Doença periodontal e aterosclerose
Periodontal disease and atherosclerosis
Jeferson Freitas Toregeani1, Carlos Augusto Nassar1, Krischina Aparecida Mendes Toregeani1,
Patrícia Oehlmeyer Nassar1
Resumo
A doença aterosclerótica (DA) constitui uma das principais causas de morbimortalidade no mundo. A sua expressão laboral pode ser através de marcadores inlamatórios, como a proteína C reativa (PCR) e/ou o espessamento da parede arterial, que pode ser analisado pela ultrassonograia com Doppler colorido. Os fatores de risco associados à DA são o diabetes mellitus, a hipertensão arterial sistêmica, a dislipidemia e o tabagismo. Mais recentemente, a doença periodontal (DP), que tem uma elevada prevalência na população mundial, tem sido considerada um fator relacionado ao desenvolvimento da DA, em que o processo inlamatório e a atividade bacteriana no periodonto parecem aumentar o risco para a DA. A motivação da higiene oral pode diminuir a expressão dos marcadores inlamatórios da DA. Com base em dados publicados em revistas eletrônicas e indexados pelos mecanismos de busca PUBMED, SCIELO e BIREME, foi realizada uma revisão de literatura sobre a DP e a DA, além dos marcadores inlamatórios expressos em ambas as doenças e suas possíveis inter-relações.
Palavras-chave: doença periodontal; aterosclerose; espessura íntima-média carotídea.
Abstract
Atherosclerotic disease (AD) is one of the most important causes of morbidity and mortality in the world. It expresses inlammatory markers such as C-reactive protein (CRP) and can provoke arterial wall thickening, which can be evaluated using Doppler ultrasound. Risk factors associated with AD include diabetes mellitus, systemic arterial hypertension, dyslipidemia and smoking. More recently, periodontal disease (PD) has been identiied as a factor related to AD. Periodontal disease has a high prevalence in the global population and the inlammatory process and bacterial activity at the periodontium appear to increase the risk of AD. Encouraging good oral hygiene can reduce expression of inlammatory markers of AD. A review of literature on PD, AD and inlammatory markers and the interrelationships between the two diseases was conducted using data published in articles indexed on the PUBMED, SCIELO and BIREME databases.
Keywords: periodontal disease; atherosclerosis; carotid intima-media thickness.
1Universidade Estadual do Oeste do Paraná – UNIOESTE, Cascavel, PR, Brasil.
Fonte de inanciamento: Nenhuma.
Conlito de interesse: Os autores declararam não haver conlitos de interesse que precisam ser informados. Submetido em: 07.11.13. Aceito em: 26.05.14.
DOENÇA ATEROSCLERÓTICA
A doença aterosclerótica (DA) representa um conjunto de alterações histopatológicas que comprometem o sistema circulatório arterial, causando obstrução da luz dos vasos sanguíneos. Tem etiologia multifatorial e caráter crônico, muitas vezes aparecendo precocemente em indivíduos com fatores de risco, como a dislipidemia, o diabetes mellitus e o tabagismo1. São exemplos de DA, o grupo de doenças
denominado doenças cardiovasculares (DCVs), tendo como principais representantes o infarto do miocárdio secundário à obstrução das artérias coronárias, o acidente vascular cerebral secundário à obstrução das artérias carótidas e seus ramos, e a doença arterial periférica, que compromete mais comumente os membros inferiores2.
A DA constitui uma das principais causas de morbimortalidade na população adulta e idosa3.
Segundo a Organização Mundial de Saúde (OMS), em 1995, as DCVs foram responsáveis por 20% das mortes no mundo, chegando à marca de 50% em alguns países em desenvolvimento. Em 1999, as DCVs corresponderam a um terço das mortes no globo terrestre. Em 2010, corresponderam a 33% de todas as mortes no mundo4. Segundo dados do
relatório anual da OMS de 20135, no mundo todo,
32% das mortes foram causadas pelas DCVs. A mortalidade relacionada às DCVs no Brasil, em 2004, foi uma das maiores da América Latina, com 286 óbitos por 100 mil pessoas6. Em 2006, 29%
das causas de óbito no Brasil foram relacionadas às DCVs. No ano de 2008, 37% das mortes foram ocasionadas pelas DVCs5.
Com relação ao gasto com a saúde pública, os custos referentes às internações hospitalares por DCVs no sistema de saúde brasileiro são elevados. Em 2007, 27,4% das internações de indivíduos de 60 anos de idade ou mais foram causadas por DCVs7.
Em 2008, 10% das internações, englobando todas as faixas etárias, foram decorrentes das DCVs, com um custo estimado em 1,6 bilhão de reais8. O baixo
nível socioeconômico de uma população inluencia
negativamente a prevalência das DVCs. Em um estudo realizado por Schmidt et al.7 em Porto Alegre,
Rio Grande do Sul, Brasil, a mortalidade – entre 45 e 64 anos de idade – por DCVs foi 163% mais alta na população mais carente, que residia na periferia da cidade7.
FISIOPATOLOGIA DA DOENÇA ATEROSCLERÓTICA
Do ponto de vista patológico, a DA é o resultado da alteração da permeabilidade do endotélio vascular da camada íntima, com a entrada de lipídios e
células inlamatórias na parede arterial, que são
elementos precursores da placa aterosclerótica9.
Normalmente, ocorre deposição de lipoproteínas de baixa densidade (LDLs) nas células endoteliais. Estas LDLs são hidrolisadas em fosfolipídios, triglicerídeos, proteínas e colesterol. Após a hidrólise, alguns receptores são expressos na membrana celular e outros produtos são utilizados na recomposição da membrana celular, como é o caso do colesterol10.
