Giliane Cristina Medeiros do Nascimento Santos
Remoção de frações de óleo leve e pesado em rocha calcária através
de sistemas microemulsionados
___________________________________________________________
Dissertação de Mestrado
Natal/RN, julho de 2013
INSTITUTO DE QUÍMICA
GILIANE CRISTINA MEDEIROS DO NASCIMENTO SANTOS
REMOÇÃO DE FRAÇÕES DE ÓLEO LEVE E PESADO DE ROCHA
CALCÁRIA ATRAVÉS DE SISTEMAS MICROEMULSIONADOS
Dissertação de mestrado apresentada ao Programa de
Pós Graduação em Química da Universidade Federal do
Rio Grande do Norte, como parte dos requisitos para
obtenção do título de Mestre em Química.
Orientadores: Profa. Dra. Tereza Neuma de Castro Dantas
Dra. Cátia Guaraciara Fernandes Teixeira Rossi
NATAL - RN
UFRN / Biblioteca Central Zila Mamede. Catalogação da Publicação na Fonte.
Santos, Giliane Cristina Medeiros do Nascimento.
Remoção de frações de óleo leve e pesado em rocha calcária através de sistemas microemulsionados. / Giliane Cristina Medeiros do NascimentoSantos. – Natal, RN, 2013.
86 f.: il.
Orientadora: Profa. Dra. Tereza Neuma de Castro Dantas.
Co-orientadora: Profa. Dra. Cátia Guaraciara Fernandes Teixeira Rossi.
Dissertação (Mestrado) – Universidade Federal do Rio Grande do Norte. Centro de Ciências Exatas e da Terra. Instituto de Química. Programa de Pós-Graduação em Química.
1. Microemulsão - Dissertação. 2. Calcário - Dissertação. 3. Óleo pesado - Dissertação. 4. Óleo leve – Dissertação. 5. Remoção de óleo – Dissertação. I. Dantas, Tereza Neuma de Castro. II. Rossi, Cátia Guaraciara Fernandes Teixeira. III. Universidade Federal do Rio Grande do Norte. IV. Título.
“A ciência nunca resolve um problema sem criar pelo menos outros dez.”
DEDICATÓRIA
À Deus por estar sempre comigo, me apoiando principalmente nos momentos mais difíceis.
À Deus, por estar presente em todos os momentos da minha vida, principalmente os mais
difíceis, me dando forças e confiança para seguir em frente.
À minha família, por estarem presentes nos momentos de dificuldades e de alegrias, pela
paciência nos momentos difíceis e pela confiança que vocês sempre depositam em mim.
Ao meu marido, André Luiz Gomes dos Santos, pelo incentivo, compreensão,
companheirismo, paciência e apoio incondicional.
A professora Dra. Tereza Neuma de Castro Dantas e a Dra. Cátia Guaraciara Fernandes
Teixeira Rossi, pela oportunidade da realização deste trabalho, pela orientação, companheirismo
e principalmente pela amizade.
Aos meus professores que facilitaram minha caminhada nesta Universidade, me
ensinando esta maravilhosa ciência.
Aos meus amigos, por tudo que pudemos compartilhar, alegrias e tristezas, certezas e
dúvidas, vitórias e derrotas, sofrimentos. Principalmente a Flávia pela bela amizade construída.
Uma irmã que a vida me deu!
Enfim, a todos que não citei, pois muitos cooperaram para o meu crescimento pessoal e
RESUMO
O presente trabalho objetivou estudar a remoção de frações de óleo leve e pesado em
rocha calcária desintegrada através de sistemas microemulsionados, comparando as eficiências de
remoção em diferentes concentrações de matéria ativa (C/T) e tempo de contato. Os sistemas
microemulsionados (SME) são constituídos por tensoativo, cotensoativo, fase oleosa e fase
aquosa. Nos sistemas estudados, três pontos ricos em água da região de microemulsão foram
utilizados para verificar a eficiência de remoção. Os sistemas foram caracterizados para avaliar a
influência do tamanho do agregado, tensão superficial e viscosidade na estabilidade micelar e
compreender como as propriedades físicas podem influenciar o processo de remoção de óleo. A
amostra de rocha calcária foi caracterizada por Termogravimetria, Área BET, Microscopia
Eletrônica de Varredura, Difração de Raios-X e Fluorescência de Raios-X. A rocha preparada foi
colocada em contato com solução de óleo leve e pesado em xileno para permitir a adsorção de
óleo. Os testes de remoção foram realizados a fim de avaliar a influência do tempo de contato (1,
30, 60 e 120 minutos), da concentração de matéria ativa (20, 30 e 40%), do cotensoativo e da fase
oleosa. Para o óleo pesado, o melhor resultado foi para o SME 1, com 20 % de matéria ativa, no
tempo de 1 minuto, com 93,33 % de eficiência. Para o óleo leve, o SME 1 no percentual de 20 %,
com 120 minutos apresentou o melhor rendimento, com 62,38 %. A partir dos resultados obtidos,
concluiu-se que os sistemas microemulsionados apresentam-se como uma alternativa eficaz para
remoção de óleo em formações calcárias.
In this research the removal of light and heavy oil from disintegrated limestone was
investigated with use of microemulsions. These chemical systems were composed by surfactant,
cosurfactant, oil phase and aqueous phase. In the studied systems, three points in the water -rich
microemulsion region of the phase diagrams were used in oil removal experiments. These
microemulsion systems were characterized to evaluate the influence of particle size, surface
tension, density and viscosity in micellar stability and to understand how the physical properties
can influence the oil recovery process. The limestone rock sample was characterized by
thermogravimetry, BET area, scanning electron microscopy and X-ray fluorescence. After
preparation, the rock was placed in contact with light and heavy oil solutions to allow oil
adsorption. The removal tests were performed to evaluate the influence of contact time (1 minute,
30 minutes, 60 minutes and 120 minutes), the concentration of active matter (20, 30 and 40 %),
different cosurfactants and different oil phases. For the heavy oil, the best result was on SME 1,
with 20 % of active matter, 1 minute of contact time, with efficiency of 93,33 %. For the light oil,
also the SME 1, with 20 % of active matter, 120 minutes of contact time, with 62,38 % of
efficiency. From the obtained results, it was possible to conclude that microemulsions can be
considered as efficient chemical systems for oil removal from limestone formations.
