www.jped.com.br
ARTIGO
ORIGINAL
Chromosomal
microarrays
testing
in
children
with
developmental
disabilities
and
congenital
anomalies
夽
Guillermo
Lay-Son
a,b,∗,
Karena
Espinoza
a,
Cecilia
Vial
a,
Juan
C.
Rivera
a,
María
L.
Guzmán
a,be
Gabriela
M.
Repetto
a,baCentrodeGenéticaHumana,FaculdadedeMedicina,ClínicaAlemanaUniversidaddelDesarrollo,Santiago,Chile bHospitalPadreHurtado,Santiago,Chile
Recebidoem20demarçode2014;aceitoem9dejulhode2014
KEYWORDS
Microarrays; Congenital anomalies; Developmental disabilities; Copynumber variants; Diagnosis
Abstract
Objectives: Clinicaluseofmicroarray-basedtechniquesfortheanalysisofmany developmen-tal disorders hasemergedduring thelastdecade.Thus, chromosomal microarrayhas been positionedasafirst-tiertest.ThisstudyreportsthefirstexperienceinaChileancohort. Methods: Chileanpatientswithdevelopmentaldisabilitiesandcongenitalanomalieswere stu-diedwithahigh-densitymicroarray(CytoScanTM HDArray,Affymetrix,Inc.,SantaClara,CA, USA).Patientshadpreviouscytogeneticstudieswitheitheranormalresultorapoorly charac-terizedanomaly.
Results: Thisstudytested40patientsselectedbytwoormorecriteria,including:major con-genitalanomalies,facialdysmorphism,developmentaldelay,andintellectualdisability.Copy numbervariants(CNVs)werefoundin72.5%ofpatients,whileapathogenicCNVwasfoundin 25%ofpatientsandaCNVofuncertainclinicalsignificancewasfoundin2.5%ofpatients. Conclusion: Chromosomalmicroarrayanalysis isa useful andpowerfultool for diagnosis of developmental diseases, by allowing accurate diagnosis, improving the diagnosis rate, and discoveringnewetiologies.Thehighercostisalimitationforwidespreaduseinthissetting. ©2014SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
DOIserefereaoartigo:http://dx.doi.org/10.1016/j.jped.2014.07.003
夽 Comocitaresteartigo:Lay-SonG,EspinozaK,VialC,RiveraJC,GuzmánML,RepettoGM.Chromosomalmicroarraystestinginchildren
withdevelopmentaldisabilitiesandcongenitalanomalies.JPediatr(RioJ).2015;91:189---95.
∗Autorparacorrespondência.
E-mail:glayson@udd.cl(G.Lay-Son).
PALAVRAS-CHAVE
Microarrays; Anomalias congênitas; Atrasode desenvolvimento; Variantedonúmero decópia;
Diagnóstico
Análisecromossômicapormicroarrayemcrianc¸ascomdeficiências dedesenvolvimentoeanomaliascongênitas
Resumo
Objetivo: Ousoclínicodetécnicasbaseadasemmicroarraysparaaanálisedetranstornosde desenvolvimentotemsurgidoduranteaúltimadécada.Assim,omicroarraycromossômicotem sidoposicionadocomoumtestedeprimeironívelclínico.Relatamosaprimeiraexperiênciaem umacoortechilena.
Métodos: Pacientes chilenos com atraso de desenvolvimentoe anomalias congênitasforam estudadoscomummicroarraydealtadensidade(CytoScanTMHDArray,Affymetrix,Inc.,Santa Clara,CA,EUA).Pacientestiveramestudoscitogenéticosanteriores,ouumresultadonormal oudeumaanomalianãobemcaracterizada.
Resultados: Foramanalisados40pacientesselecionadospordoisoumaiscritérios,incluindo: anomalias congênitasmaiores, dismorfismofacial, atraso de desenvolvimentoe deficiência intelectual.Uma variante do número decópia (CNV) foi encontrada em 72,5% dos pacien-tes,enquantoqueumaCNVpatogênicafoiencontradaem25%dospacienteseumaCNVde significadoclínicoincertofoiencontradaem2,5%dospacientes.
Conclusões: Aanálisecromossômicamicroarrayéumaferramentaútilepoderosaem trans-tornosdedesenvolvimento,permiteumdiagnósticopreciso,melhoraataxadediagnósticoe descobrenovasetiologias.Ocustomaiselevadoéumalimitac¸ãoparaum usodifundidoem nossarealidade.
©2014SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduc
¸ão
Anomaliascongênitasmúltiplasafetamdedoisatrês indiví-duosemcada100nascidosvivosesãoaprincipalcausade mortalidadeedeficiênciainfantil.1,2Apesardesuamaioria serisolada e deorigemmultifatorial, pacientescom ano-maliasmúltiplasprecisamdeumaavaliac¸ãoparaidentificar umacausagenéticasubjacente.