Na presença de hipercolesterolemia ou de situações que agravem a deposição dos LDLs, ocorre maior consumo de óxido nítrico (ON) na célula endotelial e maior produção de radicais livres, acarretando uma disfunção do metabolismo dos ácidos graxos, das apoproteínas, da lecitina e
da proteína G. O resultado inal é a incapacidade do
endotélio em responder adequadamente às agressões sistemáticas10. Ocorre diminuição da produção de
ON pelas células endoteliais, a qual é provocada pela redução na expressão da ON sintetase, pelo aumento da síntese de nicotinamida adenina dinucleotídeo fosfato oxidase (NADPH oxidase) e pelo aumento da expressão das moléculas de adesão, como a molécula de adesão intercelular do tipo 1 (ICAM‑1), a selectina endotelial e a molécula de adesão de células vasculares do tipo 1 (VCAM‑1), com maior migração de monócitos para a parede arterial e com formação das células espumosas, que são ricas em gordura, evoluindo progressivamente para placa aterosclerótica8,11.
A aterosclerose é um processo que tem início na infância, quando se evidenciam as estrias gordurosas, que são as alterações precursoras das placas de ateroma, que normalmente aparecerão em uma idade mais avançada12. A evolução é variada, com
períodos de quiescência intercalados com períodos de progressão rápida, evoluindo ao longo da vida com deposição de lipídeos ao mesmo tempo em que
o processo inlamatório cresce descontroladamente.
A placa aterosclerótica pode progredir para uma estenose significativa, após diversos anos de evolução silenciosa9. Em seu estágio inal, pode se
tornar instável e evoluir para a rotura, expondo a camada subendotelial que contém lipídeos, colágeno e elastina, o que pode levar à formação do trombo,
que costuma ser o evento agudo e inal da maior parte
das DAs obstrutivas6,8,10.
arterial coronariana (DAC); distúrbios neurológicos, como afasia, paresia, paralisia e amaurose, no caso da doença cerebrovascular, e dor em membros inferiores (claudicação intermitente, dor em repouso e gangrena), no caso da doença arterial periférica (DAP)13,14.
Fatores externos, como a dieta, o tabagismo, o exercício físico e os medicamentos, representados principalmente pelas estatinas e pelos antiagregantes
plaquetários, podem modiicar a evolução da DA e
melhorar o controle dos fatores de risco, diminuindo a expressão das moléculas de adesão e melhorando a resposta endotelial nos pacientes com aterosclerose11.
Desse modo, sabendo‑se que a DA tem caráter evolutivo e que a modificação dos fatores de risco desacelera a sua evolução, devem‑se utilizar mecanismos que permitam encontrar pessoas mais susceptíveis e que apresentarão formas mais graves. Com este objetivo, alguns marcadores podem ser utilizados para avaliar o risco de indivíduos ainda assintomáticos do ponto de vista clínico, mas que terão probabilidade maior de desenvolver DCVs.
Marcadores da doença aterosclerótica
A avaliação da DA pode ser feita através de parâmetros clínicos, laboratoriais e radiológicos. Do ponto de vista clínico, a percepção da DA inicial é falha, pois não se consegue detectar precocemente situações de risco, tendo em vista que os sintomas usualmente costumam aparecer em casos nos quais a obstrução da luz do vaso já ultrapassou 70%15.
Do ponto de vista laboratorial, podem ser obtidas amostras de sangue com o objetivo de se dosarem as concentrações plasmáticas de LDL colesterol, triglicerídeos, glicemia de jejum, hemoglobina glicada e as concentrações de PCR, que são marcadores associados à progressão da DA16.
A PCR é uma proteína de fase aguda pertencente ao grupo das pentraxinas17. Foi descoberta por
Tillett e Francis18. Na época, acreditava‑se que era
uma secreção patogênica. Tem peso molecular de
25.106 Daltons e a sua síntese é codiicada pelo
cromossomo 1. Está presente no sangue e é produzida
pelo fígado em resposta às citocinas pró-inlamatórias
liberadas em qualquer processo inflamatório sistêmico19. Após agressão tecidual, as concentrações
da PCR sobem rapidamente em duas horas e, após
cessar o estímulo inlamatório, regridem ao normal
em até 18 horas20. Em uma revisão sistemática,
Balan3 mostrou que a concentração da PCR é maior
em pacientes com processo inlamatório, como a
periodontite, geralmente acima de 2,1 mg/L.
A PCR, ao se ligar aos fosfolipídios expressos na superfície celular, pode estimular células endoteliais,
musculares lisas e inlamatórias. A proliferação das
células musculares lisas pode causar espessamento nos vasos sanguíneos e o aumento da atividade macrofágica pode levar à formação das células espumosas, que originam o processo aterosclerótico. A estimulação das células endoteliais pode promover
a expressão de receptores inlamatórios, como visto
na Tabela 121,22. Esta proteína também atua em
bactérias e outros agentes patogênicos, ligando‑se à fosfocolina e facilitando, assim, a fagocitose macrofágica. O sistema complemento é responsável
pelo processo inlamatório que leva à destruição
de células e patógenos através da destruição das membranas celulares pela deposição de proteínas chamadas de opsoninas23.
Existem vários mediadores que regulam a expressão da PCR. A IL‑6 é um importante ativador da produção da PCR24. É uma citocina
imunomodulatória e pró‑inflamatória secretada principalmente pelos monócitos, macrófagos e linfócitos T, ativados nos locais de inflamação.
Além da atividade pró-inlamatória, a IL-6 tem ação
pró‑coagulante, que pode agravar as manifestações trombóticas das DCVs25.
Os indivíduos sadios que apresentam valores de PCR acima do normal costumam ter aumento da incidência de DCVs26. A associação entre as
concentrações elevadas das proteínas de fase aguda,
como a PCR, o ibrinogênio e o amiloide sérico tipo
A, e as moléculas de adesão ICAM‑1, E‑Selectina e VCAM‑1 está relacionada com a progressão da aterosclerose e das DCVs8,11.
