www.jped.com.br
ORIGINAL
ARTICLE
Chromosomal
microarrays
testing
in
children
with
developmental
disabilities
and
congenital
anomalies
夽
Guillermo
Lay-Son
a,b,∗,
Karena
Espinoza
a,
Cecilia
Vial
a,
Juan
C.
Rivera
a,
María
L.
Guzmán
a,b,
Gabriela
M.
Repetto
a,baCenterforHumanGenetics,FacultaddeMedicina,ClínicaAlemanaUniversidaddelDesarrollo,Santiago,Chile
bHospitalPadreHurtado,Santiago,Chile
Received20March2014;accepted9July2014 Availableonline30October2014
KEYWORDS
Microarrays; Congenital anomalies; Developmental disabilities; Copynumber variants; Diagnosis
Abstract
Objectives: Clinicaluseofmicroarray-basedtechniquesfortheanalysisofmany developmen-tal disorders hasemergedduring thelastdecade.Thus, chromosomal microarrayhas been positionedasafirst-tiertest.ThisstudyreportsthefirstexperienceinaChileancohort. Methods: Chileanpatientswithdevelopmentaldisabilitiesandcongenitalanomalieswere stud-iedwithahigh-densitymicroarray(CytoScanTM HDArray, Affymetrix,Inc., SantaClara, CA,
USA).Patientshadpreviouscytogeneticstudieswitheitheranormalresultorapoorly charac-terizedanomaly.
Results: Thisstudytested40patientsselectedbytwoormorecriteria,including:major con-genitalanomalies,facialdysmorphism,developmentaldelay,andintellectualdisability.Copy numbervariants(CNVs)werefoundin72.5%ofpatients,whileapathogenicCNVwasfoundin 25%ofpatientsandaCNVofuncertainclinicalsignificancewasfoundin2.5%ofpatients. Conclusion: Chromosomalmicroarrayanalysis isa useful andpowerfultool for diagnosis of developmental diseases, by allowing accurate diagnosis, improving the diagnosis rate, and discoveringnewetiologies.Thehighercostisalimitationforwidespreaduseinthissetting. ©2014SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
夽
Pleasecitethisarticleas:Lay-SonG,EspinozaK,VialC,RiveraJC,GuzmánML,RepettoGM.Chromosomalmicroarraystestinginchildren withdevelopmentaldisabilitiesandcongenitalanomalies.JPediatr(RioJ).2015;91:189---95.
∗Correspondingauthor.
E-mail:[email protected](G.Lay-Son).
http://dx.doi.org/10.1016/j.jped.2014.07.003
PALAVRAS-CHAVE
Microarrays; Anomalias congênitas; Atrasode desenvolvimento; Variantedonúmero decópia;
Diagnóstico
Análisecromossômicapormicroarrayemcrianc¸ascomdeficiênciasde desenvolvimentoeanomaliascongênitas
Resumo
Objetivo: Ousoclínicodetécnicasbaseadasemmicroarraysparaaanálisedetranstornosde desenvolvimentotemsurgidoduranteaúltimadécada.Assim,omicroarraycromossômicotem sidoposicionadocomoumtestedeprimeironívelclínico.Relatamosaprimeiraexperiênciaem umacoortechilena.
Métodos: Pacientes chilenos com atraso de desenvolvimentoe anomalias congênitasforam estudadoscomummicroarraydealtadensidade(CytoScanTMHDArray,Affymetrix,Inc.,Santa
Clara,CA,EUA).Pacientestiveramestudoscitogenéticosanteriores,ouumresultadonormal oudeumaanomalianãobemcaracterizada.
Resultados: Foramanalisados40pacientesselecionadospordoisoumaiscritérios,incluindo: anomaliascongênitasmaiores,dismorfismofacial,atrasodedesenvolvimentoedeficiência int-electual. Uma variantedo número decópia (CNV) foi encontrada em 72,5% dos pacientes, enquantoqueumaCNVpatogênicafoiencontradaem25%dospacienteseumaCNVde signifi-cadoclínicoincertofoiencontradaem2,5%dospacientes.
Conclusões: A análise cromossômica microarray é uma ferramenta útil e poderosa em transtornos de desenvolvimento,permitindoum diagnóstico preciso, melhorando a taxade diagnóstico,edescobrindonovasetiologias.Ocustomaiselevadoéumalimitac¸ãoparaumuso difundidoemnossarealidade.
