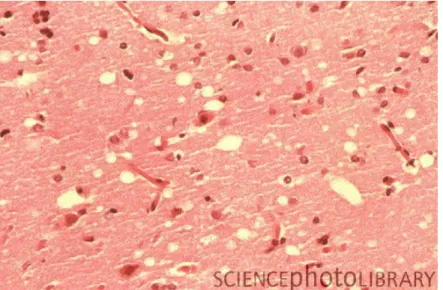
Agradeço a Deus por me permitir chegar onde estou e por ser abençoado com pessoas maravilhosas que sempre estão ou estiveram ao meu lado. Toda a minha motivação se deve a quem sempre me deu conselhos e nunca tentou me levar onde cheguei. As encefalopatias espongiformes transmissíveis (EET) são doenças cerebrais degenerativas que ocorrem em vários mamíferos.
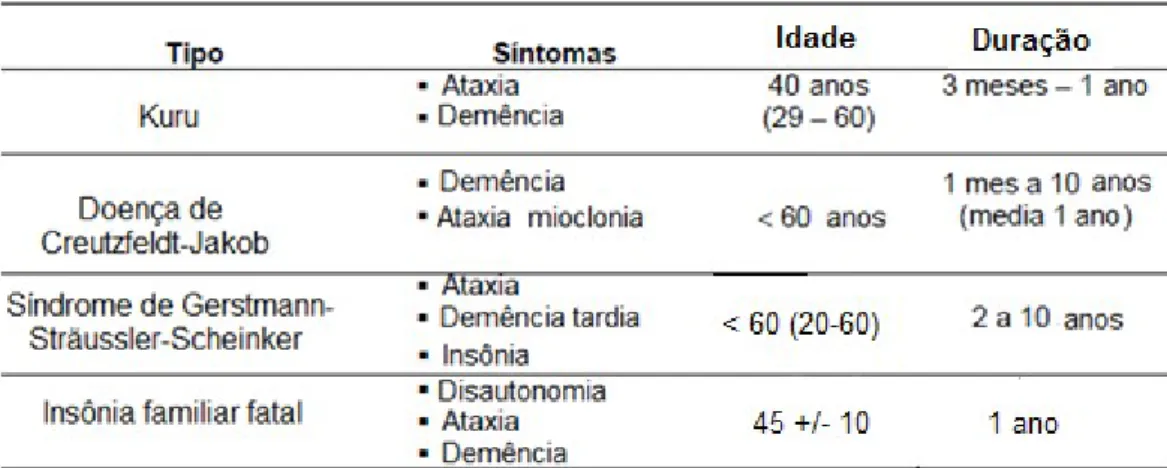
As doenças humanas mais citadas na literatura são o kuru, a insônia familiar fatal, a síndrome de Gerstmann-Sträussler-Scheinker e a doença de Creutzfeldt-Jakob. Estas doenças são caracterizadas por uma aparência esponjosa de degeneração cerebral devido à sua degeneração gradual. As doenças humanas mais frequentemente mencionadas na literatura são o kuru, a insónia familiar fatal, a doença de Gerstmann-Sträussler-Scheinker e a doença de Creutzfeldt-Jakob.
The results of this study show that TSEs are diseases characterized by a spongy appearance of the brain due to its gradual degeneration, and that each of these diseases has an impact on society and is causing a stir within the scientific community.
INTRODUÇÃO 7
A forma mais comum de EET em humanos é a doença de Creutzfeldt-Jakob (DCJ), que é classificada como esporádica (esCJD), familiar (fCJD), iatrogênica (iCJD) e nova variante (nvCJD). Após concluir a graduação e ingressar na especialização, tive a oportunidade de escrever esta tese focada no tema e assim pude ler e compreender a estrutura da proteína príon e o que acontece na principal doença descrita. na literatura, Doença de Creutzfeldt-Jakob. MARCONI, LAKATOS, 2009, relatam que toda pesquisa envolve a coleta de dados de diferentes fontes, independentemente dos métodos ou técnicas utilizadas.
A primeira geralmente consiste na coleta de dados no local onde ocorrem os fenômenos. O outro utiliza fontes de dados coletadas por outras pessoas, que podem ou não consistir em material já elaborado. Divide-se assim em pesquisa documental (ou fontes primárias) e pesquisa bibliográfica (ou fontes secundárias).
Com base nos dados acima, podemos afirmar que a pesquisa é exploratória.
REVISÃO DA LITERATURA 11
PRÍONS E PRPC 11
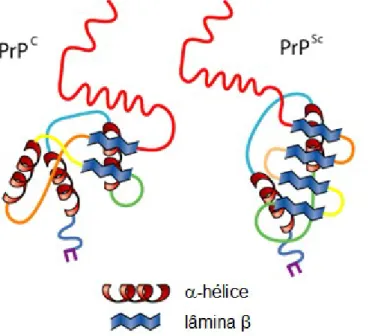
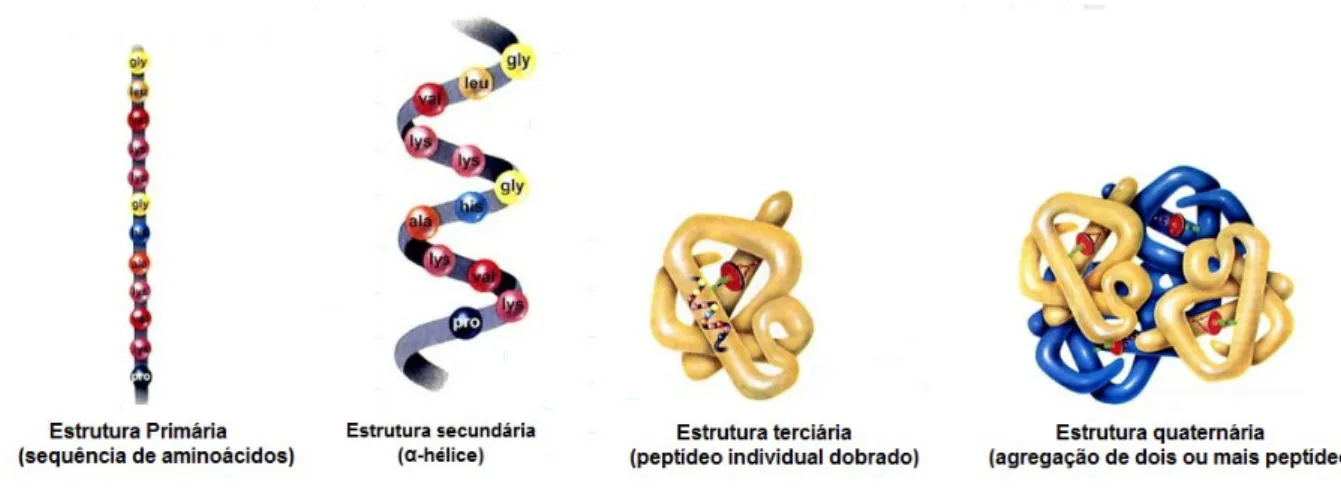
LINDEN, CORDEIRO, LIMA, 2012, mencionam que estudos apontam o papel central da EET como uma conformação anormal da PrPc causada por alterações na estrutura secundária desta proteína. MANDUJANO, et al., 2006 relatam que Stanley Prusiner sugere que a presença de aminoácidos incorretos pode desestabilizar a estrutura terciária da proteína. Também serviu para identificar cepas de acordo com sua origem, pois a glicosilação varia dependendo da origem da proteína príon, portanto a glicosilação é diferente na proteína príon dependendo se é de origem genética ou se é infecciosa (LANAU, 2010) .
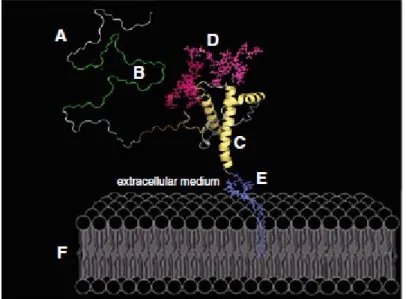
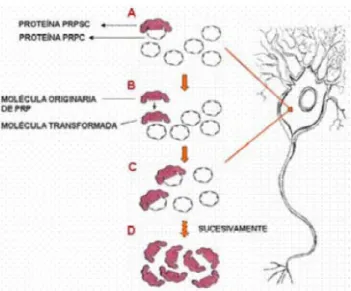
Desenho em escala aproximada da proteína príon, mostrando (a) o domínio N-terminal flexível; (b) domínio de repetição octapeptídeo; (c) domínio globular C-terminal; (d) cadeias de glicosilação, representando uma forma diglicosilada; (e) IGP; (f) membrana plasmática. Em humanos, a PrPSc pode originar-se através de mutações no PRNP, através da conversão espontânea de PrPc em PrPSc ou através do processamento pós-traducional de PrPc causado por príons exógenos (HERNANDEZ F., CESPEDES C., GONZALEZ A., 2002). Uma delas é a teoria de Prusiner, onde existe uma interação direta entre uma molécula de PrPc e uma molécula de PrPSc.