Tabela 1. Atividade biológica da Proteína ‘C’ Reativa24 (p. 544).
Atividade biológica da Proteína ‘C’ Reativa Fígado
Produção da PCR
• Estimulação por citocinas inlamatórias (IL-6) Célula Endotelial
• Diminuição da produção de óxido nítrico • Aumento da produção de Endotelina 1
• Expressão de moléculas de adesão (ICAM-1 e VCAM-1) • Liberação de Inibidor do ativador do plasminogênio (PAI-1) Célula Muscular Lisa
• Proliferação e migração de células musculares lisas • Produção de Radicais Livres
Monócitos e Macrófagos
• Aumento da captação de colesterol • Expressão de citocinas: IL-1, IL-6 e TNF-α • Expressão de MCP-1
Para avaliação do sistema arterial, servem como
métodos de imagem a ultrassonograia com Doppler colorido, a angiotomograia computadorizada e a
angiorressonância nuclear magnética27. Por se tratar
de um método mais acessível e barato, o ultrassom tem sido utilizado como método preferido de rastreamento da DA28.
O eco‑Doppler colorido é um exame não invasivo, que pode ajudar na detecção das variações morfológicas das artérias quando expostas aos fatores
que propiciam a aterosclerose, além de identiicar
alterações patológicas propriamente ditas, como as
placas ateroscleróticas, as calciicações, as tromboses
e os aneurismas28.
O ultrassom é obtido através de um transdutor formado por cristais (cerâmica ou piezoelétricos), que formam uma onda mecânica oscilatória que varia entre 2 e 17 mHz. Há um hardware que emite informações pré‑programadas aos cristais por meio de softwares especiicamente desenvolvidos. Ao mesmo tempo, o aparelho tem a capacidade de captar
os ecos gerados pela relexão deste mesmo ultrassom
nos tecidos. Ao ser analisado por um software, as ondas são convertidas em uma imagem29,30.
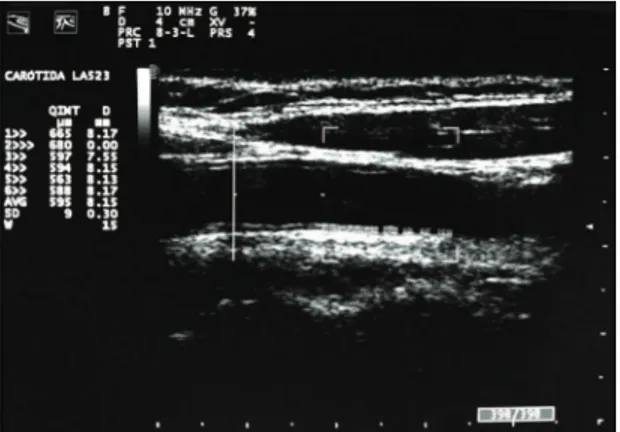
A radiofrequência é uma onda elétrica oscilatória que varia entre 3 kHz e 300 gHz. A aferição da espessura da camada íntima e média da artéria carótida comum, espessamento médio‑intimal (EMI), pode ser realizada pelas duas técnicas; contudo, a radiofrequência tem uma precisão ligeiramente maior, pois o ultrassom interage com os tecidos e pode
sofrer modiicações dependendo das propriedades de
cada órgão analisado, como a densidade e o índice de
relexão. Embora existam pequenas diferenças entre
as técnicas, ambas podem ser utilizadas para avaliar com acurácia o EMI (Figura 1)31‑33.
Em uma artéria normal, existem duas camadas com impedâncias acústicas distintas: a transição entre o sangue e a camada íntima, e a transição entre a camada média e a adventícia. A distância entre estas duas interfaces acústicas forma o complexo médio‑ intimal (CMI). Dentro das manifestações da DA, pode ocorrer o EMI, que normalmente tem caráter difuso, e também a formação da placa aterosclerótica,
que é deinida como uma estrutura focal que invade
o lúmen arterial em pelo menos 0,5 mm ou 50% do valor do CMI adjacente. Os sinais de radiofrequência utilizados para aferir o CMI da artéria carótida comum são capazes de detectar variações precoces do EMI que podem estar relacionadas com as DCVs, servindo também como monitor para avaliar o sucesso do tratamento e da redução dos fatores de risco para as DCVs33.
A aferição do EMI é recomendada pela American Heart Association para avaliação do risco cardiovascular34 e foi delineada pelo consenso de
Mannheim, na Alemanha, em 2004, e revisado em Bruxelas, na Bélgica, em 200635. A artéria carótida
comum tem sido utilizada para a aferição do EMI baseando‑se nas recomendações do American Society for Echocardiography Report36,37.
Doença periodontal
Os dentes são estruturas rígidas que têm a
inalidade de triturar os alimentos, facilitando a sua
digestão no estômago. São constituídos por tecido conjuntivo e matriz mineral, e estão inseridos nos ossos alveolares das arcadas dentárias. O dente é formado também pelo cemento, que é um material rígido, constituído em 50% por material orgânico, em especial o colágeno tipo I, e 50% por apatita. O cemento promove a cobertura da raiz dos dentes, na
qual se inserem as ibras que formam o ligamento
periodontal e que efetivamente dão sustentação ao dente. Além do cemento, os dentes possuem em sua porção mais externa um revestimento com aspecto de porcelana chamado de esmalte, que é constituído em 70% de matriz mineral e 30% de matriz orgânica e água38.
Cada dente recebe um revestimento em sua base, constituído pela diferenciação da mucosa oral em três compartimentos funcionais: o epitélio gengival, o epitélio sulcular e o epitélio juncional. O epitélio juncional tem maior importância por funcionar como protetor dos tecidos dentários profundos e, quando
sofre inlamação, expõe as estruturas que sustentam os dentes, podendo levar a processos inlamatórios,
infecciosos e até mesmo à perda do dente38.
