude oscillatory shear, as with a small amplitude the material will behave in a linear viscoelastic manner (see Figure 11 B). Within the linear viscoelastic range the polymer network is not destroyed. Another typical application example for time sweep studies would be to monitor the thickening of a solution by starch gelatinization. The shear stress output, that is produced by a sinusoidal strain input and measured by the rheometer, is described as , with as shear stress (Pa), as frequency (rad/s) and as shear strain. The phase angle or mechanical loss angle expresses the phase shift, in other words the delay of resulting shear stress as an answer to a shear strain. G’, the shear storage modulus, describes the elastic nature of a substance, whereas G’’, the shear loss modulus, describes its viscous nature. In a graph that pictures time sweep results G’, G’’ as well as δ are plotted versus time, and their development allows to draw conclusions about properties of the matrix as a gel. (Steffe, 1996;
Rao, 2010)
4 Material and Methods
4.1 Material
4.1.1 Biological material
The following strains were obtained from the culture collection of the Institute of Food Biotechnology, University of Natural Resources and Life Sciences, Vienna:
Table 5 Biological Material
Institutional code Host Plasmid Gene Reference
B230 Escherichia
coli, BL21* pET21a PcPOx Pisanelli et al.,
2009
Y58
Pichia pastoris,
X33
pPICZB BaLac Kittl et al., 2012
4.1.2 Chemicals
All chemicals used for the preparation of media, buffers and various working solutions were of the highest grade available and purchased from Sigma Aldrich Chemie (Steinberg, Germany);
Carl Roth GmbH (Karlsruhe, Germany) and Merck (Darmstadt, Germany), unless otherwise mentioned for certain applications.
4.1.3 Media
All components were weighed in according to the specifications below, but for liquid media preparation no Agar-Agar was added. The appropriate volume of RO-H2O was added to dissolve the components and finally, the medium was autoclaved.
4.1.4 Antibiotics
The following antibiotics were added to autoclaved media for selection purposes whenever necessary in the following concentrations: