Neste trabalho, esses problemas são abordados através da criação do programa computacional GapBlaster, uma ferramenta gráfica que visa reduzir a fragmentação da montagem do genoma e acelerar sua finalização. O desempenho do GapBlaster foi avaliado com base em dados NGS obtidos de um grupo de bactérias Staphylococcus aureus e Rhodobacter sphaeroides, ambos obtidos do projeto GAGE.
Contexto
No entanto, estes programas não conseguem colmatar todas as lacunas deixadas pelos montadores e muitas vezes não fornecem informações sobre as alterações do genoma. Assim, este trabalho propõe o desenvolvimento de uma ferramenta gráfica de fechamento de lacunas in silico que permite ao usuário escolher quais alterações serão feitas no genoma.
Justificativa
Além disso, genomas fragmentados podem prejudicar estudos posteriores de genômica comparativa e funcional, pois torna-se difícil identificar sequências contaminadas até que o projeto de sequenciamento do genoma seja concluído. 2002), esta contaminação varia entre 5% e 10% do total de leituras sequenciadas de micróbios cultivados em células animais. Em resumo, é importante que as montagens do genoma sejam concluídas para que os estudos genômicos comparativos e funcionais possam caracterizar adequadamente as variações na estrutura genômica e no conteúdo genético.
Objetivos
Objetivo Geral
Desta forma, espera-se que haja um aumento na precisão da montagem do genoma, uma vez que não será necessário depender da automatização completa da tarefa.
Objetivos Específicos
Metodologia
- Ambiente de Desenvolvimento
- Dados de Teste
- Parâmetros de Entrada
- Comparação do Fechamento de Gaps
- Avaliação dos Resultados
Assim, apenas foram aceitos alinhamentos cujas regiões flanqueadoras fossem altamente semelhantes ao genoma de referência. Genomas de referência e arquivos FASTA contendo estruturas originais ou fechadas foram usados como entrada para o script.

Estrutura do Trabalho
Uma Breve História do Sequenciamento de DNA
Visão Geral dos Sequenciadores de Nova Geração
Outra característica importante é que as amplificações das plataformas NGS são produzidas in vitro em vez de serem clonadas em vetores, como foi feito no sequenciamento capilar (AIRD et al., 2011). Segundo Mardis (2008), o processo de produção de bibliotecas para sequenciadores de última geração é muito mais prático do que no Método Sanger, pois os fragmentos de DNA são preparados para sequenciamento simplesmente ligando adaptadores em ambas as extremidades de cada fragmento de DNA. Mardis (2008) observa que, importante, apenas alguns microgramas de DNA são necessários para produzir uma biblioteca de fragmentos.
No entanto, as plataformas NGS também têm a capacidade de sequenciar os pares ligados de um determinado fragmento utilizando um processo sutilmente modificado para criação de biblioteca (MARDIS, 2008). Este recurso significa que as leituras produzidas pelas plataformas NGS requerem uma cobertura de aproximadamente 25 a 30 réplicas de cada amostra para capturar toda a informação genética (MARDIS, 2008). Esta economia proporcionada pelas plataformas NGS acabou por tornar o sequenciamento do genoma acessível a pequenos laboratórios.
Após a realização do sequenciamento do genoma, é necessário organizar as leituras para obter a informação genética original.
Montagem de Genomas
Mapeamento e Montagem Comparativa
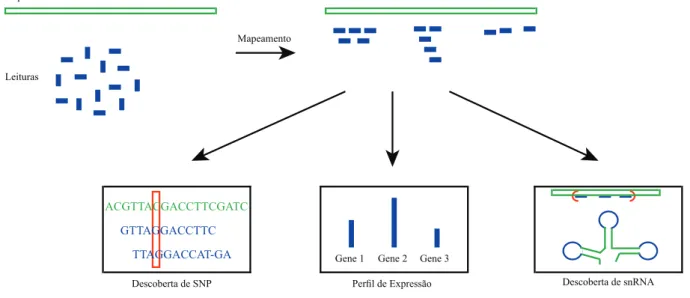
Na Figura 3 é possível observar como funciona o processo de mapeamento e composição comparativa na descoberta do polimorfismo de base única, na descoberta do perfil de expressão gênica e na descoberta do snRNA. Nesta ilustração, as leituras são mapeadas em relação a um genoma de referência para determinar o posicionamento das leituras em relação ao genoma de referência. Este processo é conhecido como descoberta de polimorfismo de base única (SNP).
Com base no mapeamento realizado em relação a um genoma de referência, também é possível descobrir qual informação genética é hereditária no genoma que está sendo montado. Essa descoberta é feita por meio do perfil de expressão gênica, que, com base nas sequências mapeadas, identifica quais genes existem no genoma de referência que também existem no genoma que está sendo montado (ver parte inferior central da Figura 3). Uma terceira aplicação de mapeamento e montagem comparativos é a descoberta de snRNAs, que estão envolvidos no splicing e outras reações de processamento de RNA, como mostrado no canto inferior direito da Figura 3.
Existe ainda outra abordagem de montagem que é utilizada quando não há genoma de referência, que é chamada de montagem de novo.
Montagem de novo
Observe que as leituras mapeadas, que são coloridas em azul, estão perfeitamente alinhadas com a sequência de referência, que é colorida em verde. Com base nesses alinhamentos é possível identificar que apenas uma base foi alterada nesta região do genoma que foi sequenciado. Essas regiões repetitivas devem ser evitadas pelo montador, pois muitas vezes são complexas demais para serem montadas a partir de leituras curtas.
Nossos andaimes são inseridos em furos quando as medições no final de um contig se sobrepõem às medições de dois ou mais contigs, pois isso é indicativo de uma região repetitiva. Os gaps podem ser calculados com base no tamanho de inserção das medidas curtas presentes nos contigs.

Algoritmo do GapBlaster
- Concatenação dos Arquivos de Contigs
- Alinhamento dos Contigs Contra os Scaffolds
- Conversão dos Alinhamentos
- Ordenação dos Alinhamentos
- Concatenação dos Alinhamentos
- Exibição dos Alinhamentos
- Fechamento dos gaps
Caso contrário, caso sejam enviados dois ou mais arquivos, estes programas não reconhecem os parâmetros enviados e consequentemente não realizam os alinhamentos necessários. Os alinhamentos de contigs aos scaffolds são feitos com uma chamada de sistema para um alinhador definido pelo usuário. No entanto, vários alinhadores podem ser adicionados convertendo a saída gerada pelo alinhador desejado para o formato usado pelo GapBlaster, conforme descrito na próxima subseção.
Os alinhamentos descritos na seção anterior devem ser convertidos para um formato utilizado pelo GapBlaster nas demais etapas do processamento. A Figura 5 mostra um exemplo de alinhamento convertido para o formato utilizado pelo GapBlast, contendo assim todas as informações descritas na lista acima. Os alinhamentos são ordenados em ordem crescente e baseados em diferentes atributos para resolver casos em que os atributos comparados são semelhantes.
Os alinhamentos identificados que fecham lacunas são apresentados ao usuário através da interface gráfica do programa, para que o usuário possa então escolher quais alinhamentos deseja utilizar para fechar lacunas.
Interface Gráfica
Após a conclusão do processo de fechamento de gaps, é apresentada ao usuário uma mensagem informando a quantidade de gaps e Ns fechados e o programa retorna à tela principal. Outra forma de o usuário selecionar scaffolds e arquivos de contaminação é através do menu de arquivos, que fica no topo da tela principal. Após selecionar os arquivos contendo os esqueletos, o botão executar é acionado na tela principal e o usuário pode clicar nele para que o GapBlaster inicie o processamento dos arquivos.
Quando o processamento for concluído, outra tela exibe os resultados dos alinhamentos (ver Figura 8) e o usuário pode então realizar o controle manual e selecionar os alinhamentos que melhor preenchem as lacunas. O usuário pode ordenar os resultados apresentados na tabela da Figura 8 em ordem crescente clicando no nome de uma das colunas. O gerenciamento manual dos alinhamentos pode ser feito clicando em uma das linhas da tabela de resultados.
Desta forma, o usuário pode fazer uma curadoria com mais segurança quando forem encontrados contigs que se alinhem com as regiões flanqueadoras do gap, fechando o gap completamente, conforme mostrado na parte inferior da Figura 8.

Dados de Teste
Análise de Fechamento de Gaps
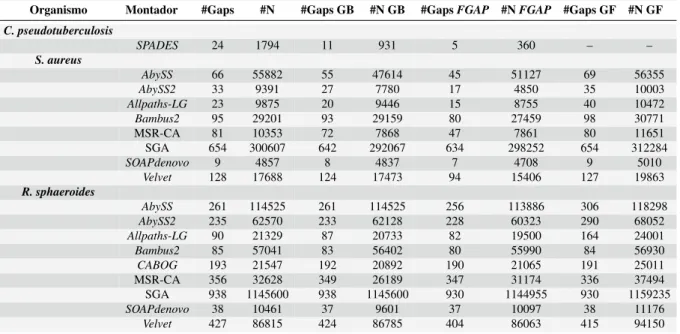
Análises com GapBlaster mostraram diminuição na quantidade de gaps e N em todas as assembleias bacterianas. Comparando o desempenho do GapBlaster com os resultados obtidos com o GapFiller (ver as últimas colunas da Tabela 4), pode-se observar que o GapFiller aumentou ou manteve a mesma quantidade de lacunas em cerca de 59% das montagens de ambos os organismos. Em outros casos, o número de bases desconhecidas representadas por Ns aumentou na maioria absoluta das montagens.
Assim, é possível argumentar que o desempenho do GapBlaster foi superior ao obtido pelo GapFiller na curadoria do genoma, pois o GapBlaster não apenas fechou mais lacunas, mas também reduziu o número de bases desconhecidas em quase todas as montagens do projeto GAGE . Continuando a análise, constatou-se que o programa FGAP fechou mais lacunas que o GapBlaster em todas as montagens obtidas do projeto GAGE. Porém, o GapBlaster conseguiu uma redução maior na quantidade de bases desconhecidas nos dados gerados pelos montadores.
Os resultados obtidos na análise de fechamento de lacunas podem ser resumidos no gráfico 2, que foi elaborado contando o total de lacunas e após a curadoria dos dados de coleta de bactérias.
Análise de uso do GapBlaster em Conjunto com Outros Programas de
Entretanto, levando em consideração todas as montagens que foram pós-fixadas com GapBlaster, um total de 22 lacunas fechadas não foram identificadas pelo FGAP antes do mostrado no Gráfico 4. Para mais detalhes sobre os resultados da análise usando GapBlaster em conjunto com FGAP, consulte Tabela 5 do Apêndice A Com base nos resultados desta análise, acredita-se que além de facilitar o processo de fechamento de lacunas através de sua interface gráfica, o GapBlaster também pode ser utilizado em conjunto com outras ferramentas para agilizar ainda mais o processo de curadoria.
Para confirmar os resultados obtidos, foi avaliado se o GapBlaster também poderia melhorar a saída gerada pelo GapFiller nos dados obtidos no projeto GAGE. No gráfico 5 demonstra-se que o GapBlaster reduziu a quantidade de gaps em 70,58% das montagens do projeto GAGE que foram previamente curadas pelo GapFiller. No entanto, a contagem global revela que foram colmatadas um total de 32 lacunas nas juntas melhoradas pelo GapBlaster, conforme mostra o Gráfico 6.
Desta forma, é possível verificar que o GapBlaster é de fato um valioso programa de código aberto que pode ser usado em conjunto com outras ferramentas de fechamento de buracos para produzir montagens de genoma mais completas.

Precisão dos Testes Realizados
Comparação das Funcionalidades
All contigs obtained in the assembly are aligned against the draft genome or the scaffold using BLAST Legacy. The choice of alignment and parameters can be defined by the user through the GapBlaster interface. To validate the hole-filling assay, an in-house script was developed to evaluate the amount of holes and Ns for each of the tests.
The results of the gap-closure process for the Corynebacterium data collected by SPADES are shown in Table 4. We performed the gap-filling analysis of the FGAP results with the original contigs of each. Gap-closure process results for the data produced by GAGE with different assemblers for S.aureus and R.sphaeroides.
Compared to the results of GapFiller, GapBlaster improved 70.58% of all assemblies of the GAGE dataset (Table 7). To assess the accuracy of closed gaps, all results produced by GapBlaster, FGAP, Gap-Filler and the original files (scaffolds) were aligned to their respective genome reference (Table 2). After you finish the selection, click the open button and the path of the scaffolding file will be displayed on the main screen.

Overview
System Requirements
Therefore, in order to run it, Java 8 or higher must be installed. For more information or installation instructions, please visit: http://java.com/en/download/manual.jsp.
Installing Dependencies on Debian
Installing Legacy Blast
Installing MUMmer
Installing Blast+
Running GapBlaster
- Editing the Preferences
- Selecting the Input Files
- Selecting alignments
- Output Files
In the preferences window you can select the local aligner and set the minimum arm length to perform alignments. To select the scaffolding file, simply click the "select" button on the main screen and a dialog box will appear. To add one or more contigs files, click the Add Contigs button on the main screen and another dialog box will appear.
After clicking the “Run” button, it may take some time for GapBlaster to complete all the required steps. However, if you are sure that you want to apply all the changes suggested by GapBlaster, you can simply click the "Select All" button at the top right of the screen. When you have finished selecting all the holes you want to close, click the button.
GapBlaster will apply any changes you've made to the scaffolding and print out how many gaps have been closed.