A doença periodontal (DP) é uma doença
inlamatória crônica complexa, com participação
de bactérias, resultando na destruição dos tecidos que sustentam os dentes39. Os sinais clínicos mais
comuns da periodontite são o sangramento e o edema gengival. As formas mais avançadas manifestam‑se
por recessão gengival, aumento do luido crevicular,
produzido ao redor do dente, aumento da mobilidade dos dentes, supuração e, eventualmente, até a perda do dente3,8.
A higiene oral precária ainda continua sendo a maior causa da DP, constituindo‑se na maior causa de perda dos dentes em indivíduos acima dos 40 anos de idade40. Segundo a OMS, a forma grave afeta 10
a 20% das pessoas de todo o mundo40. A prevalência
das formas leve e moderada varia de 20 a 50% na população, podendo chegar a 85% na população mais idosa3. As formas moderadas a graves afetam
cerca de 100 milhões de indivíduos, correspondendo a 15% dos adultos entre 21 e 50 anos de idade, e a 30% dos adultos acima de 50 anos de idade8,11. Nos
EUA, a prevalência de gengivite é de 50% entre adultos e a periodontite afeta 35% da população norte‑americana9.
A saúde bucal no Brasil tem caráter heterogêneo, sendo que, nas regiões menos desenvolvidas, os indivíduos com menores níveis de escolaridade e pertencentes às classes sociais menos favorecidas são os mais afetados. Além disso, são poucas as regiões do Brasil que mantêm um monitoramento constante
e coniável dos indicadores de saúde bucal.
FISIOPATOLOGIA DA DOENÇA PERIODONTAL
Vários micro‑organismos já foram descritos como causadores da DP, particularmente o Porphyromonas gingivalis. As bactérias produzem endotoxinas em forma de lipopolissacarídeos (LPS) capazes de desencadear a liberação de diversas citocinas
pró-inlamatórias implicadas na imunopatologia da
periodontite, bem como nas respostas sistêmicas ao
processo inlamatório bucal39. Podem ser citados
como exemplos: interleucina 1 beta (IL 1β), IL‑6,
fator de necrose tumoral alfa (TNF-α), PCR e neutróilos41,42.
A inlamação crônica associada à placa bacteriana
tem um predomínio de bactérias Gram negativas.
Após o estímulo inlamatório, ocorre um aumento da
produção de prostaglandinas E2 (PGE2) e da matriz de metaloproteinases (MMP), que levam à destruição extracelular da gengiva e do ligamento periodontal, estimulando a reabsorção do osso alveolar8. O
crescimento concomitante de bactérias anaeróbicas pode promover a formação de bolsas periodontais8.
O processo inlamatório do periodonto leva a
um aumento das concentrações da PCR e de outros mediadores, como o fibrinogênio, gerando uma resposta sistêmica40. A DP também é capaz de
inluenciar a ocorrência e a severidade de algumas
doenças sistêmicas, como as DCVs43.
Além destas citocinas, ocorre também aumento das MMPs. Todas as MMPs têm importância na resposta imune, contudo, a MMP‑8 é a única proteinase que tem a capacidade de fragmentar os colágenos tipo I e III, que são importantes na manutenção da estrutura dos dentes. Assim, quanto maior a concentração de MMP‑8, pior o prognóstico da DP. Existe correlação também entre o grau da periodontite e as concentrações de MMP‑13, MMP‑3, MMP‑2 e MMP‑13. Os efeitos da proliferação bacteriana e
da liberação das MMPs são a ativação de múltiplas
células, como os ibroblastos, os queratinócitos,
os macrófagos e as células endoteliais. Ocorre reabsorção óssea através da fragmentação dos elementos da matriz extracelular pelos osteoclastos3.
O processo inlamatório é dependente de múltiplos
fatores internos e externos. Fatores de risco controláveis incluem o hábito de fumar, o estresse, a higiene oral precária e visitas não frequentes ao Dentista. Os fatores de risco não controláveis incluem a hereditariedade, as doenças sistêmicas e a idade. O componente genético não causa a doença; no entanto, torna os pacientes mais susceptíveis a um início ou a um desenvolvimento mais grave da patologia periodontal44. Alguns patógenos, como
a Porphyromonas gingivalis, a Tanerella forsythia e o Fusobacterium nucleatum, também podem
inluenciar a gravidade da doença3.
O estresse também contribui para o aumento da incidência da DP por criar uma condição de resistência aos glicocorticoides e um aumento da produção de IL‑1, IL‑6 e TNF‑α45, como resultado
de uma disfunção dos monócitos CD11b em resposta aos produtos microbianos46. O estresse causa baixa
regulação de genes ativados pelos glicocorticoides que servem para suprimir a resposta imune e alta regulação de genes que causam exacerbação do processo inflamatório, fatores que explicam a íntima relação entre o estresse e a DP47. Pacientes
estressados tendem a adotar hábitos que pioram a
saúde dos dentes, como a higiene oral deiciente, o
tabagismo acima da média e as mudanças negativas nos hábitos alimentares48.
A relação entre a DP e a DA pode ser explicada
que estimulam a aterogênese, ou através da ação direta de bactérias patogênicas, que penetram no
sistema circulatório pelas gengivas inlamadas. Um
modelo hipotético como base biológica sugere que os indivíduos com doenças cardíacas e periodontais possuem uma resposta imunológica exacerbada mediante as infecções bacterianas49. Essa resposta é
explicada por uma capacidade secretória de células monocíticas alteradas, liberando níveis elevados de
mediadores pró-inlamatórios, como PGE-2, IL-1β e TNF‑α. As pessoas com este fenótipo monocítico
hiperinlamatório secretam cerca de três a dez vezes
mais mediadores em resposta aos lipopolissacarídeos bacterianos, quando comparadas com pessoas fenotipicamente normais50.