Nosúltimosanos,oestudoetiológicodetranstornosno desenvolvimentotemsidoenriquecidocomousoclínicode técnicasem microarray.Em paísesdesenvolvidos, o carió-tipomolecular,ou aanálise cromossômicapor microarray (CMA),éconsideradoatécnicadeprimeiralinhaparaa aná-lisedepacientescomanomaliascongênitasmúltiplas,atraso dedesenvolvimento/deficiênciaintelectualnãosindrômicos etranstornosdoespectroautista.3-7
Em contrapartida, em países em desenvolvimento, como da América Latina, a detecc¸ão de anomalias cro-mossômicas ainda é feita principalmente por técnicas citogenéticas convencionais. O cariótipo com banda-mento GTG (banda G pela tripsina e corante giemsa) em linfócitos tem sido amplamente usado principal-menteparaidentificaranomalias cromossômicascomuma resoluc¸ão maior ouigual a 5-10 megabases (5-10 Mb).8-11 A hibridizac¸ão in situ por fluorescência (FISH) está dis-ponível para um número limitado de doenc¸as causadas pormicrodelec¸ões/microduplicac¸õescromossômicasetem umaresoluc¸ão de2-5 Mbem núcleos metafásicose entre 50-150 Kb em núcleos interfásicos.8,9,11-13 Outras técnicas moleculares foram desenvolvidas para procurar pequenas microdelec¸ões/microduplicac¸ões, como a Amplificac¸ão de MúltiplasSondasDependentesdeLigac¸ão(MLPA).14Em con-trapartidaaessastécnicasconvencionais,aCMAtemmaior
resoluc¸ão, que atinge 50 Kb, dez vezes maior do que a resoluc¸ãodocariótipoconvencional.13,15Essabusca desequi-líbriosgenéticos(ganhosouperdasdesegmentos cromossô-micos)emtodoogenomaetempermitidoaidentificac¸ãode novassíndromesnãoprontamente detectadaspelos méto-dosdescritos anteriormente.16-18 Adescobertadavariante normalemvariantesdonúmerodecópias(CNVs)representa umdesafioàinterpretac¸ãoclínica.15
Embora osestudos de diagnóstico paraindivíduos com anomalias congênitas oudeficiência intelectual com base nacitogenéticaconvencionalapresentemrendimento diag-nóstico próximo de 3%, a CMA apresenta rendimento de aproximadamente 15-20%, ou seja, mais de cinco vezes acimadocariótipocombandamentoG,6oquejustificaseu usocomotestedediagnósticodeprimeiralinhapara pacien-tescomdiagnósticosclínicosdesconhecidos.Estima-seque a CMAconsiga detectar, individualmente,mais de99% de todasasanomaliasdocariótipo.5
Este relatório apresenta nossa experiência pioneira no usodaCMAemumacoortedepacienteschilenoscom ano-maliascongênitasmúltiplassemdiagnósticoetiológico.
Métodos
Pacientes
Foramselecionados40pacientesdasClínicasGenéticasdo HospitalPadreHurtado(Santiago,Chile)entremaiode2012 enovembrode2012.
desenvolvimento (AD) ou deficiência intelectual (DI). Nenhum dos pacientes apresentou uma causa definida da doenc¸a.
Dentre todosos pacientes, 36apresentaram um carió-tipo normal, dois apresentaram um pequeno cromossomo marcador supranumerário (sSMC) descaracterizado, um apresentou um cromossomo derivado, um apresentou translocac¸ão robertsoniana herdada e um apresentou monossomia docromossomoX, porém comcaracterísticas adicionaisincomuns.
Ocomitêdeéticalocalaprovouesteestudoeo consenti-mentoinformadoporescritofoiobtidodetodosospacientes e/oupais.
Processamentodasamostras
ODNAgenômicofoipurificadoapartirdecélulas mononu-clearesdosangueperiféricocomoKitMiniprepdaAxyPrep paraextrac¸ãodeDNAgenômicodosangue (Axygen Biosci-ences,UnionCity,CA,EUA)deacordocomasinstruc¸õesdo fabricante. ODNA genômicodecada paciente foi hibridi-zado parao CytoScanTM HD Array(Affymetrix, Inc.,Santa
Clara,CA,EUA)deacordocomasinstruc¸õesdofabricante. Essa hibridizac¸ão genômicacomparativa porarray dealta densidadepersonalizadacomquase2,7milhõesde marcado-resgenéticosinclui700.000marcadoresdepolimorfismode nucleotídeosimples(SNP)emaisde1,9milhãodesondasde oligonucleotídeosnãopolimórficasparaadetecc¸ãodeCNVs.
