Simone Fernanda Nedel Pértile(1), Fabyano Fonseca e Silva(2), Mayara Salvian(1) e Gerson Barreto Mourão(1)
(1)Universidade de São Paulo, Escola Superior de Agricultura Luiz de Queiroz, Departamento de Zootecnia, Avenida Pádua Dias, no 11, CEP 13418‑900 Piracicaba, SP, Brasil. E‑mail: s.pertile@zootecnista.com.br, mayara.salvian@usp.br, gbmourao@usp.br (2)Universidade Federal de Viçosa, Departamento de Zootecnia, Rua P.H. Rolfs, Centro, CEP 36570‑000 Viçosa, MG, Brasil. E‑mail: fabyanofonseca@ufv.br
Resumo – O objetivo deste trabalho foi avaliar a eficiência do método ssGBLUP quanto à seleção e à associação genômica ampla, com atribuição de pesos a marcadores genéticos e uso de informações de genótipos e fenótipos, com ou sem informações de pedigree, com diferentes coeficientes de herdabilidade. A população estudada foi obtida por simulação de dados com 8.150 animais, 5.850 dos quais eram genotipados. Utilizou-se o método ssGBLUP para a análise dos dados, com duas matrizes de relacionamento – com ou sem informações de pedigree –, e pesos para os marcadores genéticos obtidos em cada iteração. O aumento do coeficiente de herdabilidade melhorou os resultados de seleção e associação genômica. O melhor desempenho quanto à habilidade preditiva foi obtido quando não se utilizaram informações de pedigree. O tipo de matriz de relacionamento utilizada não influenciou a identificação de regiões associadas a características de interesse. O método ssGBLUP é eficiente tanto para a seleção quanto para a identificação de regiões associadas às características estudadas.
Termos para indexação: coeficiente de parentesco, habilidade preditiva, herdabilidade, validação cruzada.
Genome‑wide selection and association in animal breeding using ssGBLUP
Abstract – The objective of this work was to evaluate the efficiency of the ssGBLUP method for genome-wide selection and association, attributing weights to genetic markers and using genotype and phenotype information, with or without pedigree information, considering different coefficients of heritability. The studied population was obtained by data simulation with 8,150 animals, 5,850 of which were genotyped. The ssGBLUP method was used for data analysis, with two relationship matrices – with or without pedigree information –, and weights for the genetic markers obtained in each iteration. Increasing heritability coefficients improved the results of genomic selection and association. The best performance for predictive ability was obtained without pedigree information. The type of relationship matrix used did not affect the identification of regions associated with traits of interest. The ssGBLUP method is efficient both for selection and identification of regions associated with the studied traits.
Index terms: kinship coefficient, predictive ability, heritability, cross-validation.
Introdução
No melhoramento genético tradicional, os valores genéticos dos animais são preditos por meio de dados
fenotípicos e de pedigree, com uso da metodologia dos
modelos mistos. Neste caso, a matriz de parentesco ou de relacionamento (denominada A) é obtida por meio do valor esperado da proporção de loci idênticos por descendência (IBD). Tal valor é geralmente denominado de coeficiente de parentesco. Porém, ao utilizar informações de marcadores genéticos de alta densidade, tal matriz pode ser substituída pela matriz de parentesco genômica (ou realizada), que é construída
apenas com as informações destes marcadores; ou seja,
ela apenas tem validade para animais genotipados.