Segundo Seymour e Steele51, existem evidências
de que pacientes com formas de periodontite
agressiva possuem esse fenótipo hiperinlamatório.
Assim, a interação entre lipopolissacarídeos bacterianos e os monócitos que levam à liberação de várias citocinas é fundamental para a iniciação e a progressão da DP, além de seus efeitos sistêmicos, como a aterogênese e a trombogênese. As elevações das concentrações de PCR aumentam o risco de eventos cardiovasculares em 1,9 vez3.
Além da PCR já discutida anteriormente, outras
proteínas também sofrem ação da inlamação gerada no periodonto. O ibrinogênio plasmático, a contagem
de células brancas e o fator von Willebrand são elevados em pacientes com problemas periodontais52.
Observe‑se que, além disso, os indivíduos com DP e com concentrações elevadas de proteínas de fase
aguda, como a PCR, o ibrinogênio, o amiloide
sérico tipo A, e as moléculas de adesão ICAM‑1, E‑Selectina e VCAM‑1 têm maiores chances de desenvolver aterosclerose e doença cardiovascular11.
O ibrinogênio é produzido nos hepatócitos em
resposta à ação de citocinas, especialmente a IL‑6.
As concentrações plasmáticas de ibrinogênio estão aumentadas durante as infecções e as inlamações
crônicas, criando um estado de hipercoagulabilidade. A trombogênese está relacionada com a aterogênese e com a amplificação da placa aterosclerótica. O fibrinogênio e a fibrina produzidos, por sua vez, interagem com os monócitos, aumentando a produção de IL‑1β, um dos importantes mediadores relacionados à DP53,54.
Segundo um estudo realizado por de Oliveira et al.40, quanto pior a higiene bucal, maiores as
concentrações de PCR e ibrinogênio, e maior o
risco de eventos cardiovasculares. Houve diferença entre os grupos que tinham boa higiene dental (PCR: 3,07 mg/L) quando comparados com os
indivíduos que escovavam os dentes menos de uma vez por dia (PCR: 4,18 mg/L; P<0,05); ocorreu, da
mesma forma, com o ibrinogênio, que apresentou
concentrações maiores no grupo de higiene oral ruim (2,86 g/L × 2,98 g/L; P<0,05).
Embora o tratamento da DP eleve transitoriamente as citocinas pró‑inflamatórias, elevação esta provavelmente relacionada à manipulação dos
tecidos inlamados e a bacteremias transitórias, após
24 a 48 horas, ocorre normalização da atividade
inlamatória e, em longo prazo, se a DP responde
ao tratamento, pode ocorrer até diminuição das concentrações destas citocinas55.
No estudo NHANES I56, constatou‑se um aumento
de 25% no risco relativo para DAC nos pacientes com periodontite. Os riscos relativos para angina e para eventos coronarianos fatais foram 1,5 e 1,9, respectivamente57. A DP está associada com um
aumento de 19% no risco de doença cardiovascular, sendo este risco mais proeminente na população abaixo dos 65 anos, na qual o aumento do risco relativo chegou a 44%58.
Os patógenos periodontais podem ser encontrados colonizando placas ateroscleróticas ao longo do sistema circulatório. A invasão da parede arterial pelo Porphyromonas gingivalis, por exemplo, promove a expressão de moléculas de adesão pelo endotélio, como a IL‑6, a IL‑1β e o TNF‑α. Ocorre, então, o recrutamento de monócitos, o aumento da expressão das moléculas de adesão endotelial e o aumento da captação de lipídeos pelos macrófagos41. Os agentes bacterianos
mais frequentemente encontrados nas placas ateroscleróticas foram: Porphyromonas gingivalis (32%); Aggregatibacter actinomycetencomitans (4%); Prevotella intermédia (20%), e Treponema denticola (32%). O uso de antibióticos sistêmicos nos pacientes com periodontite mostrou diminuição dos marcadores sistêmicos da inflamação40. Em
uma revisão englobando cerca de 90 mil pacientes com periodontite, o risco relativo para DCV foi 120% maior no grupo afetado, quando comparado ao controle59. Segundo Hung et al.60, a periodontite
aumenta o risco relativo da DAP em 1,4 a 2,6 vezes. Segundo Balan3, a DP pode ser considerada um fator
de risco isolado fraco para a DCV, com risco relativo entre 24 e 35% acima do normal.
Entretanto, a relação da periodontite com as DCVs ainda é discutível para alguns autores61. Os fatores
DP quanto da DA, podendo dar uma falsa impressão de que a aterosclerose aumenta a incidência da periodontite ou vice‑versa62.
Deste modo, montar uma estratégia para o combate ao processo de adoecimento dos dentes
e seus relexos em outros sistemas orgânicos vai
demandar uma mudança nos costumes e hábitos, que podem representar maiores gastos com os próprios
dentes naturais. Os proissionais da saúde, portanto,
terão a tarefa de mostrar à população a importância de manter os dentes naturais mesmo que isto represente mais cuidados e mais gastos com a saúde de cada indivíduo. Neste ponto, os serviços de saúde
devem estabelecer uma estratégia com a inalidade
de facilitar o acesso aos serviços odontológicos, em especial para as populações mais carentes, nas quais a prevalência das doenças periodontais é maior. Lidar com este paradigma, às vezes, se torna uma tarefa difícil e até a literatura pode se opor à preservação dos dentes naturais, pois quem não tem mais dentes não tem mais DP, e assim, corre menos risco de ter os
relexos sistêmicos da periodontite. Partindo-se deste
princípio, talvez se comece a indicar a retirada dos rins, para evitar cálculos renais, e do coração, para evitar infarto do miocárdio, entre outros.