SUMÁRIO
1. INTRODUÇÃO ... 17
2. ASPECTOS TEÓRICOS ... 21
2.1. TENSOATIVOS... 21
2.1.1. Definição ... 21
2.1.2. Classificação ... 21
2.1.2.1. Tensoativos Iônicos ... 22
2.1.2.2. Tensoativos Catiônicos ... 22
2.1.2.3. Tensoativos Aniônicos ... 22
2.1.2.4. Tensoativos Anfóteros ... 23
2.1.2.5. Tensoativos Não-Iônicos... 23
2.1.3. Balanço hidrofílico-lipofílico (BHL) ... 24
2.1.4. Concentração Micelar Crítica (c.m.c.) ... 24
2.2. MICELAS ... 25
2.3. MICROEMULSÕES ... 26
2.3.1. Definição ... 26
2.3.2. Formação das Microemulsões ... 26
2.3.3. Estrutura das Microemulsões ... 27
2.4. SISTEMAS DE WINSOR ... 28
2.5. REPRESENTAÇÃO DE WINSOR EM DIAGRAMA DE FASES ... 29
2.5.1. Sistemas Ternários e Quaternários ... 29
2.5.2. Sistemas Pseudoternários ... 29
2.6. ASPECTOS GERAIS SOBRE ADSORÇÃO ... 30
2.6.1. Fatores determinantes do Processo de Adsorção ... 30
2.6.1.1. Temperatura ... 30
2.6.1.2. Natureza do Solvente ... 31
2.6.1.3. Velocidade de Adsorção ... 31
2.6.1.4. Estrutura do Poro ... 31
2.6.1.5. Área Superficial do Adsorvente ... 31
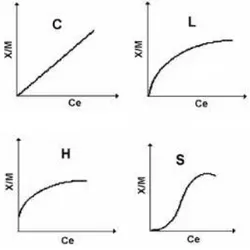
2.7. CLASSIFICAÇÃO E MODELAGEM DAS ISOTERMAS ... 33
2.7.1. As Principais Isotermas ... 33
2.7.1.1. A Isoterma “C” ... 34
2.7.1.2. A Isoterma “L” ... 34
2.7.1.3. A Isoterma “H” ... 34
2.7.1.4. A Isoterma “S” ... 35
2.7.1.5. Modelo BET (Brunauer, Emmett & Teller)... 35
2.8. CALCÁRIO ... 38
2.8.1. Movimento de Fluidos em rochas ... 38
2.8.1.1. Porosidade ... 38
2.8.1.2. Permeabilidade ... 39
2.8.2. Remoção de contaminantes ... 40
2.8.3. Remoção de frações leves e pesadas em rocha calcária através de Sistemas Microemulsionados ... 41
2.9. ESTADO DA ARTE ... 41
2.9.1. Calcário ... 41
2.9.2. Adsorção ... 43
2.9.3. Tensoativos e microemulsão ... 45
3. MATERIAIS E MÉTODOS ... 49
3.1. METODOLOGIA... 49
3.1.1. Materiais ... 51
3.1.2. Obtenção e determinação de sistemas microemulsionados através dos diagramas de fases...51
3.1.3. Preparação e caracterização da rocha calcária. ... 51
3.1.4. Adsorção de óleo leve e pesado em Rocha Calcária moída... 52
3.1.5. Remoção de óleo leve e pesado de Rocha Calcária moída ... 53
3.2. TÉCNICAS UTILIZADAS ... 54
3.2.1. Análise Térmica ... 54
3.2.2. Microscopia Eletrônica de Varredura ... 55
3.2.4. Difratometria de Raios-X ... 57
3.2.5. Área BET ... 58
4. RESULTADOS E DISCUSSÃO ... 61
4.1. CARACTERIZAÇÃO DA ROCHA CALCÁRIA ... 61
4.1.1. Análise Termogravimétrica da Rocha Calcária ... 61
4.1.2. Microscopia Eletrônica de Varredura (MEV) ... 61
4.1.3. Fluorescência de Raios-X (FRX) ... 63
4.1.4. Difratometria de Raios-X (DRX) ... 63
4.1.5. Área BET (ABET) ... 63
4.2. ADSORÇÃO DE ÓLEO LEVE E PESADO NA ROCHA CALCÁRIA MOÍDA ... 64
4.3. CRITÉRIO DE ESCOLHA DOS SISTEMAS MICROEMULSIONADOS ... 64
4.4. ESCOLHA E CARACTERIZAÇÃO DOS PONTOS NOS SISTEMAS MICROEMULSIONADOS. ... 66
4.5. REMOÇÃO DE ÓLEO DE ROCHA CALCÁRIA MOÍDA UTILIZANDO SISTEMAS MICROEMULSIONADOS... 69
4.5.1. Influência do percentual de C/T e tempo de contato. ... 69
4.5.1.1. Remoção de óleo pesado com sistemas microemulsionados ... 69
4.5.1.2. Remoção de óleo leve com sistemas microemulsionados ... 72
4.5.2. Considerações Finais ... 75
5. CONCLUSÕES... 77
Figura 1 - Representação de um tensoativo (molécula anfifílica) ... 21
Figura 2 - Exemplo de tensoativo catiônico ... 22
Figura 3 - Exemplo de tensoativo aniônico ... 22
Figura 4 - Exemplo de tensoativo anfótero ... 23
Figura 5 - Exemplo de tensoativo não-iônico ... 23
Figura 6 - Representação da formação de micelas ... 25
Figura 7 - (a) Micela direta e (b) Micela inversa ... 26
Figura 8 - Exemplos de fases em Sistemas Microemulsionados ... 28
Figura 9 - Classificação de Winsor ... 29
Figura 10 - Representação gráfica de diagramas de fases ... 30
Figura 11 - Quatro principais modelos de isoterma para adsorção em sólidos. ... 34
Figura 12 - Isotermas de adsorção de BET ... 35
Figura 13 - Classificação das histereses de acordo com formato do mesoporos ... 37
Figura 14 - Fluxogramas da metodologia experimental ... 49
Figura 15 - Rocha calcária após desintegração, aquecimento e peneiramento. ... 52
Figura 16 - Rocha calcária após contaminação ... 53
Figura 17 - Equipamento DTG50 – Shimadzu... 55
Figura 18 - Equipamento de MEV (XL-30 - ESEM) ... 56
Figura 19 - Equipamento de FRX (modelo XRF-1800) ... 57
Figura 20 - Equipamento de DRX (modelo XRD-6000 - Shimadzu) ... 58
Figura 21 - Equipamento de Área BET (NOVA V. 8.0 – Quantachrome) ... 59
Figura 22 - MEV da rocha calcária calcinada a 400°C ... 62
Figura 23 - MEV da rocha calcária aquecida a 250°C ... 62
Figura 24 - Adsorção de óleo leve e pesado em Rocha Calcária moída... 64
Figura 25 - Diagramas do sistema microemulsionado... 65
Figura 26 - Eficiências de remoção obtidas para o Óleo Pesado ... 70
LISTA DE TABELAS
Tabela 1 - Valores de BHL em função da aplicação do tensoativo ... 24
Tabela 2 - Tamanho médio das micelas em emulsões ... 27
Tabela 3 - Classificação dos poros de acordo com os diâmetros ... 36
Tabela 4 - Porosidade de alguns materiais ... 39
Tabela 5 - Propriedade físicas medidas em análises térmicas ... 54
Tabela 6 - Fluorescência de Raios-X da Rocha Calcária moída ... 63
Tabela 7 - Sistemas microemulsionados escolhidos ... 65
Tabela 8 - Caracterizações dos sistemas microemulsionados ... 67
Tabela 9 - Eficiências de remoção obtidas (por sistema) ... 69
RN - Rio Grande do Norte
PR - Paraná
CE - Ceará
C/T - Cotensoativo/ tensoativo
pH - Potencial hidrogeniônico
BHL - Balanço hidrofílico-lipofílico
c.m.c - Concentração micelar crítica
O/A - Micelas óleo em água
A/O - Micelas água em óleo
km - Quilômetro
nm - Nanômetro
WI - Winsor I
WII - Winsor II
WIII - Winsor III
WIV - Winsor IV
Fo - Fase óleo
Fa - Fase água
ME - Microemulsão
BET - Isotermas determinadas por Brunauer, Emmet e Teller
IUPAC - International Union of Pure and Applied Chemistry
Å - Angstrom
g - Grama
mL - Mililitro
C - Constant partition
L - Langmuir
H - High affinity
S - Spherical
ɸ - Diâmetro de poro P - Pressão
∆adsH - Variação de entalpia de adsorção
∆ desH - Variação de entalpia de dessorção %P - Permeabilidade
Vporo - Volume de poro
Vtotal - Volume total
Q - Vazão do fluido (cm3/s)
K - Permeabilidade do meio poroso (Darcy)
ƞ - Viscosidade absoluta (cP)
L - Comprimento do meio poroso A - Área do meio poroso (cm2)
ΔP - Diferencial de pressão (atm)
mN - Milinewton
m - Metro
CTAB - Brometo de cetiltrimetilamônio
SDS - Dodecil sulfato de sódio
mg - Miligrama
DCB - Diclorobifenilo
UV - Ultravioleta
λ - Comprimento de onda FID - Flame Ionization Detector
S/L - Razão sólido/líquido
W/V - Razão water/volume
TPH - Total de hidrocarboneto de petróleo
TG - Termogravimetria
MEV - Microscopia Eletrônica de Varredura
FRX - Fluorescência de Raios-X
DRX - Difração de Raios-X
ABET - Área BET
SME - Sistema Microemulsionado
min - Minutos
ºAPI - Grau API
DTG - Análise Termogravimétrica Diferencial
DTA - Análise Térmica Diferencial
DSC - Calorimetria Diferencial de Varredura
TMA - Análise Termomecânica
DMA - Análise Dinâmico-mecânica
PV - Pressão de vapor
PN - Pressão de Nitrogênio V - Volume
m2 - Metro quadrado
ADT - Água de Torneira
1. INTRODUÇÃO
Derramamentos de petróleo sempre ocupam as manchetes dos veículos de comunicação
ao redor do mundo. Os incidentes com petróleo sempre despertam alerta sobre os riscos e os
impactos destas atividades, principalmente devido a crescente preocupação com o meio ambiente
global. Com isto, o desenvolvimento de técnicas de limpeza mais eficazes é necessário e tem sido
objeto de diversas pesquisas atuais, como processo de tratamento biológico, dessorção térmica,
extração a vapor e lavagem de solos (Lee et al., 1998; Mulligan, Yong & Gibbs, 2001; Virkutyte,
Sillanpää & Latostenmaa, 2002; Chu & Kwan, 2003; Paria, 2008; Lai et al., 2009; Castro Dantas
et al., 2010; Dantas Neto et al., 2011; Hanna et al., 2012).
Os métodos de limpeza que devem ser utilizados nos derramamentos de petróleo
dependem de onde ocorreu o derramamento, se em solo ou em água. A principal prioridade é
sempre conter o derramamento a fim de evitar seu espalhamento e agravar a situação. A
identificação do contaminante e seu teor, bem como a destinação após a limpeza, são fatores
determinantes para a escolha da metodologia a ser empregada para a remediação do local onde
houve o derramamento (Oliveira, 2004).
O petróleo é constituído por uma complexa mistura de hidrocarbonetos, encontrada em
diversas rochas reservatório ao redor do mundo. Estas rochas podem ser formadas por arenitos,
como a formação Assu (RN) ou Botucatú (PR), calcários (Trairí – CE), e mais recentemente, foi descoberto petróleo na camada de Pré-Sal (Espírito Santo ao Paraná).
Os gastos com a recuperação do meio ambiente após um derramamento pode ultrapassar a
barreira de bilhões de dólares, como o caso da Exxon Valdez em 1989. Na época, o desastre
levou ao derramamento de mais de 10 milhões de galões de óleo que vazaram próximo ao Alasca,
causando grande dano à fauna local (Paine et al., 1996; Plater, 2010). Estima-se que a limpeza da
área custou mais de 2 bilhões de dólares.
Além da busca incessante por técnicas de limpeza mais eficazes e acessíveis, as pesquisas
buscam novos materiais a fim de diminuir o custo final de operação. Neste contexto, o uso de
substâncias tensoativas tem se tornado uma alternativa eficaz, pois substâncias tensoativas podem
ser recuperadas após o tratamento e reutilizadas (Oliveira, 2004; Rossi et al., 2006; Sabatini et
18
As substâncias tensoativas apresentam características hidrofílicas e lipofílicas, e por isto
migram para a interface óleo/água, levando à redução da tensão interfacial, facilitando a remoção
do contaminante. Estas substâncias podem ser utilizadas em sistemas microemulsionados com a
mesma finalidade. A obtenção de sistemas microemulsionados data de 1943, quando Schulman
obteve microemulsão ao adicionar alcoóis de cadeia média a emulsões de sabão.
O uso de tensoativos em sistemas microemulsionados com a finalidade de limpeza de
contaminantes em rochas tem sido alvo de algumas pesquisas. Os resultados mostram eficiências
superiores a 90% na operação (Oliveira, 2004; Pinheiro, 2005; Bonaparte et al., 2010; Dantas
Neto et al., 2010).
A constante busca por novas reservas de petróleo e gás levaram a descoberta dos vastos
campos situados no litoral brasileiro, mais especificamente nas bacias de Santos, Campos e
Espírito Santo. Nesta região, o petróleo e o gás foram encontrados na camada de pré-sal.
O pré-sal apresenta aproximadamente 800 km de extensão, acerca de 400 km da costa
brasileira, na faixa situada do Estado do Espírito Santo até Santa Catarina, a uma profundidade de
aproximadamente 7 km. Estima-se que o pré-sal foi formado há 115 milhões de anos, durante a
separação dos continentes. Estas rochas são constituídas praticamente por carbonatos de cálcio
que formam as rochas carbonáticas (Melo et al., 2011). Diante desse desafio, faz-se necessário o
conhecimento das propriedades físicas e químicas desta rocha, principalmente seu
comportamento na camada de pré-sal.
Estas descobertas, quando colocadas em produção com capacidade total, poderão deixar o
Brasil entre os cinco países que mais produzem petróleo no mundo. Atualmente o Brasil
encontra-se na 18ª posição no ranking de países produtores de petróleo, com 14 bilhões de barris
de óleo, sendo a maior parte de óleo pesado. Algumas estimativas iniciais sobre estas jazidas
preveem a produção de cerca de 300 mil barris de óleo leve por dia, que apresenta melhor
qualidade e é bem mais valorizado no mercado (Pré-sal: Perguntas e Respostas, 2009).
Portanto, será necessário o desenvolvimento de estudos envolvendo rochas carbonáticas,
de modo a propor algumas soluções para a produção no pré-sal, principalmente quanto a
geometria da rocha, porosidade, permeabilidade, capacidade de absorção do óleo e a melhor
forma de posicionar os poços, visando diminuir o tempo de perfuração, bem como o custo de
Neste trabalho, foi estudada a remoção de frações de óleo leve e pesado de rocha calcária
através de sistemas microemulsionados, comparando as eficiências de remoção em diferentes
2 ASPECTOS TEÓRICOS E
2. ASPECTOS TEÓRICOS
Neste capítulo são apresentadas teorias e definições necessárias para o entendimento do
trabalho desenvolvido, como tensoativos, micelas, microemulsões, sistemas de Winsor, sua
representação em diagramas de fases, aspectos gerais sobre adsorção, classificação e modelagem
de isotermas, e por fim o calcário. Após os aspectos teóricos, segue-se o estado da arte sobre
calcário, adsorção, e tensoativos e microemulsões, justificando a relevância do trabalho e seus
objetivos.
2.1. TENSOATIVOS
2.1.1. Definição
Tensoativo é uma molécula que apresenta em sua estrutura dois grupos com afinidades
antagônicas, sendo uma cabeça polar (hidrofílica) ligada a uma cauda apolar (hidrofóbica)
(Schramm, 2000). A presença destes dois grupos funcionais em uma mesma molécula a
caracteriza como anfifílica (Figura 1).
Figura 1 - Representação de um tensoativo (molécula anfifílica)
Fonte: Elaborado pela autora
2.1.2. Classificação
As moléculas de tensoativos podem ser classificadas segundo a região polar em iônicos e
22
2.1.2.1. Tensoativos Iônicos
Os tensoativos iônicos, quando em solução aquosa, apresentam a formação de íons na
parte polar, podendo se subdividir em catiônicos, aniônicos ou anfóteros.
2.1.2.2. Tensoativos Catiônicos
Ao se dissociarem em solução aquosa, estes tensoativos formam íons carregados
positivamente. Geralmente estes tensoativos são derivados de sais de amônio quaternários
(Figura 2).
Figura 2 - Exemplo de tensoativo catiônico
Fonte: Elaborado pela autora
2.1.2.3. Tensoativos Aniônicos
Tensoativos aniônicos formam íons carregados negativamente quando em solução aquosa.
Geralmente são derivados de grupos carboxílicos, sulfônicos e sulfatos (Figura 3).
Figura 3 - Exemplo de tensoativo aniônico
2.1.2.4. Tensoativos Anfóteros
São aqueles que apresentam caráter anfótero, ou seja, podem apresentar tanto carga
negativa quanto positiva na mesma molécula, mostrando ora caráter catiônico, ora aniônico
(Figura 4). Esta predominância da carga ocorrerá em função do pH da solução. Caso o pH seja
ácido, a predominância será do caráter catiônico, caso o pH seja básico, o caráter predominante
será o aniônico.
Figura 4 - Exemplo de tensoativo anfótero
Fonte: Elaborado pela autora
2.1.2.5. Tensoativos Não-Iônicos
Esta classe de tensoativos não formam íons em soluções aquosas, por isso são chamados
de não-iônicos (Figura 5). Seus radicais mais comuns são éter, hidroxi e éster (nonilfenol
etoxilado, álcoois graxos etoxilados e o propilenoglicoletoxilado).
Figura 5 - Exemplo de tensoativo não-iônico
24
2.1.3. Balanço hidrofílico-lipofílico (BHL)
O balanço hidrofílico-lipofílico visa quantificar as contribuições das partes polares e
apolares de um tensoativo, o que determina suas características específicas (Griffin, 1954) e suas aplicações, definindo que quanto mais hidrofílico for o tensoativo maior seu valor de BHL
(Bouchemal et al., 2004). A Tabela 1 mostra alguns valores de BHL característicos para alguns
tensoativos.