©2014SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduction
Majorcongenitalanomaliesaffecttwotothreeofevery100 livenewborns, andarealeadingcauseofinfantmortality anddisability.1,2Althoughmostareisolatedand
multifacto-rialinorigin,patients withmultiple abnormalitiesrequire
anassessmenttoidentifyanunderlyinggeneticcause.
In recent years, the etiological study of
developmen-tal disorders has been enriched with the clinical use
of microarray-based techniques. In developed countries,
molecular karyotyping or chromosomal microarray (CMA)
is considered the first-line technique for the analysis of
patientswithmultiplecongenitalanomalies,nonsyndromic
developmental delay/intellectual disability, and autism
spectrumdisorders.3---7
In contrast, in developing nations such asLatin
Amer-ican countries, detection of chromosomal anomalies is
still performed mainly by conventional cytogenetic
tech-niques. GTG (G-bands by trypsin using Giemsa) banding
karyotypinginlymphocytes hasbeen mainlyusedto
iden-tify chromosomal abnormalities with a resolution equal
or greater than 5-10 megabases (5-10 Mb).8---11
Fluores-centin situ hybridization(FISH) is available for a limited
number of diseases caused by chromosomal
microdele-tions/microduplications and has a resolution of 2-5 Mb
in metaphase and between 50-150 Kb in interphase
nuclei.8,9,11---13Othermoleculartechniqueshavebeen
devel-oped to look for small microdeletions/microduplications,
such as multiplex ligation-dependent probe amplification
(MLPA).14Incontrasttotheseconventionaltechniques,CMA
hasahigherresolution, whichreaches upto50Kb, aten
timeshigherresolutionthanconventionalkaryotyping.13,15
Itseeksgeneticimbalances(gainsorlossesofchromosomal
segments)acrossthegenomeandhasallowedthe
identifi-cation ofnewsyndromesthat arenot readilydetected by
themethodsdescribedabove.16---18 Thediscoveryofnormal
variationascopynumbervariations(CNVs)posesachallenge
fortheclinicalinterpretation.15
Whilstdiagnosticstudiesfor individualswithcongenital
anomalies or intellectual disability basedon conventional
cytogeneticshaveadiagnosticyieldcloseto3%,CMAhasa
yieldofaround15%to20%,overfivetimesgreaterthan
G-bandedkaryotype,6justifyingitsuseasafirstlinediagnostic
test for patients withan unknown clinical diagnosis. It is
estimatedthatCMAaloneiscapableofdetectingover99%
ofallkaryotypeabnormalities.5
Thisreportpresentstheauthors’pioneeringexperience
intheuseofCMAinacohortofChileanpatientswith
mul-tiplecongenitalanomalieswithoutetiologicaldiagnosis.
Methods
Patients
FortypatientswereselectedfromtheGeneticClinicsat Hos-pitalPadreHurtado(Santiago,Chile),betweenMayof2012 andNovemberof2012.
This study included patients who had at least two of thefollowingclinicalfeatures:majorcongenitalanomalies (MCAs),facialdysmorphism(FD),developmentaldelay(DD), orintellectualdisability(ID).Allpatientslackedadefinite causeforthedisorder.
inheritedRobertsonian translocation,andone patienthad monosomyX,butwithunusualadditionalfeatures.
Thelocalethicscommitteeapprovedthisstudy,and writ-teninformedconsentwasobtainedfromallpatientsand/or parents/guardians.
SampleProcessing
Genomic DNA was purified from peripheral blood mono-nuclearcellswithAxyPrepBloodGenomicDNAMiniprepKit (AxygenBiosciences,UnionCity,CA,USA)following manu-facturer’s instructions. Genomic DNAof each patientwas hybridizedwiththeCytoScanTMHDArray(Affymetrix,Inc.,
SantaClara,CA,USA)accordingtomanufacturer’s instruc-tions. This is a custom high-densitycomparative genomic hybridization array with almost 2.7 million of genetic markers,includes 700,000singlenucleotidepolymorphism (SNP)markersandover1.9oligonucleotidenon-polymorphic probesforCNVdetection.