REFERÊNCIAS
1. Dumitrescu AL. Inluence of periodontal disease on cardiovascular diseases. Rom J Intern Med. 2005;43(1-2):9-21.
2. Bongard V, Cambou JP, Lezorovcz A, et al. Comparison of cardiovascular risk factors and drug use in 14,544 French patients with a history of myocardial infarction, ischaemic stroke and/ or peripheral arterial disease. Eur J Cardiovasc Prev Rehabil. 2004;11(5):394-402.
3. Balan H. Do cardio-vascular and periodontal diseases have a
close, causal relationship? Rom J Intern Med. 2010;48(2):121-9. 4. World Health Organization - WHO. NCD Brazil proiles. Genebra:
World Health Organization – WHO; 2011.
5. World Health Statistics [site na Internet]. 2013. Genebra: World Heath Organization – WHO. [atualizado 2014; citado 2013 maio 29]. www.who.int
6. Fischer RG. Doença periodontal e doenças cardiovasculares. In: Paiva JS, Almeida RV, editors. Periodontia: a atuação clínica baseada em evidências cientíicas. São Paulo: Artes Médicas; 2005. p. 285-9.
7. Schmidt MI, Duncan BB, Azevedo e Silva G, et al. Chronic non-communicable diseases in Brazil: burden and current challenges. Lancet. 2011;377(9781):1949-61. http://dx.doi.org/10.1016/S0140-6736(11)60135-9. PMid:21561658
8. Paizan ML, Martin JFV. Association between periodontal disease, cardiovascular disease and hypertension. Rev Bras Hipertens. 2009;16(3):183-5.
9. Almeida CSL, Dias LZS. The importance of inflammatory citokynes in the causal relationship between periodontal and cardiovascular diseases. Rev ABO Nac. 2008;16(5):294-8.
10. Jorge PAR. Endotélio, lípides e aterosclerose. Arq Bras Cardiol. 1997;68(2):129-34. PMid:9433841.
11. Ramírez JH, Arce RM, Contreras A. Periodontal treatment efects on endothelial function and cardiovascular disease biomarkers in subjects with chronic periodontitis: protocol for a randomized clinical trial. Trials. 2011;12(1):46. http://dx.doi.org/10.1186/1745-6215-12-46. PMid:21324167
12. Napoli C, Glass CK, Witztum JL, Deutsch R, D’Armiento FP, Palinski W. Inluence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet. 1999;354(9186):1234-41. http://dx.doi.org/10.1016/ S0140-6736(99)02131-5. PMid:10520631
13. Teixeira J, Morado Pinho M. Association between periodontitis and stroke (CVA). Rev Port Estomatol Med Dent Cir Maxilofac. 2011;52(2):115-21.
14. Nguyen TT, Liao Y, Gildengorin G, Tsoh J, Bui-Tong N, McPhee SJ. Cardiovascular risk factors and knowledge of symptoms among Vietnamese Americans. J Gen Intern Med. 2009;24(2):238-43. http://dx.doi.org/10.1007/s11606-008-0889-1. PMid:19089498 15. Hara T, Takamura N, Akashi S, et al. Evaluation of clinical markers
of atherosclerosis in young and elderly Japanese adults. Clin Chem Lab Med. 2006;44(7):824-9. http://dx.doi.org/10.1515/ CCLM.2006.149. PMid:16776627
16. Kanoh Y. [Biochemical markers and physiological tests of atherosclerosis—changes and usefulness of markers in anti-atherosclerotic therapy]. Rinsho Byori. 2011;59(1):80-8. PMid:21404587
17. hompson D, Pepys MB, Wood SP. he physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7(2):169-77. http://dx.doi.org/10.1016/S0969-2126(99)80023-9. PMid:10368284
18. Tillett WS, Francis T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J Exp Med. 1930;52(4):561-71. http://dx.doi.org/10.1084/jem.52.4.561. PMid:19869788
19. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inlammation. N Engl J Med. 1999;340(6):448-54. http://dx.doi.org/10.1056/NEJM199902113400607. PMid:9971870 20. Pepys MB, Hirschield GM. C-reactive protein: a critical update.
J Clin Invest. 2003;111(12):1805-12. http://dx.doi.org/10.1172/ JCI200318921. PMid:12813013
21. Torzewski M, Rist C, Mortensen RF, et al. C-reactive protein in the arterial intima: role of C-reactive protein receptor-dependent monocyte recruitment in atherogenesis. Arterioscler hromb Vasc Biol. 2000;20(9):2094-9. http://dx.doi.org/10.1161/01. ATV.20.9.2094. PMid:10978254
22. Volp AC, Alfenas RC, Costa NM, Minim VP, Stringueta PC, Bressan J. [Inlammation biomarkers capacity in predicting the metabolic syndrome]. Arq Bras Endocrinol Metabol. 2008;52(3):537-49. http://dx.doi.org/10.1590/S0004-27302008000300015. PMid:18506280
23. Torzewski M, Bhakdi S. Complement and atherosclerosis-united to the point of no return? Clin Biochem. 2013;46(1-2):20-5. PMid:23010447
24. Steel DM, Whitehead AS. he major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today. 1994;15(2):81-8. http:// dx.doi.org/10.1016/0167-5699(94)90138-4. PMid:8155266 25. Biasucci LM, Vitelli A, Liuzzo G, et al. Elevated levels of
26. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286(3):327-34. http://dx.doi.org/10.1001/ jama.286.3.327. PMid:11466099
27. Rudd JH, Myers KS, Sanz J, Fayad ZA. Multimodality imaging of atherosclerosis (magnetic resonance imaging/computed tomography/positron emission tomography-computed tomography). Top Magn Reson Imaging.
2007;18(5):379-88 . http://dx.doi.org/10.1097/rmr.0b013e3181598db0.
