Role of Autophagy in the Control of Body Metabolism
Texto
Imagem
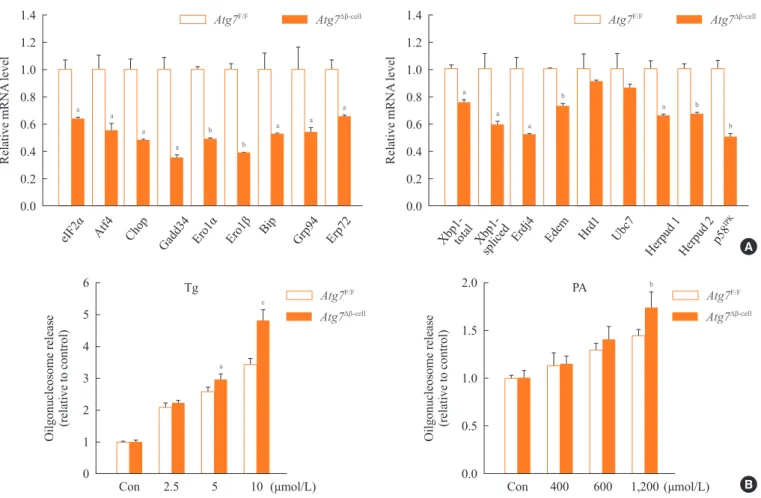
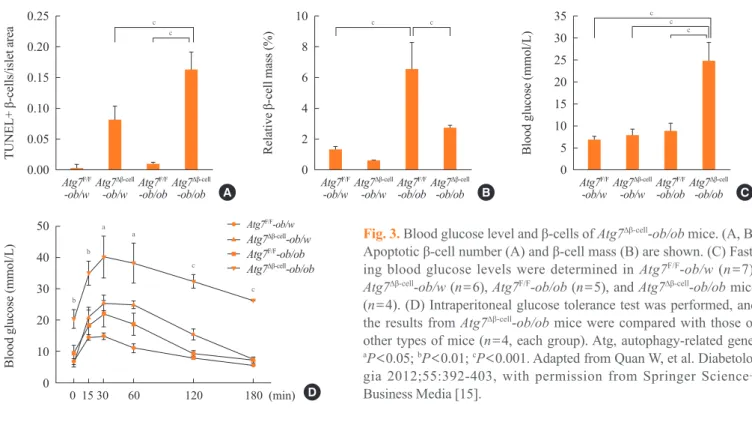
Documentos relacionados
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
Nosso objetivo neste parágrafo é introduzir alguns conceitos relacionados com condições de finitude sobre G, definir grupos e pares de dualidade e enunciar
The variables included in the regression are: Tobin’s Q (measured as the total market value of the firm divided by its total asset value), ACSIZE (the number of AC members),
Yet in the second chapter, dealing with the relations between HIR and po- litical theories, Robert Frank reminds us of the importance of the 1964’s work to consolidate the relevance
Para Cohn (1997), trata-se de ajudar o cliente a libertar-se das conse- quências perturbadoras da negação e evasão no seu confronto com os dados da existência, acedendo a uma forma
Primeiramente, observamos como dito na introdução do Capítulo 2 que o ramo atualmente denominado de cálculo fracionário, historicamente, é tão antigo quanto o cálculo tradicional
No campo das invalidades, por vezes colidem a preponderância do princípio da legalidade com a segurança jurídica, principalmente diante da ampliação da esfera de direitos
El feminismo denunció el lugar privilegiado del sujeto varón en la enunciación del mundo, la construcción de la epistemología científica, la normatividad, etc. Pero así