DataAnalysis
Array data were analyzed using the Affymetrix®
Chromo-some Analysis Software Suite (ChAS) v.1.2.2 (Affymetrix, Inc.,SantaClara,CA,USA)basedonthereferencegenome sequence of the UCSC Genome Browser hg19, Feb. 2009 (GRCh37/hg19). The authors analyzed CNVs over 400 Kb as recommended.6 With this higher-resolution platform,
it was possible to evaluate smaller CNVs, but no patient
hadclinicallyrelevant abnormalitiesbetween100and400
Kb, so this limit was kept for purposes of this report.
CNVs over 400 Kb were categorized by clinical
signifi-canceasCNVofclearclinicalrelevance(group1),CNVof
unclear relevance or uncertain significance (group 2), or
benign or polymorphic CNV (group 3), using the publicly
available databases ISCA (International Standard
Cytoge-nomic Array),19 DGV (Database of Genomic Variants),20,21
OMIM (Online Mendelian Inheritance in Man),22
DECI-PHER(DatabaseofChromosomalImbalanceandPhenotypein
HumansUsingEnsemblResources)23,24 andECARUCA
(Euro-pean Cytogeneticists Association Register of Unbalanced
ChromosomeAberrations).25
Results
Of the 40 patients analyzed, 16 (41%) were female.Ages ranged from 1 month to 25 years, with a median age of 4.2 years. As selected, the vast majority of the patients havemultipleanomalies,includingstructuralandfunctional developmentaldisorders.Clinicaldetailsaresummarizedin
Table1.
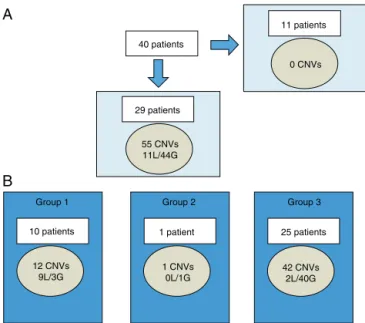
CNVcharacterization(Fig.1)
Fifty-five CNVs over 400 Kb were observed in 29 of 40 patients(72.5%),rangingbetween0and4CNVsperpatient. Ofthetotal,11werelossesand44weregains(Fig.1A).The
size of chromosomalimbalances ranged from420.9 Kb to
25.2 Mb. The latter corresponded toone patient withan
sSMCdetectedbykaryotype.
These55CNVswereclassifiedintothreegroupsbasedon
theirclinicalinterpretation.
Accordingtotheclassificationadopted (Fig.1B), 21.8%
belongedtogroup1(clearclinicalrelevancerelatedtothe
phenotype),1.8%togroup2(unclearrelevance),and76.4%
togroup3 (benign or polymorphic).Losses were
predom-inant in group 1, while gains were predominant in group
3.
Diagnosticyield
In Group 1, two patients had two terminal chromosome imbalances,eachprobablyderivedfromunbalancedcryptic translocations.Sevenpatientshadamicrodeletionandone patienthadamosaicpartialtrisomy20(oneofthepatients hadansSMC).MoredetailsaresummarizedinTable1.
InGroup2,onlyonepatienthadavariant ofuncertain
significance(VOUS).26 This patienthad a2 Mbtriplication
at 9p23 [arr 9p23(9,323,653-11,359,708)x3], but withthe
informationavailable in the databases and in biomedical
literature,adefinitivepathogeniceffectcouldnotberuled
outorassigned.
The authors failed to find true genetic imbalances in
the second patient with an sSMC and the patient with a
derivativechromosome16.Inthepatientwiththe
Robert-soniantranslocation,nogenomicimbalancethatexplained
thephenotypewasfound.Finally,inthepatientwith
mono-somyX,thedeletionofoneXwasconfirmed,butadditional
genomicimbalanceswerenotfound(Table1).
Clinicalinterpretation
ThisstudyfoundapathogenicCNVthatwasnotpreviously observedby(n=9)ornotwellcharacterized(n=1)by con-ventionalkaryotypeintenof40patientsofgroup1(25.0%). Inaddition,aknowncytogeneticanomalyinonepatientwas corroborated(monosomyX).
Ifonlythepatientswithnormalkaryotypeareconsidered (35patients),thediagnosisrateincreasedto28.5%.
Additionally,thisstudyfoundaCNVofuncertainclinical significanceorVOUS(group2)inoneadditionalpatient.This patientis waiting for additional testing and follow up to definetheclinicalrelevanceofthisfinding.Itisplannedto studybothparentswithFISHand/orMLPA.
Discussion
This is the first report of the CMAtesting in a cohort of Chilean patients with developmental disabilities, consid-ering that there are few studies of the clinical use of chromosomalmicroarraysinLatinAmerica,asexperiences of individual cases are the norm. Within South Amer-ica, a similar experience was reported with a group of patients from Brazil.27,28 Those authors analyzed 95
syn-dromic patients with normal karyotypes and reported a
diagnosticyieldof17%.28
The presentstudy detected over25% pathogenic
alter-ations in this cohort, which is in the upper range that
hasbeen reported in theliterature. In patients with
Table1 Summaryofclinicalfeaturesandgeneticimbalancesfoundinthiscohort.
Patient ID Developmental Delay Intellectual Disability Generalized hypotonia Seizures Central nervous system malformation Microcephaly Facial dysmorphism Congenital eye anomaly Visual anomaly External ear anomaly Hearing defect Cleft lip +/-palate Cleft palate/Robin sequence Congenital heart disease Hemivertebrae/Scoliosis Gastrointestinal malformation Abdominal hernia Kidney malformation Genital/Urinary tract defect Limb anomaly Generalized ligamentous hyperlaxity Skeletal anomaly Short stature Failure to thrive Obesity Congenital Skin Defect Cytogenetic studies CMA results Clinical interpretation
1 N/A 46,XX arr(1-22,X)x2
2 N/A 46,XX arr(1-22,X)x2
3 N/A 46,XX arr(1-22,X)x2
4 N/A 46,XY arr(1-22)x2,(XY)x1
5 46,XY arr(1-22)x2,(XY)x1
6 N/A 46,XY arr 9p23(9,323,653
-11,359,708)x3
Variant of unknown significance (VUOS)
7 N/A 46,XY arr(1-22)x2,(XY)x1 No genomic imbalances
8 46,XX; FISH22q(-) arr 6p25.1-p23(6,389,847
-15,124,349)x1 6p25.1p23 deletion
9 N/A 46,XX arr(1-22,X)x2
10 46,XY,der(16) arr(1-22)x2,(XY)x1 Pericentric inversion 16
11 N/A 46,XY arr(1-22)x2,(XY)x1
12 N/A 46,XY arr(1-22)x2,(XY)x1
13 46,XX; FISH22q(-) arr(1-22,X)x2
14 46,XX arr(1-22,X)x2
15 46,XY; FISH22q(-) arr(1-22)x2,(XY)x1
16 N/A 46,XY arr(1-22)x2,(XY)x1
17 46,XY arr(1-22)x2,(XY)x1
18 46,XX arr(1-22,X)x2
19 47,XX,+mar/46,XX
arr
20p12.3q11.22(7,180,552-32,402,789)x2~3
Partial trisomy 20 mosaicism
20 N/A 46,XY arr
2q23.2q24.2(150,216,033- 2q23.2q24.3 deletion 161,228,203)x1
21 46,XY;FISH 22q(-) arr(1-22)x2,(XY)x1 No genomic imbalances
22 46,XY;FISH22q(-) arr 15q25.2(83,08
3,418-84,834,123)x1 15q25.2 deletion
23 46,XY;FISH22q(-) arr(1-22)x2,(XY)x1 No genomic imbalances
No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances No genomic imbalances
24 N/A 46,XY arr(1-22)x2,(XY)x1 No genomic imbalances
25 46,XX; FISH 17p(-) arr(1-22,X)x2 No genomic imbalances
26 46,XX arr(1-22,X)x2 No genomic imbalances
27 N/A 46,XY arr(1-22)x2,(XY)x1 No genomic imbalances
28 N/A
46,XY;FISH22q(-); Chromosome breakageanalysis(-)
arr(1-22)x2,(XY)x1 No genomic imbalances
29 46,XX arr(1-22,X)x2 No genomic imbalances
30 45,XY,der
(14;15)(q10;q10) arr(1-22)x2,(XY)x1
Robertsonian translocation, no genomic imbalances