PMid:18025992
28. Casella IB, Presti C, Porta RM, Sabbag CR, Bosch MA, Yamazaki Y. A practical protocol to measure common carotid artery intima-media thickness. Clinics (São Paulo). 2008;63(4):515-20. http:// dx.doi.org/10.1590/S1807-59322008000400017. PMid:18719764 29. de Korte CL, Hansen HH, van der Steen AF. Vascular ultrasound for atherosclerosis imaging. Interface Focus. 2011;1(4):565-75. http://dx.doi.org/10.1098/rsfs.2011.0024. PMid:22866231
30. Tola M, Yurdakul M, Cumhur T. Combined use of color
duplex ultrasonography and B-low imaging for evaluation of patients with carotid artery stenosis. AJNR Am J Neuroradiol. 2004;25(10):1856-60. PMid:15569764
31. ESAOTE SpA. Discover the extraordinary potential of RF-data Technology, the unexplored side of ultrasound. Geneva: WHO Press; 2009 [citado 2009 abr. 03]. http://www.esaote.com.ar/ media/docs/ProductNews_RF%20Technology.pdf
32. Ciccone MM, Scicchitano P, Zito A, et al. Correlation between coronary artery disease severity, left ventricular mass index and carotid intima media thickness, assessed by radio-frequency. Cardiovasc Ultrasound. 2011;9(1):32. http://dx.doi. org/10.1186/1476-7120-9-32. PMid:22087814
33. Schreuder FH, Graf M, Hameleers JM, Mess WH, Hoeks AP.
Measurement of common carotid artery intima-media thickness in clinical practice: comparison of B-mode and RF-based technique. Ultraschall Med. 2009;30(5):459-65. http://dx.doi. org/10.1055/s-0028-1109187. PMid:19544231
34. Smith SC Jr, Amsterdam E, Balady GJ, et al. Prevention Conference V: Beyond secondary prevention: identifying the high-risk patient for primary prevention: tests for silent and inducible ischemia: Writing Group II. Circulation. 2000;101(1):E12-6. http://dx.doi. org/10.1161/01.CIR.101.1.e12. PMid:10618317
35. Touboul PJ, Hennerici MG, Meairs S, et al. Mannheim
carotid intima-media thickness consensus (2004-2006). An update on behalf of the Advisory Board of the 3rd and 4th Watching the Risk Symposium, 13th and 15th European Stroke Conferences, Mannheim, Germany, 2004, and Brussels, Belgium, 2006. Cerebrovasc Dis. 2007;23(1):75-80. http://dx.doi. org/10.1159/000097034. PMid:17108679
36. Roman MJ, Naqvi TZ, Gardin JM, Gerhard-Herman M, Jaf M, Mohler E, and the American Society of Echocardiography, and the Society for Vascular Medicine and Biology. American society of echocardiography report. Clinical application of noninvasive vascular ultrasound in cardiovascular risk stratiication: a report from the American Society of Echocardiography and the Society for Vascular Medicine and Biology. Vasc Med. 2006;11(3):201-11. http://dx.doi.org/10.1177/1358863x06070511. PMid:17288128 37. Gerhard-Herman M, Gardin JM, Jaf M, Mohler E, Roman M,
Naqvi TZ, American Society of Echocardiography, Society for Vascular Medicine and Biology. Guidelines for noninvasive vascular laboratory testing: a report from the American Society of Echocardiography and the Society for Vascular Medicine and Biology. Vasc Med. 2006;11(3):183-200. http://dx.doi. org/10.1177/1358863x06070516. PMid:17288127
38. Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontol. 2000;40:11-28.
39. Buhlin K, Gustafsson A, Pockley AG, Frostegård J, Klinge B. Risk factors for cardiovascular disease in patients with periodontitis. Eur Heart J. 2003;24(23):2099-107. http://dx.doi.org/10.1016/j. ehj.2003.09.016. PMid:14643270
40. de Oliveira C, Watt R, Hamer M. Toothbrushing, inlammation, and risk of cardiovascular disease: results from Scottish Health Survey. BMJ. 2010;340(may27 1):c2451. http://dx.doi.org/10.1136/ bmj.c2451. PMid:20508025
41. Chen YW, Umeda M, Nagasawa T, et al. Periodontitis may increase the risk of peripheral arterial disease. Eur J Vasc Endovasc Surg. 2008;35(2):153-8. http://dx.doi.org/10.1016/j. ejvs.2007.08.016. PMid:17964192
42. Iacopino AM, Cutler CW. Pathophysiological relationships between periodontitis and systemic disease: recent concepts involving serum lipids. J Periodontol. 2000;71(8):1375-84. http:// dx.doi.org/10.1902/jop.2000.71.8.1375. PMid:10972656
43. Mattila KJ, Nieminen MS, Valtonen VV, et al. Association
between dental health and acute myocardial infarction. BMJ. 1989;298(6676):779-81. http://dx.doi.org/10.1136/ bmj.298.6676.779. PMid:2496855
44. Newman MG, Takei HH, Klokkevold, PR. Carranza FA, Carranza periodontia clínica. Rio de Janeiro: Elsevier; 2006.
45. Powell CA, Bannister SR, Mackey SA, Maller SC, McDonnell HT, Deas DE. Periodontal wound healing with and without platelet-rich plasma: histologic observations and assessment of lap tensile strength. J Periodontol. 2009;80(6):985-92. http://dx.doi. org/10.1902/jop.2009.080626. PMid:19485830
46. Bailey MT, Kinsey SG, Padgett DA, Sheridan JF, Leblebicioglu B. Social stress enhances IL-1beta and TNF-alpha production by Porphyromonas gingivalis lipopolysaccharide-stimulated CD11b+ cells. Physiol Behav. 2009;98(3):351-8. http://dx.doi. org/10.1016/j.physbeh.2009.06.013. PMid:19560480
47. Hilgert JB, Hugo FN, Bandeira DR, Bozzetti MC. Stress, cortisol, and periodontitis in a population aged 50 years
and over. J Dent Res. 2006;85(4):324-8 . http://dx.doi.