Tabela 1 - Valores de BHL em função da aplicação do tensoativo
APLICAÇÃO BHL (teórico)
Anti-espumante 1,5 – 3,0
Espumante 7,0 – 9,0
Emulsificante (A/O) 3,0 – 6,0 Emulsificante (O/A) 8,0 – 18,0 Detergente 13,0 – 15,0
Fonte: Adaptado de Griffin, 1954.
2.1.4. Concentração Micelar Crítica (c.m.c.)
Em soluções com baixas concentrações de tensoativos, eles tendem a ficar separados,
como monômeros e migram para as interfaces existentes, como água/óleo, água/ar ou água/solo.
Quando ocorre a saturação das interfaces e a concentração de tensoativos continua a ser
incrementada, ocorre a formação de micelas (Figura 6). A concentração em que ocorre a
formação de micelas é conhecida por Concentração Micelar Crítica (Attwood & Florense, 1985;
Figura 6 - Representação da formação de micelas
Fonte: Elaborado pela autora
2.2. MICELAS
Uma das características mais importantes dos tensoativos é a capacidade de organizar-se
em estruturas conhecidas como micelas. As micelas podem ser definidas como agregados
moleculares, que apresentam uma região hidrofóbica, e outra hidrofílica, podendo formar
agregados moleculares de dimensões coloidais (West & Harwell, 1992).
Durante o processo de formação das micelas em água, ocorre o balanço dos impedimentos
estéricos, das forças intermoleculares e interações de van der Waals. A força de atração resulta da
cauda apolar do tensoativo e a força repulsiva se dá devido aos efeitos estéricos e interações
eletrostáticas entre as cabeças polares. A formação da micela só ocorre quando estas forças
encontram-se em equilíbrio (Tanford, 1980; Israelachvili, 1991; Lange, 1999).
As micelas podem estar dispostas de dois modos diferentes: de forma direta ou inversa
(Figura 7). As micelas diretas formam-se em solventes polares e apresentam a parte apolar do
tensoativo no centro do agregado, isolando-a do solvente. Já as micelas inversas formam-se em
solventes apolares, e a parte polar do tensoativo encontra-se no centro do agregado (Evans &
26
Figura 7 - (a) Micela direta e (b) Micela inversa
(a) (b)
Fonte: Elaborado pela autora
2.3. MICROEMULSÕES
2.3.1. Definição
As microemulsões são sistemas formados espontaneamente pela auto-organização de
moléculas tensoativas nas interfaces óleo-água, formando microestruturas dispersas em um meio
contínuo formado por três ou mais constituintes, tais como tensoativo, fase óleo e fase água
(Kumar & Mittal, 1999; Oliveira et al., 2004; Rossi et al., 2007).
Os sistemas microemulsionados podem ser definidos como sendo sistemas
termodinamicamente estáveis, dispersos, monofásicos, transparentes ou translúcidos, com baixa
tensão interfacial e que possuem capacidade de combinar grandes quantidades de dois líquidos
imiscíveis em uma única fase homogênea, na presença de tensoativo e/ou cotensoativo (Robb,
1982; Attwood & Florense, 1985; Paul & Moulik, 1997; Oliveira et al., 2004; Rossi et al., 2007).
2.3.2. Formação das Microemulsões
Para que uma microemulsão seja formada é necessária a mistura de alguns constituintes
tais como tensoativo, fase aquosa, fase oleosa e, se necessário um co-tensoativo. O sistema
poderá ser do tipo óleo em água (O/A) ou água em óleo (A/O), tudo dependerá das propriedades
Ao se misturar dois líquidos imiscíveis, sob agitação constante, eles tendem a formar
gotículas uma dentro da outra; porém, quando a agitação cessa, eles tendem a coalescer. Ao se
adicionar um tensoativo ao sistema e agitá-lo, ele torna-se homogêneo e apresenta estabilidade
termodinâmica. Isto se deve ao fato de o tensoativo agir na interface do sistema, diminuindo a
tensão interfacial, facilitando assim a solubilização destes líquidos.
2.3.3. Estrutura das Microemulsões
De acordo com a composição dos sistemas microemulsionados, eles podem apresentar
diversas configurações, como micelas diretas (óleo em água) ou inversas (água em óleo). O
tamanho médio das micelas em emulsões estão apresentados na Tabela 2.
Tabela 2 - Tamanho médio das micelas em emulsões
Tipo de emulsão Tamanho da partícula
Macroemulsões > 400 nm
Microemulsões < 100 nm
Nanoemulsões 100 – 400 nm Fonte: Adaptado de Rosen, 2004.
Podem ainda ocorrer outras estruturas nas microemulsões como os cristais líquidos.
28
Figura 8 - Exemplos de fases em Sistemas Microemulsionados
(a)Hexagonal (b) Lamelar (c) Cúbica.
(a) (b) (c)
Fonte: Adaptado de Rosen, 2004.
2.4. SISTEMAS DE WINSOR
Winsor (1948) estudou sistemas contendo microemulsões em equilíbrio com fases aquosa
e oleosa e propôs uma classificação. Em função das fases em equilíbrio, foram estabelecidos
quatro sistemas (Figura 9):
Winsor I (WI): Equilíbrio entre a fase de microemulsão (do tipo O/A) e a fase oleosa
em excesso.
Winsor II (WII): Equilíbrio entre a fase microemulsão (do tipo A/O) e uma fase
aquosa em excesso.
Winsor III (WIII): Coexistência das três fases em equilíbrio – oleosa, microemulsão
(geralmente do tipo bicontínua) e aquosa.
Winsor IV (WIV): Apenas a fase de microemulsão (sistema monofásico), que pode
Figura 9 - Classificação de Winsor
Fonte: Elaborado pela autora
2.5. REPRESENTAÇÃO DE WINSOR EM DIAGRAMA DE FASES
Os diagramas de fases são ferramentas utilizadas para representar os sistemas formados,
de modo que as regiões de miscibilidade possam ser delimitadas e visualizadas (Silva de Araújo,
2004).
2.5.1. Sistemas Ternários e Quaternários
Os diagramas ternários são formados por três constituintes, um em cada vértice, sendo sua
representação feita em um triângulo equilátero (Figura 10 a). Os diagramas quaternários (Figura
10 b) é obtido em uma pirâmide equilátera, com quatro constituintes, um em cada vértice. Uma
vez que há quatro constituintes, faz-se necessário uma representação tetraédrica (Paul & Moulik,
2001).
2.5.2. Sistemas Pseudoternários
Devido à difícil interpretação envolvendo construção tridimensional dos diagramas
quaternários, utilizam-se diagramas pseudoternários como alternativa (Friberg, 1977). Dois componentes são agrupados em um único vértice, sendo considerado como um
30
Figura 10 - Representação gráfica de diagramas de fases
a) Ternário b) Quaternário c) Pseudoternário
Fonte: Rossi et al, 2007.
2.6. ASPECTOS GERAIS SOBRE ADSORÇÃO
Adsorção pode ser definida como a interação entre um fluido (líquido ou gasoso) e uma
superfície (sólida). Ela pode ser classificada de duas maneiras: quimissorção e fisissorção. A
quimissorção é baseada em ligações químicas formadas devido à afinidade do adsorvente com o
substrato (superfície), em que ocorrem alterações no estado eletrônico (compartilhamento ou
troca de elétrons). Já a fisissorção é relacionada às interações de forças fracas de Van Der Walls
entre o adsorvente e a superfície (Gregg & Sing, 1982).
2.6.1. Fatores determinantes do Processo de Adsorção
O processo de adsorção pode ser afetado por diversos fatores. Abaixo encontram-se
detalhados alguns destes fatores.
2.6.1.1. Temperatura
Usualmente o processo de adsorção é exotérmico (Gregg & Sing, 1982). Como é
necessário que as moléculas mantenham uma interação com a superfície para que a adsorção seja
aumentada, o que ocorre com o aumento da temperatura, as moléculas podem ser dessorvidas da
superfície, diminuindo a eficiência da adsorção. Porém, existem casos em que a adsorção pode
ser favorecida com o aumento da temperatura, que pode ser atribuído ao aumento no tamanho do
poro (Gupta, 1998).
2.6.1.2. Natureza do Solvente
A utilização do solvente no processo de adsorção visa aumentar a interação na interface
líquido-sólido, logo ele não deve competir com a superfície sólida pela matéria a ser adsorvida.