31 46,XX
arr
22q12 .1q12.3(28,966,333-34,541,402)x1
22q12.1q12.3 deletion
32 N/A 47,XX,+mar(mat);
FISH 15q(-) arr(1-22,X)x2
SMC bisatellited (NOR staining), heterochromatin maternally derived (healthy mother)
33 N/A 46,XX; FISH 22q(-) arr(1-22,X)x2 No genomic imbalances
34
46,XY;FISH7q(-), 22q(-)subtelomeres (-)
arr 6p25.3-p25.2(156
,974-2,400,023)x1 6p25.3p25.2 deletion
35 N/A 46,XY
arr6p25.3p25.2(294 ,910-3,403,875)x3,12p13.33(173, 786-3,283,049)x1
12p13.33 deletion; 6p25.3p25.2 triplication
36 46,XY
arr
14q32.12q32.33(94,17 4,317-107,285,437)x3,22q13.31q13 .33(47,852,611-51,197,838)x1
22q13 deletion; 14q32.12q32.33 triplication
37 N/A 46,XX; FISH 22q(-)
arr4q34 .1-q35.2(174,40 9,811-190,957,473)x1
4q34.1q35.2 deletion
38 N/A 45,X arr Xp22.33q28(16
8,546-155,233,731)x1
Monosomy X, without other imbalances 39 46,XY arr(1-22)x2,(XY)x1 No genomic imbalances
40 46,XY arr 19p13.3(3,78
8,725-5,346,511)x1 19p13.3 deletion
Group 1 Group 2 Group 3 29 patients
40 patients
12 CNVs 9L/3G
1 CNVs 0L/1G
42 CNVs 2L/40G 55 CNVs
11L/44G
0 CNVs
A
B
10 patients 1 patient
11 patients
25 patients
Figure 1 Characterization of CNVs larger than 400 Kb in Chileancohort.
A,among40patients,CNVswerenotfoundin11individuals.In theremaining29patients,55CNVswerefound(11lossesand44 gains).B,eachpatientmayhaveCNVofmorethanonegroupat atime.CNVswerecategorizedbyclinicalsignificanceasCNVof clearclinicalrelevance(group1),CNVofuncertainsignificance (group2),orbenignorpolymorphicCNV(group3).
CNVs,copynumbervariants;G,gains;L,losses.
disabilitywithnormalkaryotype/FISH,meta-analysisshows adiagnosticyieldof7.8%-13.8%,rangingfrom5%to50%.5,29
This is largely explained by heterogeneity in the design
ofstudies,especiallyinpatientselection,previoustesting
realized,andarrayplatformsused.Inthepresentcase,the
highrateofdiagnosiscanbeexplainedbythefactthatthe
studied cohort wasrelatively small,had the bias of very
selected patients,many of whom have remainedfor long
timewithoutdiagnosis,andfinally,usedahigh-density
plat-form.
Over time, different onlinepublic databases have
col-lected phenotypic and genomic information of thousands
of anonymous patients, which allow elucidating the vast
majorityofCNVsasbenignorpolymorphic,withoutfurther
analysis.Infact,themajorityofthisstudy’sfindingswere
polymorphic CNVs (copy number polymorphisms, [CNPs]),
corroboratedinthosecytogenomicdatabasesandfromthe
authors’owncohortdata.RecurrentCNVsover400Kbwere
observed in half of these patients, mainly involving the
followingchromosomalloci:10q11.22,14q32.22,16p11.2,
17q21.31,Yq11.223,and Yq11.23.Only onecase that had
a VOUS required further analysis to determine possible
pathogenicity. This patient was one year and 11 months,
with DD, language delay, hearing deficit, inguinal hernia,
andshortstature.Hehada46,XYkaryotypeandthearray
showed a 2 Mb gain in chromosome 9p23, including
par-tial triplication of onegene: PTPRD. The protein Ptprdis
areceptor-typeprotein-tyrosinephosphatase,isexpressed
incertainregionsofthebrain,suchasthehippocampus,and
couldhavearoleinlearningandmemory30andthesynaptic
organization.31Nophenotypehasbeenassignedtothefull
triplicationofthisgene.