org/10.1177/154405910608500408. PMid:16567552
48. Deinzer R, Förster P, Fuck L, Herforth A, Stiller-Winkler R, Idel H. Increase of crevicular interleukin 1beta under academic stress at experimental gingivitis sites and at sites of perfect oral hygiene. J Clin Periodontol. 1999;26(1):1-8. http://dx.doi. org/10.1034/j.1600-051X.1999.260101.x. PMid:9923503 49. Friedewald VE, Kornman KS, Beck JD, et al, and the American
Journal of Cardiology, and the Journal of Periodontology. he American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol. 2009;104(1):59-68. http:// dx.doi.org/10.1016/j.amjcard.2009.05.002. PMid:19576322 50. Beck JD, Eke P, Heiss G, et al. Periodontal disease and
coronary heart disease: a reappraisal of the exposure.
Circulation. 2005;112(1):19-24. http://dx.doi.org/10.1161/
CIRCULATIONAHA.104.511998. PMid:15983248
51. Seymour RA, Steele JG. Is there a link between periodontal disease and coronary heart disease? Br Dent J. 1998;184(1):33-8. http://dx.doi.org/10.1038/sj.bdj.4809536. PMid:9479812 52. Mattila K, Vesanen M, Valtonen V, et al. Effect of treating
periodontitis on C-reactive protein levels: a pilot study. BMC Infect Dis. 2002;2(1):30. http://dx.doi.org/10.1186/1471-2334-2-30. PMid:12475397
coronary heart disease]. Hua Xi Kou Qiang Yi Xue Za Zhi. 2008;26(3):262-6. PMid:18705507
54. Ge S, Wu YF, Liu TJ, He QM, Zhao L, Meng S. [Correlation between levels of ibrinogen, beta455 g/A ibrinogen gene polymorphism and chronic periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. 2008;43(2):87-91. PMid:18683729
55. Tonetti MS, Van Dyke TE, and the Working group 1 of the joint EFP/AAP workshop. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/ AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol. 2013;40(Suppl 14):S24-9. http://dx.doi.org/10.1111/ jcpe.12089. PMid:23627332
56. DeStefano F, Anda RF, Kahn HS, Williamson DF, Russell CM. Dental disease and risk of coronary heart disease and mortality. BMJ. 1993;306(6879):688-91. http://dx.doi.org/10.1136/ bmj.306.6879.688. PMid:8471920
57. Beck J, Garcia R, Heiss G, Vokonas PS, Ofenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. 1996;67(10, Suppl):1123-37. http://dx.doi.org/10.1902/jop.1996.67.10.1123. PMid:8910831
58. Janket SJ, Baird AE, Chuang SK, Jones JA. Meta-analysis of periodontal disease and risk of coronary heart disease and stroke. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.
2003;95(5):559-69. http://dx.doi.org/10.1067/moe.2003.107.
PMid:12738947
59. homopoulos C, Tsiouis C, Soldatos N, Kasiakogias A, Stefanadis C. Periodontitis and coronary artery disease: a questioned association between periodontal and vascular plaques. Am J Cardiovasc Dis. 2011;1(1):76-83. PMid:22254188
60. Hung HC, Willett W, Merchant A, Rosner BA, Ascherio A, Joshipura KJ. Oral health and peripheral arterial disease. Circulation. 2003;107(8):1152-7. http://dx.doi.org/10.1161/01. CIR.0000051456.68470.C8. PMid:12615794
61. Lane JS, Vittinghof E, Lane KT, Hiramoto JS, Messina LM. Risk factors for premature peripheral vascular disease: results for the National Health and Nutritional Survey, 1999-2002. J Vasc Surg. 2006;44(2):319-24, discussion 324-5. http://dx.doi.org/10.1016/j. jvs.2006.04.015. PMid:16890861
62. Genco R, Offenbacher S, Beck J. Periodontal disease and cardiovascular disease: epidemiology and possible mechanisms. J Am Dent Assoc. 2002;133(1, Suppl):14S-22S. http://dx.doi. org/10.14219/jada.archive.2002.0375. PMid:12085720
Correspondência
Jeferson Freitas Toregeani Rua Dom Pedro II, 2359 CEP 85812-120 – Cascavel (PR), Brasil Fone: +55 (45) 3225-1288 E-mail: [email protected]
Informações sobre os autores
JFT - Cirurgião Vascular, TSBACV, Área de Atuação em Ecograia Vascular, Professor Assistente da Disciplina de Cirurgia Vascular da Universidade Estadual do Oeste do Paraná (UNIOESTE) e da Faculdade Assis Gurgacz (FAG). Mestre em Biociências e Saúde - UNIOESTE. CAN e PON - Mestres e Doutores em Periodontia pela Universidade Estadual Paulista Júlio de Mesquita Filho (UNESP), Professores Titulares de Periodontia da Universidade Estadual do Oeste do Paraná (UNIOESTE). KAMT - Nutricionista, Graduanda do Curso de Ciências Biológicas na Universidade Estadual do Oeste do Paraná (UNIOESTE). Mestre em Nutrição pela Universidade Estadual de Maringá (UEM).
Contribuições dos autores
Concepção e desenho do estudo: JFT, CAN Análise e interpretação dos dados: JFT, CAN, KAMT, PON Coleta de dados: JFT, CAN, PON Redação do artigo: JFT, CAN Revisão crítica do texto: CAN, PON Aprovação inal do artigo*: JFT, CAN, KAMT, PON Análise estatística: JFT, PON Responsabilidade geral do estudo: JFT, CAN Informações sobre inanciamento: Não houve.