Uma vez formada na membrana plasmática da célula, a PrPSc segue a rota metabólica da PrPc: entra no citoplasma dos neurônios através de endocitose, continua a replicar-se através do sistema endocítico-lisossomal e não é degradada nos lisossomos secundários, onde eventualmente se acumula para ter uma vida infinita, embora parte do PrPSc também possa entrar no aparelho de Golgi e transportar vesículas para retornar à membrana plasmática. Existem evidências científicas da existência do transporte de PrPC e PrPSc ao longo do axônio do neurônio, e em sistemas de cultura celular foi demonstrado que PrPSc se acumula fundamentalmente em lisossomos secundários de células ou em estruturas relacionadas (corpos multivesiculares e estruturas túbulo-reticulares) (HERNANDEZ F., CESPEDES C., GONZALEZ A., 2002). A única doença priônica animal à qual os humanos são suscetíveis é a BSE, o que pode ser explicado pelo fato de a PrPc bovina conter áreas estruturalmente muito semelhantes aos domínios-chave da PrPc humana (HERNANDEZ F., CESPEDES C., GONZALEZ A., 2002).
Dependendo do polimorfismo da metionina ou valina no códon 129 da proteína príon, os genótipos são Met/Met (metionina), Met/Val (valina) e. Onda/Onda; e de acordo com o padrão de migração eletroforética da proteína, a proteína príon patológica também foi diferenciada em dois tipos: tipo 1 com peso molecular de 21 kDa e sítio de clivagem no resíduo 82 e tipo 2 com peso molecular de 19 kDa e sítio de clivagem no resíduo 97. 10% dos casos relatados de doenças por príons são de origem hereditária, portanto a maioria delas são esporádicas e contagiosas e não representam mutações no gene ou alterações na sequência de aminoácidos da proteína (MANDUJANO, et al., 2006 ).
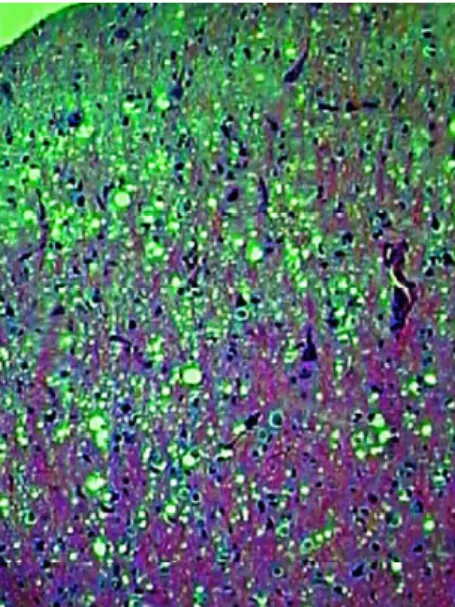
Ensaios clínicos para novos tratamentos para doenças por príons foram desenvolvidos com base em efeitos terapêuticos robustos em células cultivadas infectadas por príons, principalmente relacionados à prevenção ou desmontagem de agregados de conformação anormal de PrPc. A confirmação post-mortem do diagnóstico é necessária pela observação de espongiose em estágio terminal e morte de células teciduais, juntamente com detecção imuno-histoquímica de agregados compactos, insolúveis e resistentes à protease, conformação anormal de PrPc. A função da PrPc tem sido alvo de grandes pesquisas, pois camundongos sem esse gene aparentemente se desenvolvem normalmente, mas nos estudos realizados foram atribuídas à PrP funções como: fator de transcrição que afeta a polaridade celular, regulação da glicosilação, modulação de neurônios entre outras cobre expressão, transdução de sinal, memória, transmissão sináptica, ritmo circadiano, respostas inflamatórias, proliferação e diferenciação celular.
Dentre eles, o mecanismo de conversão de PrPc em príons; os eventos que causam disfunção e degeneração neuronal; moléculas de transdução de sinal que transmitem sinais patogênicos derivados da agregação progressiva de espécies tóxicas; o papel da disfunção e da morte de neurônios e células gliais nos vários sintomas associados à EET; a topografia diferencial das lesões cerebrais associadas às diferentes EET; por que mutações específicas no PRNP levam a uma conversão conformacional; o possível papel dos ácidos nucléicos na composição dos príons; e o último.