Uma solução de soluto e solvente orgânicos não seria adsorvida tão bem quanto se o soluto fosse
solubilizado, por exemplo, em um solvente mais polar (Gregg & Sing, 1982).
2.6.1.3. Velocidade de Adsorção
A adsorção em fase líquida ocorre bem mais lentamente que a em fase gasosa. Um dos
fatores pra que isso ocorra é a viscosidade do líquido, já que quanto mais viscoso, maior as
interações entre suas moléculas, e isto demanda a necessidade de mais energia para que a
adsorção ocorra, já que esta interação deverá ser maior entre as moléculas do adsorvato e a
superfície sólida que entre as moléculas do adsorvato entre si.
2.6.1.4. Estrutura do Poro
A estrutura do poro é determinante para o processo de adsorção, já que se a molécula a ser
adsorvida for maior que o diâmetro do poro, este atuará como um limitante do processo de
adsorção, visto que a adsorção neste caso só ocorrerá na superfície externa do sólido (Do, 1998).
2.6.1.5. Área Superficial do Adsorvente
A adsorção deve ser proporcional à área superficial (externa e interna aos poros) da
superfície sólida disponível (Weber Jr, 1974). A área superficial é dada através da adsorção de N2
32
superficial total com que ela conseguirá entrar em contato será menor, pois ela poderá não
penetrar no poro, devido ao fato de o diâmetro do poro ser bem menor que o diâmetro da
molécula.
2.6.1.6. Tipos de Adsorventes
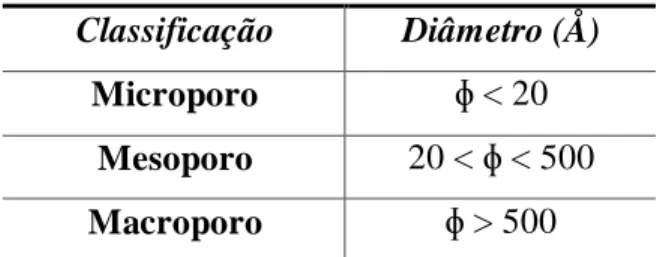
A natureza da superfície sólida, assim como o tamanho e distribuição dos poros, afetam
as propriedades adsortivas (Curbelo, 2002). Segundo a International Union of Pure and Applied
Chemistry (IUPAC), os poros são definidos de acordo com o seu tamanho em: microporo, com
menos de 20 Å; mesoporo, de 20 a 500 Å; e macroporo, maior que 500 Å (Seader & Henley,
2011).
Os microporos são ligeiramente maiores do que a molécula a ser adsorvida. Neles,
considera-se que todas as moléculas encontram-se na fase adsorvida, já que o campo de forças da
superfície sólida prende a molécula, mesmo que ela esteja no centro do poro.
Em adsorventes cristalinos, como as zeólitas, o tamanho dos microporos são definidos
pela estrutura da rede cristalina, e por isso não ocorre uma distribuição de tamanho. Já
adsorventes, como a sílica gel e a alumina ativada (produzidos através da precipitação de
partículas coloidais, seguida por desidratação), ou adsorventes como carbono (obtidos pela
queima controlada de materiais carbonáceos), permitem uma distribuição do tamanho do poro
bem maior.
Por outro lado, mesoporos contribuem mais efetivamente para a capacidade adsortiva,
mas seu papel principal é o de fornecer acesso aos microporos. Nestes mesoporos, a difusão pode
ocorrer por diferentes mecanismos.
2.6.1.7. Natureza das Superfícies
Uma substância polar é mais bem adsorvida em superfícies polares. Estas substâncias são
ditas hidrofílicas, ou seja, tem maior afinidade com a água, que é uma molécula bastante polar. Já
superfícies não polares apresentam maior interação com substâncias apolares, e por isso são
2.6.1.8. Razão Sólido/Solução
Apesar de este parâmetro geralmente não influenciar a forma das isotermas, alguns
estudos têm observado uma significância e dependência não linear da razão sólido/solução e a
efetividade da adsorção (Porro, Newman & Dunnivant, 2000; Limousin et al., 2007).
Geralmente, em experimentos em batelada com materiais porosos naturais, a razão
sólido/solução deverá estar entre 1 g de sólido para 2 mL de solução até 1 g de sólido para 4 mL
de solução (Porro, Newman & Dunnivant, 2000; Limousin et al., 2007).
2.7. CLASSIFICAÇÃO E MODELAGEM DAS ISOTERMAS
A isoterma de adsorção é função da pressão (gases) ou da concentração (líquido), da
temperatura, do gás (ou líquido) e do sólido, mostrando que a isoterma depende também da
massa e do tipo do adsorvente e da superfície. Em temperaturas constantes a adsorção dos gases
podem ser determinadas pelas curvas, chamadas isotermas, que será em função da pressão (Giles,
Smith & Huitson, 1974; Guimarães, 2006).
2.7.1. As Principais Isotermas
Em 1974, Giles, Smith & Huitson, propuseram modelagens para adsorção de soluções
contendo sólidos. As isotermas foram então classificadas de acordo com seu declive inicial em
34
Figura 11 - Quatro principais modelos de isoterma para adsorção em sólidos.
Fonte: Adaptado de Falone & Vieira, 2004
2.7.1.1. A Isoterma “C”
Sua denominação vem do inglês (“Constant partition”). Como o próprio nome já diz, esta isoterma equivale a uma divisão constante entre o soluto e a solução com o adsorvente, o que
implica em uma reta. Estas isotermas podem ser favorecidas por substratos porosos flexíveis
(Falone & Vieira, 2004; Avelino, 2009).
2.7.1.2. A Isoterma “L”
A isoterma L é a isoterma de “Langmuir”. Esta isoterma apresenta inclinação não linear e
côncava em relação à abcissa. Isto implica numa diminuição da disponibilidade dos sítios
adsortivos com o aumento da concentração da solução adsorvente (Falone & Vieira, 2004;
Avelino, 2009).
2.7.1.3. A Isoterma “H”
Esta isoterma é um caso especial da isoterma do tipo L. Ela ocorre quando a superfície do
adsorvente possui alta afinidade pelo soluto adsorvido. Por isso sua denominação do inglês é
2.7.1.4. A Isoterma “S”
Do inglês, “Spherical”, esta isoterma apresenta inclinação linear e convexa em relação à
abcissa. No início, a adsorção é baixa, porém ela tende a aumentar à medida que o número de
moléculas adsorvidas aumenta (Falone & Vieira, 2004; Avelino, 2009). Isto implica em uma
adsorção cooperativa, ou seja, a adsorção ocorre muito mais quando há uma associação entre as
moléculas adsortivas (Hinz, 2001).
2.7.1.5. Modelo BET (Brunauer, Emmett & Teller)
Baseado nos estudos da fisissorção, Brunauer, Emmett & Teller (1938) classificaram as isotermas de adsorção em seis tipos, como mostrado na Figura 12.
Figura 12 - Isotermas de adsorção de BET
Fonte: Adaptado de Coutinho et al., 2001.
As isotermas de adsorção podem definir o tipo de poro de cada superfície. Os tipos de
36
Tabela 3 - Classificação dos poros de acordo com os diâmetros
Classificação Diâmetro (Å)
Microporo ɸ < 20
Mesoporo 20 < ɸ < 500
Macroporo ɸ > 500
Fonte: Adaptado de Coutinho, Teixeira & Gomes, 2001.
Para a obtenção da equação da isoterma de BET foi necessário definir algumas hipóteses,
sendo elas as seguintes:
1. A adsorção é feita através de interações de Van der Waals;
2. Podem existir infinitas camadas;
3. A primeira camada adsorve-se seguindo o modelo de Langmuir;
4. Cada camada possui o tamanho da superfície e a quantidade de sítios ativos constantes;
5. Não há interações laterais entre as moléculas adsorvidas;
6. A energia de adsorção é constante na primeira camada;
7. A energia de adsorção nas demais camadas é igual à energia de condensação.
A isoterma do tipo I representa sólidos microporosos (ɸ < 20Å), em que a isoterma de adsorção é semelhante à quimissorção, ou seja, a de Langmuir, responsável pela formação da
primeira camada (a única no caso de Langmuir – monocamada). A facilidade da adsorção para a formação de monocamada é visível por apresentar no início da curva uma inclinação quase
vertical, mostrando a facilidade da adsorção em materiais microporosos. Logo após, atinge-se um
platô (quase horizontal) representando a saturação da monocamada. Diferente da quimissorção, o
processo de fisissorção ocorre em temperaturas inferiores e poderão ser formadas posteriormente
outras camadas (Figura 12 I).