Although CMA is a very robust and reliable technique,
ithaslimitations: itisunabletodetectbalanced genomic
abnormalities such as inversions, reciprocal, and
Robert-sonian translocations. Depending on the platform used,
low-level mosaicism and some polyploidies cannot be
detected.When CMAtechnique isusedasthefirstline of
study,ithasbeendescribedthatthissituationcanoccurin
0.78%ofcases.5Finally,CMAdoes notgiveinformationon
positionoftherearrangement,FISHisoftenusedas
comple-mentarymethod toidentifypossible rearrangements with
implicationsforgeneticcounseling.7,32
Inthepresent cohort,it wasfound thatthreepatients
withpreviously known cytogenetic abnormalities resulted
inanormalmolecularkaryotype.Asexpected,thepatient
whoiscarrierfor a Robertsoniantranslocation hada
nor-mal/balancedaCGHresult. Inthe case of oneof the two
patientswithsSMC,thecoverageofthearray,thegenomic
sequences involved, and the size sSMC may explain why
that geneticmaterial were undetected by this method.19
Thus,thissmallbisatellitedchromosomelikelycorresponds
tohighlyrepetitivesequencestypicalofacrocentric
chro-mosomesnotincludedinthearraythatwasdemonstrated
bynucleolusorganizerregion(NOR)banding.Inthepatient
withaderivativechromosome16,withan abnormal
band-ingpatternandmorphology(moremetacentricthanusual),
apericentricinversionisthemostplausibleexplanation.
Thus, karyotype remains more suitable to evaluate
potentialcarriersofchromosomalrearrangements,couples
withrecurrent miscarriage, or patients with a distinctive
aneuploidyphenotype.However,FISHismoresuitableifa
specificmicrodeletionsyndromeishighlysuspected.7
Chromosomemicroarraysareahighlyaccurate,robust,
andhigh-throughput method.This study useda combined
oligonucleotide-basedarrayplusSNParray,whichhasmany
advantages, where the SNP array significantly improves
the accuracy and sensitivity of the CNV detection and
mosaicism, also allowing the detection of copy-neutral
variants.33 In the case of the chip used in this report
(Cytoscan HD, Affymetrix), the platform chemistry and
itsalgorithms analyzethe oligonucleotideandSNPprobes
independently, and thus each CNV can be detected and
confirmedat thesametime,without requiringanyfurther
confirmation.32,33Inthiscase,insteadofaconfirmationof
thearrayfinding,FISHanalysisallowsfordeterminationof
thetypeofrearrangement.32
Alimitationforwidespreaduseofmolecularkaryotype
isthehighcostofthetest.InChile, CMAisapproximately
fourtoseventimesmoreexpensivethanakaryotypeand/or
FISH,andiscurrentlynotcoveredbyhealthinsurance.
How-ever,toreachanearlydiagnosis inselectedpatients with
multiple congenitalanomalies and/or global
developmen-taldelay,itcanavoidunnecessarytesting(the‘‘diagnostic
odyssey’’)andallowfocusingonspecificissues,whichinthe
longtermcanbecost-effective.34---38Itmaybeexpectedthat
costswilldecreaseovertime,enablingitsmorewidespread
use. Finally, it should bementioned that thereare other
platforms that have lower resolution but at a relatively
reducedcost.
Recognizingitslimitations,thereisgrowingevidenceof
the clinical impact of this technology. In many patients,
a definitivediagnosis can impactnot only on information
onactivesurveillance insearchof possiblecomplications,
amongothertypesofmedicalinterventions.37,38Thepresent
datashowtheusefulnessofCMA, allowingimproved
diag-nosticcapabilitywithremarkableprecisionandoptimization
ofthemanagementandsupervisionofhealthinthisgroup
ofpatientswithspecialneeds.
Funding
GrantsupportfromChildHealthFoundationinBirmingham, Alabama.
Conflicts
of
interest
Theauthorsdeclaretohavenoconflictsofinterest.
Acknowledgments
TheauthorsaregratefultotheChildHealthFoundationin Birmingham,Alabamafortheirgrantsupportforthiswork, totheparticipatingpatientsandfamilies,andtoDr.Silvia CastilloandcytogeneticistAnaMaríaFuentesforreviewof theconventionalkaryotyperesults.
References
1.Christianson A, HowsonM,Modell B.March ofDimes: global reportonbirthdefects.Thehiddentollofdyinganddisabled children.WhitePlains,NY:MarchofDimesBirthDefects Foun-dation;2006.