Análisededados
Os dados de array foram analisados com o software da Affymetrix® Chromosome Analysis Software Suite (ChAS)
v.1.2.2(Affymetrix,Inc.,SantaClara,CA,EUA)combasena sequênciagenômicadereferênciadoUCSCGenomeBrowser hg19,fevereirode2009(GRCh37/hg19).Analisamos varian-tesdonúmerodecópias(CNV)demaisde400Kb,conforme recomendado.6 Com essa plataforma de maiorresoluc¸ão, conseguimosavaliarumaCNVmenor,porém nenhum paci-ente apresentou anomalias clinicamente relevantes entre 100e400Kbe,dessaforma,mantivemosesselimitepara finsdesterelatório.AsCNVsdemaisde400Kbforam catego-rizadasporrelevânciaclínicacomoCNVderelevânciaclínica clara(grupo1),CNVderelevâncianãoclaraourelevância incerta(grupo2)ouCNVbenignaoupolimórfica(grupo3), comasbasesdedadospublicamentedisponíveisIsca (Inter-nationalStandardCytogenomicArray),19 DGV(Databaseof GenomicVariants),20,21Omim(OnlineMendelianInheritance in Man),22 Decipher (Database of Chromosomal Imbalance andPhenotype in HumansUsing Ensembl Resources)23,24 e Ecaruca (European Cytogeneticists Association Registerof UnbalancedChromosomeAberrations).25
Resultados
Dos40 pacientesanalisados,16 (41%)eramdosexo femi-nino.As idadesvariavamdeummês a25 anos, comuma idademédiade4,2anos.Comoselecionado,avasta maio-riadenossospacientestemanomaliasmúltiplas,incluindo
Grupo 1 Grupo 2 Grupo 3
29 pacientes 40 pacientes
12 CNVs 9L/3G
1 CNVs 0L/1G
42 CNVs 2L/40G 55 CNVs
11L/44G
0 CNVs
A
B
10 pacientes 1 paciente
11 pacientes
25 pacientes
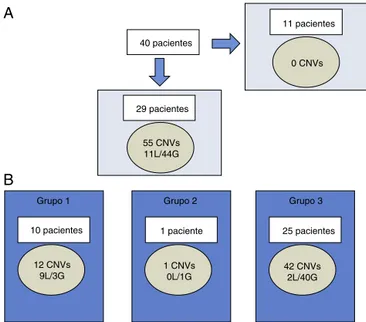
Figura1 Caracterizac¸ãodasCNVsmaioresdoque400Kbna coortechilena.
A,nãoforamencontradasCNVsem11de40pacientes.Nos29 pacientesrestantes,foramencontradas55CNVs (11perdase 44ganhos).B,cadapacientepoderáterCNVdemaisumgrupo ao mesmotempo. AsCNVs foram categorizadas por relevân-ciaclínicacomoCNVderelevânciaclínicaclara(grupo1),CNV derelevânciaincerta(grupo2)ouCNVbenignaoupolimórfica (grupo3).
CNVs:variantesdonúmerodecópias;G:ganhos;L:perdas.
transtornosdedesenvolvimentoestruturaisefuncionais.Os detalhesclínicosestãoresumidosnatabela1.
Caracterizac¸ãodavariantedonúmerodecópias (CNV)(fig.1)
Encontramos55CNVsdemaisde400Kbem29de40 paci-entes(72,5%),variac¸ãoentre0e 4CNVsporpaciente. Do total,11foramperdase44foramganhos(fig.1A).O tama-nhodos desequilíbrios cromossômicos variava entre420,9 Kbe25,2Mb.Esseúltimovalorcorrespondeaumpaciente comsSMCdetectadoporcariótipo.
Esses 55 CNVs foram classificados em três grupos com baseemsuainterpretac¸ãoclínica.
Deacordocom nossaclassificac¸ão (fig. 1B), 21,8% per-tenciamaogrupo1(relevânciaclínicaclararelacionadaao fenótipo),1,8%aogrupo2(relevância incerta)e 76,4%ao grupo3 (benigno oupolimórfico). As perdas aconteceram predominantemente no grupo 1, ao passo que os ganhos ocorreramnogrupo3.
Rendimentodiagnóstico
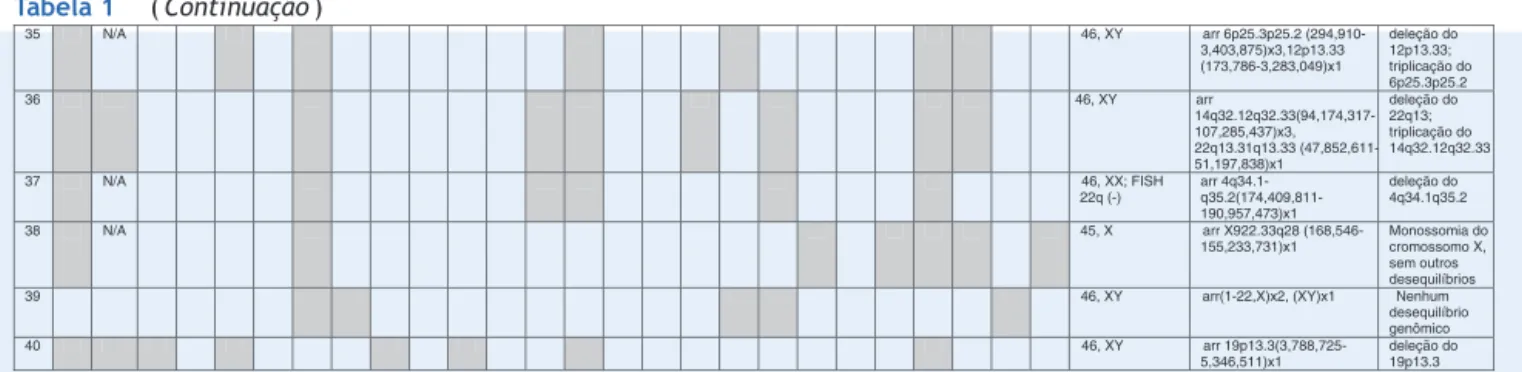
Tabela1 Resumodascaracterísticasclínicasedosdesequilíbriosgenéticosencontradosnessacoorte N º do Pa ci e nt e At raso no De se nv o lv ime nt o Def ic iên cia In te le ct ua l H ip o ton ia ge ner al iza da Co nv u ls õe s
Malformação do sistema nervoso Microcefalia Dismorfismo facial An
om al ia con g êni ta d o o lh o An om al ia v isu al An om al ia do o uv id o e xt er no Def ic iên cia au di ti va Fi ss ur a la bi o pa la tin a (+/-) Fi ss ur a pa lati na /Seq uê nc i Doença ca rdíaca con g êni ta He mi vé rt eb ra s/ Es co lios e Mal for m aç ão g a st ro int es ti nal Hér n ia abd om in al Mal for m aç ão no s rins De fei to s na Ge ni tá lia/ Tr at o An om al ia do s membro s Hi pe rfrou xi dã o liga me nt a r An om al ia es qu el ét ic a Baix a es ta tu ra Dé fi c it de cres ci m e nto Obesidade Def eit o Con g êni to d a Es tu do s ci to ge n ét ic os Res ul ta do s d a Análi s e Cro m ossô m ica po r Mi c ro a rr ay (CMA )
Interpretação clínic
a
1 N/A 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
2 N/A 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
3 N/A 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
4 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
5 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
6 N/A 46, XY arr 9p23 (9,323, 653-
11,359,708)x3
Variante de relevância incerta (VOUS)
7 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
8 46, XX; FISH
22q (-)
arr 6p25.1-p23 (6,389,84 7-15,124,349)x1
deleção do 6p25.1p23
9 N/A 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
10 46, XY, der (16) arr(1-22,X)x2, (XY)x1 Inversão
pericêntrica do cromossomo 16
11 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
12 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
13 46, XX; FISH
22q (-)
Nenhum arr(1-22,X)x2
desequilíbrio genômico
14 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
15 46, XY; FISH
22q (-)
Nenhum arr(1-22,X)x2, (XY)x1
desequilíbrio genômico
16 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
17 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
18 46,XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
19 47, XX,
+mar/46, XX arr 20p1(7,180,552.3q112-32,40.222,789)x2~3 Trissomia mosaico parcial do cromossomo 20
20 N/A 46, XY arr
2q23.2q24.2(150,2016,0 3-161,228,203)x1
deleção do 2q23.2q24.3
21 46, XY; FISH
22q (-)
Nenhum arr(1-22,X)x2, (XY)x1
desequilíbrio genômico
22 46, XY; FISH
22q (-)
arr 15q25.2 (83,083,418 -84,834,123)x1
deleção do 15q25.2
23 46, XY; FISH
22q (-)
Nenhum arr(1-22,X)x2, (XY)x1
desequilíbrio genômico
24 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
25 46, XX; FISH
17P (-)
Nenhum arr(1-22,X)x2
desequilíbrio genômico
26 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
27 N/A 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
28 N/A 46, XY; FISH
22q (-); Análise de quebras cromossômicas (-)
Nenhum arr(1-22,X)x2, (XY)x1
desequilíbrio genômico
29 46, XX arr(1-22,X)x2 Nenhum
desequilíbrio genômico
30 45, XY, der
(14;15)(q10;q10)
arr(1-22,X)x2, (XY)x1 Translocação Robertsoniana, nenhum desequilíbrio genômico
31 46, XX arr
22q12.1q12.3(28,966, 333-34,541,402)x1
deleção do 22q12.1q12.3
32 N/A 47, XX,
+mar(mat); FISH
15q (-)
arr(1-22,X)x2 SMC bissatélite (RON manchadas), heterocromatina de origem materna (mãe saudável)
33 N/A 46, XX; FISH
22q (-)
Nenhum arr(1-22,X)x2
desequilíbrio genômico
34 46, XY; FISH 7q
(-); 22q(-) subtelômeros (-)
arr 6p25.3-p25.2 (156,97 4-2,400,023)x1
Tabela1 (Continuação)
35 N/A 46, XY arr 6p25.3p25.2
(294,910-3,403,875)x3,12p13.33 (173,786-3,283,049)x1
deleção do 12p13.33; triplicação do 6p25.3p25.2
36 46, XY arr
14q32.12q32.33(94,174,317 -107,285,437)x3, 22q13.31q13.33 (47,852,611-51,197,838)x1
deleção do 22q13; triplicação do 14q32.12q32.33
37 N/A 46, XX; FISH
22q (-)
arr 4q34.1-q35.2(174,40 9,811-190,957,473)x1
deleção do 4q34.1q35.2
38 N/A 45, X arr X922.33q28 (168
,546-155,233,731)x1
Monossomia do cromossomo X, sem outros desequilíbrios
39 46, XY arr(1-22,X)x2, (XY)x1 Nenhum
desequilíbrio genômico
40 46, XY arr 19p13.3(3,788,72
5-5,346,511)x1
deleção do 19p13.3
CMA,análisecromossômicapormicroarray;FISH,hibridizac¸ãoinsituporfluorescência;N/A,nãoaplicávelpelaidade;NOR,região
organizadoradonúcleo;sSMC:pequenocromossomomarcadorsupranumerário.
No Grupo 2, apenas um paciente apresentou uma variac¸ãoderelevânciaincerta(VOUS).26Essepacientetinha uma triplicac¸ão de 2 Mb na posic¸ão 9p23 [array 9p23 (9.323.653-11.359.708)x3],porém,comasinformac¸ões dis-poníveisnasbasesdedadosenaliteraturabiomédica,não poderíamos descartar nem atribuir um efeito patogênico definitivo.
Nãoconseguimosencontrardesequilíbriosgenéticos ver-dadeirosnosegundopacientecomsSMCenopacientecom cromossomo 16 derivado. No paciente com translocac¸ão robertsoniana,nãoencontramosdesequilíbriogenômicoque explicasseofenótipo.Porfim,nopacientecommonossomia docromossomo X, confirmamos a delec¸ão deum cromos-somoX, porém nãoencontramos desequilíbrios genômicos adicionais(fig.1).
Interpretac¸ãoclínica
EncontramosumaCNVpatogênicanãovistaanteriormente (n=9)ounãobemcaracterizada (n=1)porcariótipo con-vencionalem10de40pacientesdogrupo1(25,0%).Além disso, corroboramosumaanomaliacitogenéticaconhecida emumpaciente(monossomiadocromossomoX).
Seconsiderarmosapenasospacientescomcariótipo nor-mal(35),nossataxadediagnósticosaumentoupara28,5%. Alémdisso,encontramosumaCNVderelevânciaclínica incertaouVOUS(grupo2)emoutropaciente.Esseaguarda testesadicionaiseacompanhamentoparadefinira relevân-ciaclínicadesseachado.Planejamosestudarambosospais pormeiodeFishe/ouMLPA.
Discussão
EsteéoprimeirorelatóriodetestedeCMAemumacoorte depacienteschilenoscomdeficiênciasdedesenvolvimento, considerandoqueexistempoucos relatóriosdousoclínico demicroarrayscromossômicosnaAméricaLatina,que rela-tamexperiênciasdecasosindividuais.NaAméricadoSul, uma experiência semelhante foi relatada em um grupo depacientes noBrasil.27,28 Foram analisados95 pacientes ‘‘sindrômicos’’’comcariótiponormalefoirelatadoum ren-dimentodiagnósticode17%.28
Detectamos maisde25%dealterac¸õespatogênicasem nossa coorte, que estão na mais alta faixa relatada na literatura.Em pacientes com atraso dedesenvolvimento/
deficiência intelectual sindrômicos e não sindrômicos e cariótipo normal/Fish, a metanálise apresenta um rendi-mentodiagnósticode7,8%-13,8%,variac¸ãode5%a50%.5,29 Issoéexplicado,emgrandeparte,pelaheterogeneidadeno modelodosestudos,principalmentenaselec¸ãodos pacien-tes,testespreviamentefeitoseplataformadearrayusada. Emnosso caso,a alta taxade diagnósticopodeser expli-cadapelo fatodeque a coorteestudadaé relativamente pequenae apresentaoviés depacientesmuitobem sele-cionados,muitosdosquaispermaneceramsemdiagnóstico pormuitotempo. Finalmente,usamosumaplataformade altadensidade.