Já a isoterma do tipo II (Figura 12 II) representa sólidos não porosos, em que ∆adsH < 0. Inicialmente o gás adsorve-se rapidamente, conforme mostrado pela inclinação quase vertical no
começo da curva. Logo após, essa adsorção passa a ser mais lenta (aumento da inclinação); isso
Depois de preenchida toda a primeira camada, passa-se a formar as múltiplas camadas,
representadas no final do gráfico. A área superficial da amostra pode ser calculada através da
primeira região da curva, em que se formou a monocamada.
A isoterma do tipo III (Figura 12 III) representa sólidos não porosos, em que o ∆adsH > 0, sendo então a adsorção desfavorável, de modo que a afinidade do adsorvente por si mesmo pode
ser maior que a do adsorvente pelo substrato, e também a afinidade do adsorvente pela superfície
ser muito fraca.
Nas isotermas dos tipos IV e V (Figura 12 IV e 12 V) estão representados sólidos
mesoporosos (entre 20 e 500 Å), sendo na Figura IV, o ∆ adsH < 0, e na Figura V. o ∆adsH > 0. Na isoterma tipo IV a monocamada é formada rapidamente, como se percebe pelo início da curva, e
logo após ocorre a histerese; já no tipo V, a adsorção é desfavorável, sendo verificada sua
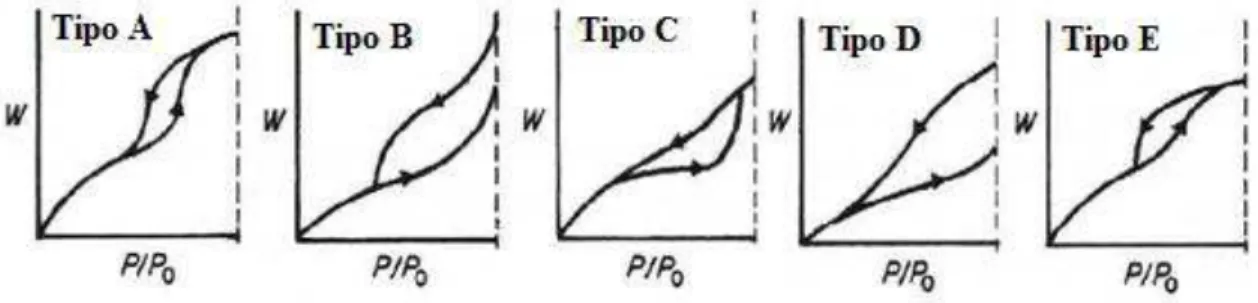
formação no final da curva. Essas duas curvas são caracterizadas por apresentarem histereses. As
histereses ocorrem devido à diferença da entalpia de adsorção e de dessorção (∆adsH ≠ ∆desH). Os fenômenos de histereses (Figura 13) podem ser classificadas de acordo com as formas
dos mesoporos, podendo ser cilíndricos (Tipo A), no formato de fenda (Tipo B), cônicos (Tipo
C), formato de cunha (Tipo D), de garrafa (Tipo E), conforme mostrado abaixo (Claudino, 2003).
Figura 13 - Classificação das histereses de acordo com formato do mesoporos
Fonte: Adaptado de Claudino, 2003.
A isoterma do tipo VI (Figura 12 IV) representa superfícies quase uniforme em sólidos
não porosos, em que visivelmente ocorre a formação de múltiplas camadas, sendo a altura de
38
2.8. CALCÁRIO
As rochas sedimentares podem ser classificadas em Clásticas (originadas de rochas
preexistentes), Químicas (oriundas de possíveis precipitações, como por exemplo, de sais
presentes em rios, lagos e mares) e Orgânicas (devido a acumulações de substâncias orgânicas
provenientes de animais ou vegetais). Dentre as rochas sedimentares de origem química
(também chamadas de autigênicas), temos as silicosas, salinas, ferruginosas e carbonáticas. A
rocha calcária é um dos principais tipos de rocha carbonática (composta por mais de 50% de
seus minerais, como a calcita e o carbonato de cálcio); um ponto relevante é que os carbonatos
também podem apresentar origem orgânica, originados do acúmulo, por exemplo, de carapaças e
esqueletos de organismos vivos (Rocha & Azevedo, 2009).
A estruturação do calcário se dá inicialmente por intemperismo e posteriormente pelo
processo de diagênese, em que ocorre a transformação dos sedimentos que são inicialmente
inconsolidados e friáveis em rocha maciça devido ao processo de litificação. Assim, a
organização dos seus grãos forma interstícios denominados poros, influenciando propriedades
físicas importantes como porosidade e permeabilidade (Thomas, 2004; Rocha & Azevedo,
2009).
2.8.1. Movimento de Fluidos em rochas
Os grãos que compõem uma rocha se tocam, deixando interstícios entre si, chamados de
poros. Os poros são caracterizados por forma, tamanho e distribuição. Raramente as rochas são
compostas por um único material e com apenas uma granulometria. Os movimentos de fluidos
são afetados, principalmente, por duas propriedades físicas: porosidade e permeabilidade
(Chiossi, 1987 apud Pinheiro, 2005).
2.8.1.1. Porosidade
A porosidade pode ser definida pela razão entre o volume do poro e o volume total do
material, sendo expressa em porcentagem conforme a Equação 1 (Chiossi, 1987). Na Tabela 4
(Equa ção 1)
Onde:
%P = permeabilidade
Vporo = Volume de poro
Vtotal = Volume total.
Tabela 4 - Porosidade de alguns materiais
Material Porosidade (%)
Argila 45 – 55 Silte 40 – 50 Arenito 10 – 20 Calcário 1 – 10
Fonte: Chiossi, 1987 2.8.1.2. Permeabilidade
A permeabilidade é a facilidade com que o fluido atravessa uma rocha, em um
determinado espaço de tempo e com certa velocidade. Isto depende da interligação entre poros,
da granulometria dos materiais que compõem a rocha e das fraturas que possam estar presentes.
A medida da permeabilidade é expressa pela Lei de Darcy, conforme a Equação 2 (Chiossi, 1987;
Dullien, 1992):
(Equa ção 2)
Sendo:
Q = Vazão do fluido (cm3/s)
K = Permeabilidade do meio poroso (Darcy)
ƞ = Viscosidade absoluta (cP)
40
A = Área do meio poroso (cm2)
ΔP = Diferencial de pressão (atm)
2.8.2. Remoção de contaminantes
A exploração das reservas de petróleo podem causar derramamentos durante o transporte
do óleo de uma região a outra. O petróleo é uma mistura complexa de hidrocarbonetos (Thomas,
2004; Cardoso, 2005). Quando o petróleo entra em contato com os materiais, além de contaminar
a porção superficial, ele pode penetrar e chegar a contaminar aquíferos, gerando problemas ainda
maiores (Oliveira, 2004).
A remoção de um contaminante consiste em eliminar ou diminuir a concentração do
contaminante. Para que esta remoção ocorra, faz-se necessário, inicialmente, a identificação do
contaminante, bem como, a determinação da melhor política de limpeza a ser adotada (Oliveira,
2004).
A remoção de contaminantes pode ser realizada de duas formas: quanto a ação de
tratamento e ao local de tratamento. Quanto a ação do tratamento, elas podem ser classificadas
em: biológica, física e química. A remoção biológica vem da ação de fungos e/ou bactérias que
consomem o material orgânico. Na remoção física são utilizados principalmente tratamentos
térmicos. A remoção química faz uso de técnicas de lavagem, utilizando desde água até
substâncias que reajam com os contaminantes a fim de eliminá-los (Oliveira, 2004; Pinheiro,
2005).