2.KumarP,BurtonBK.Dysmorphology. In:KumarP,BurtonBK, editors.Congenitalmalformations:evidence-basedevaluation andmanagement.NewYork:McGraw-HillProfMed/Tech;2007. p.3---11.
3.RauchA,HoyerJ,GuthS,ZweierC,KrausC,BeckerC,etal. Diagnosticyieldofvariousgeneticapproachesinpatientswith unexplaineddevelopmentaldelayormentalretardation.AmJ MedGenetA.2006;140:2063---74.
4.VermeeschJR,FieglerH,deLeeuwN,SzuhaiK,SchoumansJ, CicconeR,etal.Guidelinesformolecularkaryotypingin consti-tutionalgeneticdiagnosis.EurJHumGenet.2007;15:1105---14. 5.Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, et al. Array analysis and karyotyp-ing: workflow consequences based on a retrospective study of36,325patientswithidiopathicdevelopmentaldelayinthe Netherlands.EurJMedGenet.2009;52:161---9.
6.Miller DT,AdamMP, Aradhya S, BieseckerLG,Brothman AR, CarterNP,etal.Consensusstatement:chromosomalmicroarray isafirst-tierclinicaldiagnostictestforindividualswith devel-opmentaldisabilitiesorcongenitalanomalies.AmJHumGenet. 2010;86:749---64.
7.Manning M, Hudgins L. Professional Practice and Guidelines Committee.Array-basedtechnologyandrecommendationsfor utilizationinmedicalgeneticspracticefordetectionof chro-mosomalabnormalities.GenetMed.2010;12:742---5.
8.ShafferLG,LedbetterDH,LupskiJR. Molecularcytogenetics ofcontiguousgenesyndromes:mechanismsandconsequences ofgenedosageimbalance.In:ScriverCR,BeaudetAL,SlyWS, ValleD,ChildsB,KinzlerKW,etal.,editors.Themetabolicand molecularbasisofinheriteddisease.NewYork:McGraw Hill; 2001.p.1291---324.
9.TraskBJ.Humancytogenetics:46chromosomes,46yearsand counting.NatRevGenet.2002;3:769---78.
10.Salman M,Jhanwar SC,Ostrer H.Will the newcytogenetics replacetheoldcytogenetics?ClinGenet.2004;66:265---75. 11.SmeetsDF.Historicalprospectiveofhumancytogenetics:from
microscopetomicroarray.ClinBiochem.2004;37:439---46. 12.TurleauC,VekemansM.Newdevelopmentsincytogenetics.Med
Sci(Paris).2005;21:940---6.
13.EdelmannL,HirschhornK.ClinicalutilityofarrayCGHforthe detectionofchromosomalimbalancesassociatedwithmental retardationandmultiplecongenitalanomalies.AnnNYAcad Sci.2009;1151:157---66.
14.Jehee FS, Takamori JT, Medeiros PF, Pordeus AC, Latini FR, BertolaDR,etal.UsingacombinationofMLPAkitstodetect chromosomalimbalancesinpatientswithmultiplecongenital anomaliesandmentalretardationisavaluablechoicefor devel-opingcountries.EurJMedGenet.2011;54:e425---32.
15.LeeC,IafrateAJ,BrothmanAR.Copynumbervariationsand clinicalcytogeneticdiagnosis ofconstitutionaldisorders.Nat Genet.2007;39:S48---54.
16.ShafferLG,BejjaniBA,TorchiaB,KirkpatrickS,CoppingerJ, BallifBC. Theidentification of microdeletionsyndromesand otherchromosomeabnormalities:cytogeneticmethodsofthe past,newtechnologiesforthefuture.AmJMedGenetCSemin MedGenet.2007;145C:335---45.
17.Schluth-BolardC,TillM,EderyP,SanlavilleD.Newchromosomal syndromes.PatholBiol(Paris).2008;56:380---7.
18.Slavotinek AM. Novel microdeletion syndromes detected by chromosomemicroarrays.HumGenet.2008;124:1---17. 19.International Collaboration for Clinical Genomics (ICCG).
International Standard Cytogenomic Array(ISCA) Consortium Database Search. [cited 4 March 2014]. Available from: http://www.iccg.org/
20.MacDonald JR, Ziman R, YuenRK, Feuk L, Scherer SW. The DatabaseofGenomicVariants:acurated collectionof struc-tural variation in the human genome. Nucleic Acids Res. 2014;42:D986---92.