DOENÇAS PROVOCADAS PELA ATIVAÇÃO DOS PRÍONS 22
- Kuru 25
- Insônia Familiar Fatal 27
- Síndrome de Gerstmann-Sträussler-Scheinker 27
- Doença de Creutzfeldt-Jakob 28
- esDCJ 32
- fDCJ 33
- iDCJ 34
- nvDCJ 34
- DCJ variante de Heidenhain
Em humanos, diversas doenças têm sido associadas a príons, tais como: DCJ, Síndrome de Gerstmann-Straussler-Scheinker, Kuru, IFF, Síndrome de Alpers (PERRONE; et al., 2003). Familiares: doença familiar de Creutzfeldt-Jakob, síndrome de Gerstmann-Sträussler-Scheinker, insônia familiar fatal, doenças priônicas atípicas. Alguns autores propuseram outras patologias nas quais o agente etiológico pode se comportar como príon, como a doença de Alzheimer, a doença de Huntington e a doença de Parkinson (LANAU, 2010).
A presença e a extensão dessas características variam de acordo com o tipo de doença priônica e de caso para caso (MANDUJANO, et al., 2006). A síndrome de Gerstmann-Sträussler-Scheinker é a única doença por príon transmitida exclusivamente de forma hereditária, resultante de mutações pontuais autossômicas no gene que codifica a proteína príon. Foi proposto que a substituição de um aminoácido por outro na sequência desta proteína altera sua estabilidade termodinâmica no meio celular, promovendo a alteração conformacional que confere seu caráter patológico (MANDUJANO, et al., 2006).
Em alguns estudos, descobriu-se que as infecções por príons são semelhantes à doença de Parkinson, à esclerose lateral amiotrófica e à doença de Alzheimer. Usando métodos imuno-histoquímicos, a presença de proteína príon anormal em uma amostra de biópsia também pode ser identificada. A comunidade científica está direcionando seus esforços para encontrar um medicamento que atenda ao princípio farmacológico de superar a barreira hematoencefálica e seja capaz de impedir a conversão da proteína príon normal em uma proteína anormal.
As estratégias farmacológicas para o tratamento de doenças por príons incluem muitos outros princípios ativos, bem como o uso de alternativas terapêuticas, tais como: anticorpos contra a proteína príon, uso de RNA de interferência e imunização contra a proteína príon patológica. Muitos pesquisadores propõem que a DCJ esporádica ocorre através da geração endógena de príons, acredita-se que o dobramento anormal aleatório da proteína príon pode dar origem a uma série de isoformas anormais, outros afirmam mutações somáticas no PRNP (VILLEGAS, VELANDIA, PAYÁN, 2008). Em um estudo realizado por HUANG et al., 2001, um paciente alemão de 57 anos, residente no Brasil, apresentou tontura, que foi rapidamente seguida por distúrbios de marcha e fala.
Foi coletado o sangue periférico do paciente e extraído o DNA, e através disso foi detectada uma mutação pontual causada pela substituição de valina por isoleucina no códon 210 (V210I) e heterozigosidade no códon 129 do gene da proteína príon. É causada pela exposição de indivíduos à proteína príon durante procedimentos neurocirúrgicos, como transplante de córnea ou dura-máter e tratamento com extratos da glândula pituitária de cadáveres humanos. Em estudo de caso realizado por ARRUDA et al., 2004, uma mulher de 54 anos foi internada no Instituto de Neurologia de Curitiba com sintomas progressivos da forma Heidenhain.
Neste estudo foi possível perceber que os príons são partículas proteicas que possuem a mesma organização estrutural de outras proteínas, que se originam da PrPc, uma isoforma da Proteína Príon Celular Normal.
Este estudo conclui que a comunidade científica está a trabalhar incansavelmente no desenvolvimento de medicamentos e terapias paliativas para que os pacientes com estas doenças possam ter uma melhor qualidade de vida quando as consequências desta doença se tornarem graves e marcantes. Ainda há muito a descobrir sobre a doença, mas os esforços de vários cientistas reconhecidos e altamente competentes têm contribuído para uma melhoria do tempo restante destes pacientes que estão condenados a um doloroso fim de vida. Doença de Creutzfeldt-Jakob forma Heidenhain: Relato de caso com ressonância magnética e achados de DWI.
GURGUL, A.; et al. Polymorphism of the prion protein gene (PRNP) in Polish cattle affected by classic bovine spongiform encephalopathy. Familial Creutzfeldt-Jakob disease associated with a point mutation at codon 210 of the prion protein gene. Provide it:
Available at: http://www.scq.ubc.ca/prions-infectious-proteins-responsible-for-mad-cow-disease/.