Já com relação ao local de tratamento, podemos classificar a limpeza de solos em: in situ
ou ex situ. Na técnica in situ, as medidas de remoção são realizadas no mesmo local onde se deu a
contaminação, sem a necessidade da remoção do material. Na técnica ex situ, o material
contaminado é retirado do local onde ocorreu o fato, e levado a um local diferente onde os
2.8.3. Remoção de frações leves e pesadas em rocha calcária através de Sistemas
Microemulsionados
Para a remoção de frações leves e pesadas de contaminantes presentes na rocha calcária,
é importante entender o mecanismo do escoamento dos fluidos em seu interior bem como sua
estrutura e composição, além da interação destas frações de petróleo com a rocha. O calcário é
composto por diversos minerais, tendo em sua predominância a presença de carbonatos, que,
dependendo do processo de decomposição, recomposição e consolidação, ao passar dos anos, em
condições geológicas definidas, podem formar reservatórios petrolíferos.
2.9. ESTADO DA ARTE
Pesquisas relacionadas a remediação em rochas calcárias foram realizadas não tendo sido
evidenciado na literatura nenhum trabalho acerca deste tema. No entanto, tem sido bastante
explorada a mudança na molhabilidade do calcário quando tratado com soluções de tensoativos.
Na literatura alguns trabalhos mencionam o uso de tensoativos como uma alternativa
eficaz na remoção de contaminantes e na recuperação avançada de petróleo seja em soluções
micelares ou como componentes de microemulsões.
2.9.1. Calcário
Standnes & Austad (2000) estudaram a alteração de molhabilidade em calcários calcíticos com baixa permeabilidade usando 14 diferentes tensoativos em soluções e duas fases óleo
diferentes – n-heptano puro e um óleo cru ácido do Mar do Norte dissolvido em n-heptano. A alteração da molhabilidade foi determinada através do ângulo de contato entre o óleo e a amostra
da rocha. A eficiência dos tensoativos está relacionada com propriedades como c.m.c.,
propriedades hidrofóbicas e efeitos estéricos relacionados ao átomo de nitrogênio. Também foi
realizado o teste de Amott, onde as amostras de rocha são submetidas a inibição com solução de
tensoativos. Elas foram preenchidas com salmoura e depois saturadas com óleo. Na rocha é então
reinjetada a salmoura para ver a quantidade de óleo que é produzida, sendo calculado o índice de
42
carboxilatos orgânicos existentes no óleo cru. Tensoativos catiônicos do tipo R – N+(CH3)3X foram capazes de modificar a molhabilidade do calcário de forma irreversível; já tensoativos
aniônicos não demonstraram esta capacidade de modificar a molhabilidade de forma irreversível
e nem de remover carboxilatos aniônicos. Os tensoativos etoxilados removeram os orgânicos de
forma espontânea em processo lento e em baixas tensões interfaciais (0,08 mN/m).
Drummond & Israelachvili (2004) buscaram o entendimento fundamental da molhabilidade das superfícies das rochas por petróleo e salmoura, através das interações
intermoleculares entre as fases em contato, medindo diretamente essas interações e estabelecendo
suas implicações para o comportamento da molhabilidade. Ao modificar uma ou ambas as
superfícies através de pré-adsorção controlada de agentes de superfície polares a partir do
petróleo bruto, a interação entre a rocha e óleo em salmoura ou rocha em óleo foram
determinadas. A partir das medidas do ângulo de contato foi traçado um “mapa de molhabilidade” que mostra as regiões em função de diferentes ângulos de contato, ou
“molhabilidade” em função do pH e da força iônica. Este “mapa de molhabilidade” foi feito para ser correlacionado independentemente com as medidas de forças de superfície e adsorção. Foi
estabelecido o conceito de uma superfície intrinsecamente molhável a óleo e molhável a água no
reservatório e isto não pode, em geral, ser corrigido: dependendo da natureza do óleo e da
salmoura, diferentes espécies de um óleo poderá se adsorver nas superfícies dos minerais e a
interface óleo-água, determinando assim o comportamento de molhabilidade de um reservatório
ou de rocha.
Jarrahian et al. (2012) estudaram a alteração da molhabilidade de rochas carbonáticas, considerando que em todo o mundo, várias reservas de petróleo encontravam-se localizadas em
reservatórios carbonáticos naturalmente fraturados, apresentando baixa eficiência de recuperação,
devido a sua molhabilidade e a matriz ser muito condensada. A eficiência de recuperação pode
ser melhorada se a molhabilidade da rocha reservatório for alterada de molhável a óleo para
molhável a água, aumentando assim, a quantidade de água embebida na rocha. Através do uso de
diferentes ferramentas analíticas, como Infravermelho com Transformada de Fourier, Análise
termogravimétrica, Microscopia atômica, Potencial Zeta e medidas de ângulo de contato, foram
estudados os efeitos de agentes tensoativos na molhabilidade das rochas. A fase orgânica
utilizada foi o ácido esteárico. Os resultados indicaram que os agentes tensoativos atuaram de
mudança na molhabilidade de forma mais efetiva, removendo irreversivelmente o ácido esteárico
da superfície do calcário dolomítico através de interação iônica. Tensoativos não-iônicos, como o
Triton X-100, são adsorvidos na superfície da rocha através da polarização de elétrons π e troca iônica; o ácido esteárico liberado é então adsorvido como uma nova camada sobre a superfície,
através de interação hidrofóbica entre a cauda do tensoativo adsorvido e a parte não polar do
ácido esteárico. Tensoativos aniônicos, como o SDS, são adsorvidos na superfície via interação
hidrofóbica entre a cauda do tensoativo e o ácido adsorvido, alterando assim a molhabilidade da
superfície para uma condição neutra.
2.9.2. Adsorção
Liu et al. (2004) realizaram um estudo de recuperação avançada de petróleo através da adsorção-dessorção em injeção química, onde o principal componente da injeção química foi o
tensoativo, já que sua perda durante o processo era uma das maiores preocupações. A perda de
tensoativo devido a adsorção nas rochas reservatório diminuiu a eficácia da injeção química na
redução da tensão interfacial óleo-água, o que torna o processo economicamente inviável. Neste
trabalho, as concentrações de tensoativo e alcalinidade no efluente do fluxo injetado e a tensão
interfacial óleo-água foram determinadas de acordo com diferentes estratégias de injeção, o que
evidenciou que uma injeção química estendida de fluxo viscoso, seguida por uma injeção de
solução alcalina viscosa de tensoativo, fez com que o tensoativo fosse dessorvido para a fase
aquosa. Os resultados mostraram que foi obtido um adicional de recuperação do óleo de 13 %
após a injeção de agente tensoativo alcalino, devido ao sinergismo entre o tensoativo dessorvido e
o teor de alcalinidade do meio. Este resultado mostrou que a eficiência e a economia de injeção
química melhoraram através da utilização do tensoativo dessorvido durante os processos de
infiltração prolongados.
Dudášová et al. (2008) investigaram a adsorção de asfaltenos em diferentes materiais minerais e calcários, determinando a adsorção através da Espectroscopia de UV. O processo de
adsorção se ajustou à isoterma de Langmuir. Os valores de saturação de asfaltenos adsorvidos em
minerais (0,26 - 3,78 mg/m2) foram da mesma ordem de grandeza que a adsorção de asfaltenos
em metais. Foi verificado que a adsorção dependia mais do tipo de partícula do que a origem dos
44
a correlação entre a quantidade adsorvida e a composição elementar, encontrando-se uma
correlação entre a quantidade de nitrogênio nas amostras de asfaltenos e sua quantidade adsorvida
na partícula.
Mendoza de la Cruz et al. (2009) realizaram um conjunto de experiências a fim de estudar os mecanismos de adsorção envolvidos na interação interfacial do sistema asfalteno/minerais em
altas concentrações de asfalteno. Isotermas de adsorção dos asfaltenos foram obtidas através do
contato de amostras de arenito Berea, calcário Bedford, e rocha calcária dolomítica Mexicana
com uma fração de asfaltenos, obtidos de um óleo pesado bruto do México, dissolvidos em
tolueno. Em baixas concentrações, as isotermas de adsorção do asfalteno foram semel hantes às
relatadas na literatura, enquanto que em elevadas concentrações verificou -se uma isoterma de
tipo passo a passo, o que sugere uma alteração qualitativa no comportamento de adsorção, que
está relacionadas com a associação de asfaltenos e a sua agregação. Ambas isotermas de
adsorção, tanto para baixas concentrações como para altas concentrações podem ser consideradas
do tipo Freundlich generalizada.