21.Database of GenomicVariants (DGV). [cited 4 March 2014]. Availablefrom:http://dgv.tcag.ca/dgv/app/home
22.Online Mendelian Inheritance in Man, OMIM®. McKusick-NathansInstituteofGeneticMedicine,JohnsHopkinsUniversity (Baltimore, MD). [cited 10 March 2014]. Available from: http://omim.org/
23.FirthHV,RichardsSM,BevanAP,ClaytonS,CorpasM,RajanD, etal.DECIPHER:DatabaseofChromosomalImbalanceand Phe-notypeinHumansUsingEnsemblResources.AmJHumGenet. 2009;84:524---33.
24.DatabaseofChromosomalImbalanceandPhenotypeinHumans usingEnsembl Resources(DECIPHER). [cited10March2014]. Availablefrom:https://decipher.sanger.ac.uk/
25.EuropeanCytogeneticists Association Register ofUnbalanced ChromosomeAberrations (ECARUCA).[cited10March 2014]. Available from: http://umcecaruca01.extern.umcn.nl:8080/ ecaruca/ecaruca.jsp
26.deLeeuwN,DijkhuizenT,Hehir-KwaJY,CarterNP,FeukL,Firth HV,etal.Diagnosticinterpretationofarraydatausingpublic databasesandinternetsources.HumMutat.2012.
27.RosenbergC,Knijnenburg J, Bakker E,Vianna-Morgante AM, SloosW,OttoPA,etal.Array-CGHdetectionofmicro rearrange-mentsinmentallyretardedindividuals:clinicalsignificanceof imbalancespresentbothinaffectedchildrenandnormal par-ents.JMedGenet.2006;43:180---6.
28.Krepischi-SantosAC, Vianna-MorganteAM, Jehee FS, Passos-Bueno MR, Knijnenburg J, Szuhai K, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromesrevisitedand newalterations. Cytogenet Genome Res.2006;115:254---61.
StandardsSubcommitteeoftheAmericanAcademyof Neurol-ogyandthePracticeCommitteeoftheChildNeurologySociety. Neurology.2011;77:1629---35.
30.UetaniN, Kato K, Ogura H, MizunoK, Kawano K, Mikoshiba K, et al. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19:2775---85.
31.TakahashiH, Craig AM. Protein tyrosine phosphatases PTP␦,
PTP, and LAR: presynaptic hubs for synapse organization.
TrendsNeurosci.2013;36:522---34.
32.BuiTH,VetroA,ZuffardiO,ShafferLG.Currentcontroversiesin prenataldiagnosis3:isconventionalchromosomeanalysis nec-essaryinthepost-arrayCGHera?PrenatDiagn.2011;31:235---43. 33.Mason-SuaresH,KimW,GrimmettL,WilliamsES,HornerVL, KunigD,etal.Densitymatters:comparisonofarrayplatforms fordetectionofcopy-numbervariationandcopy-neutral abnor-malities.GenetMed.2013;15:706---12.
34.Wordsworth S, Buchanan J, Regan R, Davison V, Smith K, Dyer S, et al. Diagnosing idiopathic learning disability: a
cost-effectiveness analysis of microarray technology in the NationalHealthServiceoftheUnitedKingdom.GenomicMed. 2007;1:35---45.
35.NewmanWG,HamiltonS,AyresJ,SangheraN,SmithA,Gaunt L,etal.Arraycomparativegenomichybridizationfor diagno-sisofdevelopmentaldelay:anexploratorycost-consequences analysis.ClinGenet.2007;71:254---9.
36.TrakadisY,ShevellM.Microarrayasafirstgenetictestinglobal developmentaldelay: a cost-effectiveness analysis.DevMed ChildNeurol.2011;53:994---9.
37.CoulterME,Miller DT,HarrisDJ, HawleyP,PickerJ,Roberts AE,etal.Chromosomalmicroarraytestinginfluencesmedical management.GenetMed.2011;13:770---6.
38.Riggs E,WainK, RiethmaierD,Smith-Packard B,Faucett W, Hoppman N,et al. Chromosomal microarrayimpacts clinical management.ClinGenet.2013.