Com o passar do tempo, diferentes bases de dados públicason-linecoletaraminformac¸õesfenotípicase genô-micasdemilhares depacientesanônimos,o que permitiu a compreensão da vasta maioria de CNVs como benignas ou polimórficas, sem análises adicionais. Na verdade, a maioria denossos achados foi de CNVs polimórficas (Poli-morfismosdoNúmero de Cópias[CNP]), corroboradasnas bases de dados citogenômicas e dos dadosde nossa pró-priacoorte.Observamos CNVsrecorrentesdemaisde400 Kbemmetadedospacientes,principalmenteenvolvendoos seguinteslocicromossômicos:10q11.22,14q32.22,16p11.2, 17q21.31,Yq11.223eYq11.23.Apenasumcasoque apresen-touumaVOUSexigiuumaanáliseadicionalparadeterminar a patogenicidade possível. Esse paciente tinha um ano e 11 meses, com DI, atraso de linguagem, deficiência de audic¸ão, hérnia inguinal e baixa estatura. Eleapresentou umcariótipo 46,XY e o array mostrou umganho de2 Mb no cromossomo 9p23, incluindo a triplicac¸ão parcial de umgene: PTPRD. A proteína PTPRD é uma proteína tiro-sinafosfatasetiporeceptor,expressaemcertasregiõesdo cérebro, como o hipocampo, e pode ter uma func¸ão no aprendizadoena memória30 ena organizac¸ãosináptica.31 Nenhum fenótipo foi atribuído à triplicac¸ão total desse gene.
paraidentificarpossíveisrearranjoscomimplicac¸õesparao aconselhamentogenético.7,32
Em nossa coorte, descobrimos que três pacientes com anomalias citogenéticas previamente conhecidas resulta-ram em um cariótipo molecular normal. Como esperado, o paciente portador de uma translocac¸ão robertsoniana apresentouumresultadodeCGH-arraynormal/equilibrado. No caso de um dos dois pacientes com sSMC, a cober-tura do array, as sequências genômicas envolvidas e o tamanho do sSMC podem explicar por que esse mate-rialgenéticonãofoi detectadoporessemétodo.19 Assim, esse pequeno cromossomo bisatélite provavelmente cor-responde a sequências altamente repetitivas, típicas de cromossomos acrocêntricosnão incluídosno arrayque foi demonstrado por bandamento da região organizadora do nucléolo(NOR). Nopacientecomumcromossomo16 deri-vado, com um padrão de bandamento anormal e uma morfologia também anormal (mais metacêntrica que o normal),umainversãopericêntricaéaexplicac¸ãomais plau-sível.
Assim, o cariótipocontinua sendo maisadequado para avaliarpossíveisportadores de rearranjoscromossômicos, casaiscomabortosrecorrentesoupacientescomum fenó-tipocaracterísticodeaneuploidia.Damesmaforma,aFish émaisadequadaemcaso desuspeitaelevadadeuma sín-dromeespecíficademicrodelec¸ão.7
A análise cromossômica por microarray é um método altamente preciso, robusto e de alto rendimento. Temos usadoumarraycombinadocombaseemoligonucleotídeos e um array de SNP, o que cria muitas vantagens, já que o array de SNP melhora de forma significativa a preci-são e a sensibilidade da detecc¸ão e mosaicismo da CNV epermite igualmente adetecc¸ão devariantes neutrasde númerode cópias.33 No caso do chip usadoneste relató-rio(CytoscanHD,Affymetrix),aplataformaquímicaeseus algoritmosanalisamooligonucleotídeoeassondasdeSNPde formaindependente.Assim,podemosdetectareconfirmar cadaCNVao mesmo tempo, o quenãoexige confirmac¸ão adicional.32,33 Nesse caso, em vezde umaconfirmac¸ãode achadodearrays,aanáliseFISHpermitedeterminarotipo derearranjo.32
Oaltocustodoexamecriaumalimitac¸ãonouso difun-dido do cariótipo molecular. No Chile, o custo da CMA é quatroasete vezes maiordoque o cariótipoe/ou Fishe atualmente não é coberto por plano de saúde. Contudo, parachegaraumdiagnósticoprecoceemalgunspacientes comanomaliascongênitas múltiplase/ouatraso globalde desenvolvimento, é possível evitar exames desnecessários (a‘‘odisseiade diagnóstico’’)e permitir foco nos proble-masespecíficos,oquepodeterumbomcusto-benefícioem longoprazo.34-38Podeseresperadaumadiminuic¸ãodos cus-toscomo passardotempo, o quepermitiráseuuso mais difundido.Porfim,devesermencionadoqueexistemoutras plataformascomresoluc¸ãomaisbaixa,porémporumcusto relativamentereduzido.
Reconhecendo suas limitac¸ões, existe cada vez mais evidência do impacto clínico dessa tecnologia. Em mui-tos pacientes, um diagnóstico definitivo pode afetar não apenas a informac¸ão e o aconselhamento de seu ambi-entefamiliar,39mastambémavigilânciaativaembuscade possíveiscomplicac¸ões,entreoutrostipos deintervenc¸ões médicas.37,38
NossosdadosmostramautilidadedaCMA,quepermite umacapacidadedediagnósticomelhoradacomprecisãoeo aprimoramentodagestãoesupervisãodasaúdedessegrupo depacientescomnecessidadesespeciais.