Castro et al. (2009) apresentaram uma abordagem molecular termodinâmica para a modelagem de isotermas de adsorção de asfaltenos em amostras de arenito Berea, calcário
Bedford, e rocha calcária dolomítica, usando um modelo de precipitação de asfaltenos e uma
abordagem bidimensional para fluidos confinados, baseados na Teoria Estatística de Associação
de Fluidos para Potenciais de Faixa de Variável. Esta teoria foi aplicada aos modelos de
isotermas de adsorção a partir de dados experimentais de asfaltenos extraídos de uma amostra de
petróleo bruto pesado de um reservatório mexicano. Os resultados teóricos indicaram o
comportamento da isoterma de Langmuir, o que foi observado experimentalmente. Embora haja
um acordo entre teoria e os experimentos, houve alguns desvios em baixas concentrações. O
modelo reproduziu dados de adsorção em altas concentrações, situação em que outros modelos
semi-empíricos falharam.
Ahmadi & Shadizadeh (2013) realizaram uma investigação experimental de adsorção de um tensoativo não-iônico em minerais carbonáticos a fim de utilizá-lo na recuperação avançada
de petróleo. O tensoativo utilizado foi extraído de Zizyphus spina christi (pequena árvore com
galhos espinhosos comumente encontrada no Jordão, Iraque, Irã e Egito) e testado em rocha
carbonática, através da análise do comportamento de adsorção. A adsorção de tensoativo foi
adsorção aumentou com o aumento da concentração de tensoativo no sistema e sua isoterma se
ajustou ao modelo de Freundlich.
2.9.3. Tensoativos e microemulsão
Deshpande et al. (2000) verificaram a solubilização de contaminantes com sistemas de tensoativo DOWFAX. A solubilização do fenantreno pelo tensoativo aumentou com o aumento
da hidrofobicidade de tensoativo. A solubilização dos contaminantes em solos através dos
sistemas de tensoativo aumentou para DOWFAX com maior potencial de solubilização e não foi
significativamente impactada pela adsorção do tensoativo do meio.
Mulligan, Yong & Gibbs (2001) revisaram a remediação de metais (cobre, cádmio e zinco) em solos contaminados através de sistemas de biotensoativos em testes de laboratório,
demonstração de campo e aplicação em escala. Três tipos de biotensoativos tiveram sua eficácia
testada, e os resultados indicaram as possibilidades de eliminar os metais com os biotensoativos
aniônicos, que após 5 dias apresentaram uma eficiência de remoção de aproximada mente 70 %
para o cobre, 24 % para o zinco e 16 % para o cádmio.
Chu & Kwan (2003) estudaram uma nova abordagem utilizando uma solução de tensoativo no processo de lavagem do solo a fim de melhorar o desempenho de descontaminação
de solos com tensoativos convencionais. Três tensoativos (Brij 35, Tween 80, e SDS) e três
solventes orgânicos (acetona, trietilamina, e esqualano) foram utilizados para avaliar os
desempenhos de dessorção de 4,4-diclorobifenilo (DCB) de três solos com diferentes
características de adsorção. A técnica utilizada para acompanhamento da quantidade de DCB
extraído foi a cromatografia gasosa com detector de elétrons e todos os espectros de absorção
foram obtidos no UV (λ = 600 nm). A melhoria de desempenho se deve a uma melhor dissolução
dos contaminantes hidrofóbicos assistida pelo solvente, e a formação de micelas do tensoativo
incorporadas no solvente, o que aumentou tanto o tamanho quanto a afinidade de micelas para
uma extração mais eficaz dos contaminantes. Julgando a partir dos dados experimentais e como
verificado pelas duas constantes no modelo de lavagem de solos proposto, o solvente orgânico
coexistiu com as micelas do tensoativo, e tanto o desempenho de lavagem de solos como a
capacidade final de lavagem do solo são aumentadas em comparação com os de um processo
46
conseguiram extrair o DCB da solução em aproximadamente 50 % para o SDS, 80 % para o
Tween 80 e 90 % para o Brij 35.
Silva, Delerue-Matos & Fiúza (2005) exploraram a possibilidade de utilizar uma técnica de remediação ex-situ para solos contaminados com hidrocarbonetos de petróleo. Verificou -se a
miscibilidade dos sistemas ternários escolhidos, constituídos por acetato de etila, acetona e água.
Este sistema provou satisfazer os requisitos anteriores que permitiam a formação de uma mistura
única de fase líquida dentro de um largo espectro de composições, e também permitindo um
contato íntimo com o solo. Contaminantes da faixa do diesel com diferentes grupos funcionais
foram selecionados, em especial o naftaleno, o xileno e o hexadecano. O controle analítico foi
feito por cromatografia gasosa acoplado a FID. A cinética da extração demonstrou ser rápida,
atingindo o equilíbrio depois de 10 min. O efeito da razão sólido-líquido sobre a eficiência da
extração também foi estudado, mostrando que baixos valores de razão S/L (1:8, m/v) são mais
eficazes, atingindo recuperações da ordem de 95 %. O solvente pode ser regenerado através de
destilação com uma perda de cerca de 10 %. Os contaminantes não são evaporados e
permanecem na fase não-volátil. Os resultados mostraram que uma extração ex-situ com solvente
é uma opção viável para a remediação de aromáticos semi-voláteis, poliaromáticos e
hidrocarbonetos linear.
Paria (2008) estudou a remediação de solos e água contaminados com materiais orgânicos através de sistemas de tensoativos, com foco na adsorção do tensoativo ao solo, na solubilização
de hidrocarbonetos orgânicos nas micelas, supersolubilização, densidade, deslocamento
modificado, degradação do hidrocarboneto orgânico na presença de tensoativos, separação de
tensoativos no solo e na fase orgânica líquida, particionamento de contaminantes no solo, e na
remoção de compostos orgânicos a partir do solo, em presença de agentes tensoativos. Sistemas
microemulsionados mostraram maior capacidade de solubilização de hidrocarbonetos do que
sistemas de tensoativos. No caso de biodegradação dos hidrocarbonetos, a taxa é muito lenta
devido à baixa solubilidade em água e velocidade de dissolução, mas a presença de agentes
tensoativos pode aumentar a biodisponibilidade de compostos hidrofóbicos para solubilização,
consequentemente aumentando a taxa de degradação.
Lai et al. (2009) verificaram a remoção de hidrocarbonetos em solos contaminados com óleo pesado através de sistemas de biotensoativos. A capacidade de remoção total de
comparada com a eficiência dos tensoativos sintéticos. Foi ainda evidenciado que biotensoativos
apresentaram uma eficiência de remoção muito maior do que tensoativos sintéticos. Usando 0,2
% em massa de ramnolipidos, surfactina, Tween 80 e Triton X-100, a remoção de TPH do solo
contaminado com 3.000 mg de TPH/kg a seco foi de 23 %, 14 %, 6 % e 4 %, respectivamente,
enquanto que a eficiência de remoção aumentou para 63 %, 62 %, 40 % e 35 %, respectivamente,
para o solo contaminado com 9000 mg de TPH/kg a seco. A eficiência de remoção de TPH
também aumentou com o aumento da concentração de biotensoativo (de 0 a 0,2 % em massa)
mas não varia significativamente durante um tempo de contato de 1 e 7 dias.
Castro Dantas et al. (2010) estudaram o uso de sistemas microemulsionados na solubilização de frações pesadas de petróleo para prevenção de formação de borra de petróleo nas
operações, sendo utilizados amostras de arenito de formação Botucatu e Assu para o processo de
contaminação/remediação, e o óleo pesado foi obtido da fazenda Belém. Os testes foram
conduzidos com sistema microemulsionado com concentração de matéria ativa de 20, 30 e 40 %
em massa. As eficiências obtidas foram acima de 80 % para todas as concentrações de matéria
3. MATERIAIS E MÉTODOS
Neste capítulo de Materiais e Métodos estão apresentados os materiais utilizados para
realização dos experimentos, bem como a metodologia experimental desenvolvida para obtenção
dos resultados.
3.1. METODOLOGIA
A metodologia experimental seguida para a realização do presente trabalho foi constituída
da obtenção, caracterização e contaminação da rocha calcária (Figura 14a), da obtenção e
caracterização dos sistemas microemulsionados (Figura 14b) e da remoção do óleo da rocha
contaminada (Figura 14c).
Figura 14 - Fluxogramas da metodologia experimental
50
(b) Obtenção e caracterização dos sistemas microemulsionados