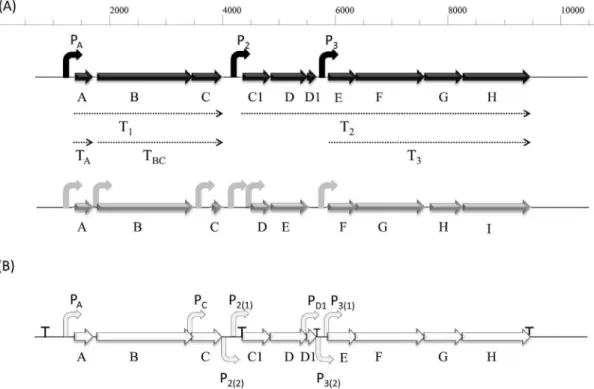
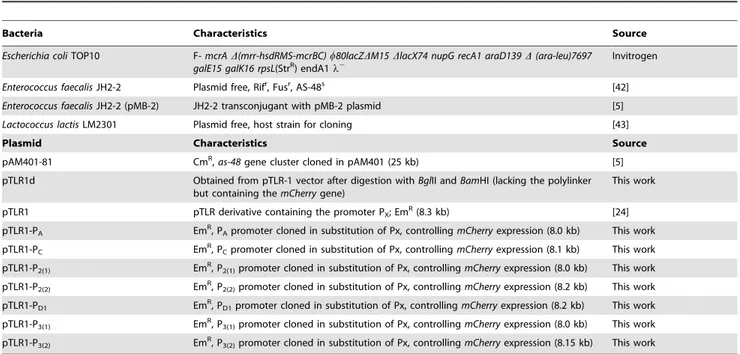
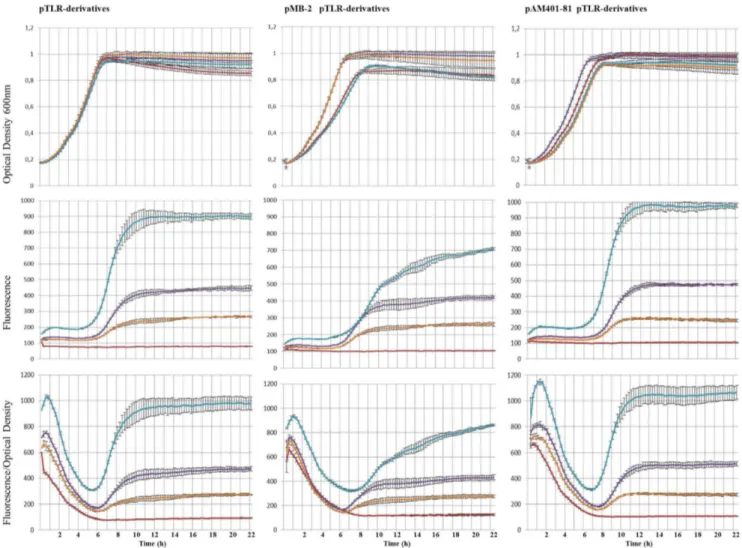
Analysis of the promoters involved in enterocin AS-48 expression.
Texto
Imagem

Documentos relacionados
Vê-se, portanto, que a influência do Estado na vida pri- vada do cidadão não deixa de ser uma forma direta de exercer o Poder, razão pela qual é extremamente relevante o tema
Ao Dr Oliver Duenisch pelos contatos feitos e orientação de língua estrangeira Ao Dr Agenor Maccari pela ajuda na viabilização da área do experimento de campo Ao Dr Rudi Arno
Neste trabalho o objetivo central foi a ampliação e adequação do procedimento e programa computacional baseado no programa comercial MSC.PATRAN, para a geração automática de modelos
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
Peça de mão de alta rotação pneumática com sistema Push Button (botão para remoção de broca), podendo apresentar passagem dupla de ar e acoplamento para engate rápido
Material sujeito a análise : Plano Nacional de Saúde (MS. DGS, 2004a, 2004b);. Método
If, on the contrary, our teaching becomes a political positioning on a certain content and not the event that has been recorded – evidently even with partiality, since the
Para tanto foi realizada uma pesquisa descritiva, utilizando-se da pesquisa documental, na Secretaria Nacional de Esporte de Alto Rendimento do Ministério do Esporte