Financiamento
Bolsa daFundac¸ãodeSaúde Infantilem Birmingham, Ala-bama.
Conflitos
de
interesse
Osautoresdeclaramnãohaverconflitosdeinteresse.
Agradecimentos
AgradecemosàFundac¸ãodeSaúdeInfantilemBirmingham, Alabama, porsua bolsa paraeste trabalho, aospacientes eparentesparticipanteseàDra.SilviaCastilloeà citoge-neticistaAna MaríaFuentespelaanálisedosresultadosde cariótipoconvencional.
Referências
1.ChristiansonA, Howson M,ModellB. Marchof Dimes:global reportonbirthdefects.Thehiddentollofdyinganddisabled children.WhitePlains,NY:MarchofDimesBirthDefects Foun-dation;2006.
2.KumarP,BurtonBK.Dysmorphology.In: KumarP,BurtonBK, editors.Congenitalmalformations:evidence-basedevaluation andmanagement.NewYork:McGraw-HillProfMed/Tech;2007. p.3---11.
3.RauchA,HoyerJ,GuthS,ZweierC,KrausC,BeckerC,etal. Diagnosticyieldofvariousgeneticapproachesinpatientswith unexplaineddevelopmentaldelayormentalretardation.AmJ MedGenetA.2006;140:2063---74.
4.VermeeschJR,Fiegler H,DeLeeuwN, SzuhaiK, Schoumans J,Ciccone R, et al. Guidelines for molecular karyotyping in constitutionalgenetic diagnosis.Eur JHum Genet.2007;15: 1105---14.
5.Hochstenbach R, Van Binsbergen E, Engelen J, Nieuwint A, PolstraA, Poddighe P,et al. Arrayanalysisand karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands.EurJMedGenet.2009;52:161---9.
6.MillerDT,AdamMP,AradhyaS,BieseckerLG,BrothmanAR, Car-terNP,etal.Consensusstatement:chromosomalmicroarrayis afirst-tierclinicaldiagnostictestforindividualswith develop-mentaldisabilitiesorcongenitalanomalies.AmJHumGenet. 2010;86:749---64.
7.Manning M, Hudgins L. Professional Practice and Guideli-nesCommittee.Array-basedtechnologyandrecommendations for utilization in medical genetics practice for detec-tion of chromosomal abnormalities. Genet Med. 2010;12: 742---5.
8.ShafferLG,LedbetterDH,LupskiJR.Molecularcytogenetics ofcontiguousgenesyndromes:mechanismsandconsequences ofgenedosageimbalance.In:ScriverCR,BeaudetAL,SlyWS, ValleD,ChildsB,KinzlerKW,etal.,editors.Themetabolicand molecularbasisofinheriteddisease.NewYork:McGrawHill; 2001.p.1291---324.
10.SalmanM, JhanwarSC, Ostrer H. Will the newcytogenetics replacetheoldcytogenetics?ClinGenet.2004;66:265---75.
11.Smeets DF. Historical prospective of human cytogenetics: from microscope to microarray. Clin Biochem. 2004;37: 439---46.
12.TurleauC,VekemansM.Newdevelopmentsincytogenetics.Med Sci(Paris).2005;21:940---6.
13.EdelmannL,HirschhornK.ClinicalutilityofarrayCGHforthe detectionofchromosomalimbalancesassociatedwithmental retardationandmultiplecongenitalanomalies.AnnNYAcad Sci.2009;1151:157---66.
14.JeheeFS, Takamori JT, Medeiros PF, Pordeus AC, Latini FR, BertolaDR,etal.UsingacombinationofMLPAkitstodetect chromosomalimbalancesinpatientswithmultiplecongenital anomaliesandmentalretardationisavaluablechoicefor deve-lopingcountries.EurJMedGenet.2011;54:e425---32.
15.LeeC,IafrateAJ,BrothmanAR.Copynumbervariationsand clinicalcytogeneticdiagnosis ofconstitutionaldisorders.Nat Genet.2007;39:S48---54.
16.ShafferLG,BejjaniBA,TorchiaB,KirkpatrickS,CoppingerJ, BallifBC. Theidentification ofmicrodeletionsyndromes and otherchromosomeabnormalities:cytogeneticmethodsofthe past,newtechnologiesforthefuture.AmJMedGenetCSemin MedGenet.2007;145C:335---45.
17.Schluth-BolardC,TillM,EderyP,SanlavilleD.Newchromosomal syndromes.PatholBiol(Paris).2008;56:380---7.
18.Slavotinek AM. Novel microdeletion syndromes detected by chromosomemicroarrays.HumGenet.2008;124:1---17.
19.International Collaboration for Clinical Genomics (ICCG),
International Standard CytogenomicArray (ISCA) Consortium
DatabaseSearch Disponível em http://www.iccg.org/.
Aces-sadoem4demarc¸ode2014.
20.MacDonald JR, ZimanR, YuenRK, Feuk L, SchererSW. The DatabaseofGenomicVariants: acurated collectionof struc-tural variation in the human genome. Nucleic Acids Res. 2014;42:D986---92.
21.Databaseof Genomic Variants (DGV),Disponível em http://
dgv.tcag.ca/dgv/app/home.Acessadoem4demarc¸ode2014.
22.McKusick-Nathans Institute of Genetic Medicine, Johns
Hop-kinsUniversity(Baltimore,MD).OnlineMendelianInheritance
inMan,OMIM®Disponívelemhttp://omim.org/.Acessadoem
10demarc¸ode2014.
23.FirthHV,RichardsSM,BevanAP,ClaytonS,CorpasM,RajanD, etal.Decipher:DatabaseofChromosomalImbalanceand Phe-notypeinHumansUsingEnsemblResources.AmJHumGenet. 2009;84:524---33.
24.Database of Chromosomal Imbalance and Phenotype in
Humans using Ensembl Resources (Decipher). Disponível em
https://decipher.sanger.ac.uk/.Acessadoem10demarc¸ode 2014.
25.EuropeanCytogeneticists AssociationRegister of Unbalanced
Chromosome Aberrations (Ecaruca). Disponível em http://
umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp.
Acessadoem10demarc¸ode2014.
26.DeLeeuwN,DijkhuizenT,Hehir-KwaJY,CarterNP,FeukL,Firth HV,etal.Diagnosticinterpretationofarraydatausingpublic databasesandinternetsources.HumMutat.2012.
27.RosenbergC, Knijnenburg J,Bakker E, Vianna-MorganteAM, SloosW,OttoPA,etal.Array-CGHdetectionofmicro rearran-gementsinmentallyretardedindividuals:clinicalsignificance of imbalancespresent both in affected children and normal parents.JMedGenet.2006;43:180---6.
28.Krepischi-Santos AC,Vianna-Morgante AM, JeheeFS, Passos-Bueno MR, Knijnenburg J, Szuhai K, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisitedandnew alterations.Cytogenet Genome Res.2006;115:254---61.
29.MichelsonDJ,ShevellMI,SherrEH,MoeschlerJB,GropmanAL, AshwalS.Evidencereport:Geneticandmetabolictestingon childrenwithglobaldevelopmentaldelay:reportoftheQuality StandardsSubcommitteeoftheAmericanAcademyof Neuro-logyandthePracticeCommitteeoftheChildNeurologySociety. Neurology.2011;77:1629---35.
30.Uetani N, Kato K, OguraH, Mizuno K, Kawano K, Mikoshiba K, et al. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19:2775---85.
31.Takahashi H, Craig AM. Protein tyrosine phosphatases PTP␦, PTP, and LAR: presynaptic hubs for synapse organization. TrendsNeurosci.2013;36:522---34.
32.Bui TH, Vetro A, Zuffardi O, Shaffer LG. Current contro-versies in prenatal diagnosis 3: is conventional chromosome analysis necessaryin the post-array CGH era?Prenat Diagn. 2011;31:235---43.
33.Mason-SuaresH,KimW,GrimmettL, WilliamsES,HornerVL, KunigD,etal.Densitymatters:comparisonofarrayplatforms fordetectionofcopy-numbervariationandcopy-neutral abnor-malities.GenetMed.2013;15:706---12.
34.Wordsworth S, Buchanan J, Regan R, Davison V, Smith K, Dyer S, et al. Diagnosing idiopathic learning disability: a cost-effectivenessanalysisofmicroarraytechnologyinthe Nati-onal Health Service of the United Kingdom. Genomic Med. 2007;1:35---45.
35.NewmanWG,HamiltonS,AyresJ,SangheraN,SmithA,Gaunt L,etal.Arraycomparativegenomichybridizationfor diagno-sisofdevelopmentaldelay:anexploratorycost-consequences analysis.ClinGenet.2007;71:254---9.
36.TrakadisY,ShevellM.Microarrayasafirstgenetictestin glo-baldevelopmentaldelay:acost-effectivenessanalysis.DevMed ChildNeurol.2011;53:994---9.
37.CoulterME,Miller DT,HarrisDJ, HawleyP,PickerJ,Roberts AE,etal.Chromosomalmicroarraytestinginfluencesmedical management.GenetMed.2011;13:770---6.
38.Riggs E,WainK, RiethmaierD,Smith-Packard B,Faucett W, Hoppman N,et al. Chromosomal microarrayimpacts clinical management.ClinGenet.2013.