Nessa matriz (denominada G), os coeficientes são dados pela proporção de loci idênticos por estado (IBS), os quais geralmente capturam mais informação do que o coeficiente tradicional. Existe, ainda, a possibilidade de se utilizar uma matriz que combina os coeficientes IBD e IBS, denominada H, que permite utilizar informações de parentesco de indivíduos genotipados e não genotipados simultaneamente. Esse tipo de matriz é atualmente utilizado na maioria dos
Nos métodos denominados multi-step, as informações genômicas e de pedigree são utilizadas separadamente nas análises. O pedigree é geralmente utilizado em uma primeira etapa, para a obtenção de pseudofenótipos, tais como os valores genéticos tradicionais e os valores genéticos desregredidos. Em uma segunda etapa, estes pseudofenótipos são considerados variáveis dependentes, em diferentes modelos, para estimar valores genético-genômicos, que são gerados apenas para animais genotipados. A utilização da matriz H é postulada de forma diferente com o método denominado single-step, ou ssGBLUP (Legarra et al., 2009; Aguilar et al., 2010), que tem-se mostrado altamente eficiente quanto à predição de valores genéticos, em comparação aos métodos
multi-step (Wang et al., 2012).
Porém, estudos com comparações para as estimativas dos efeitos de marcadores (além dos valores genéticos) ainda são escassos, principalmente em situações que envolvam diferentes valores de herdabilidade e proporções de animais genotipados. Paralelamente a isto, é importante que se investigue o peso de informações de pedigree para estudos com dados de marcadores genéticos, já que elas apresentam
alto custo de coletas de dados.
O objetivo deste trabalho foi avaliar a eficiência do método ssGBLUP para a seleção e associação genômica, com uso de informações fenotípicas e genotípicas – com ou sem as informações de pedigree –, e diferentes coeficientes de herdabilidade.
Material e Métodos
Os dados foram simulados com o auxílio do programa
QMSim (Sargolzaei & Schenkel, 2009), tendo-se utilizado parâmetros semelhantes aos considerados por Brito et al. (2011). Para a abordagem de simulação de dados, utilizou-se o método forward-time, pelo qual as forças evolutivas e o desequilíbrio de ligação são simulados da geração mais antiga até a mais recente
(Calafell et al., 2001).
A população histórica constituiu-se inicialmente de 1.000 indivíduos por geração, número que foi mantido até a geração 1.000, seguido de uma redução gradual do tamanho da população até a geração 2.020, a qual foi composta por 200 indivíduos. Além disso, esta população foi gerada tendo-se considerado
acasalamentos aleatórios, sem que tenha havido
migração ou sobreposição de gerações. As frequências
alélicas na primeira geração foram fixadas em 0,5, e o número de machos e fêmeas foi o mesmo em cada uma das gerações.
No passo seguinte da simulação, os animais da última geração da população histórica foram considerados como fundadores (100 machos e 100 fêmeas) da população de expansão, que foi composta por oito gerações, tendo-se considerado: cinco progênies por fêmea, por geração; o crescimento exponencial do número de fêmeas; a união aleatória de gametas; e a ausência de seleção.
A população recente foi simulada em dois passos. No primeiro, 640 machos e 32 mil fêmeas da última geração da população de expansão foram selecionados
e acasalados de forma aleatória, tendo-se mantido no
rebanho, a cada geração, os animais com maiores valores genéticos. Considerou-se uma taxa de substituição de 60% de machos e 20% de fêmeas. Os animais com menores valores genéticos, em cada geração, foram descartados. O número de progênies considerado foi de um descendente por fêmea, por geração. No segundo passo da simulação da população recente, a geração de fundadores foi formada a partir da seleção aleatória de 50 machos e 1.350 fêmeas, na última geração da população recente obtida no primeiro passo. Esses
indivíduos formaram um rebanho constituído de cinco
gerações, com seleção para valores genéticos altos e, como na população anterior, os acasalamentos foram realizados de forma aleatória, tendo-se considerado uma taxa de substituição de 20% dos machos e 5% das fêmeas, e descarte dos animais com valores genéticos
mais baixos.
O genoma foi simulado com 5 cromossomos de
100 cM cada, dos quais 7.500 marcadores bialélicos
foram distribuídos de forma equidistante, com aproximadamente 1.500 marcadores segregantes por
cromossomo. Levou-se em consideração a presença de 30 QTLs (quantitative trait loci), distribuídos
aleatoriamente ao longo dos 5 cromossomos, e os
efeitos destes foram simulados a partir da distribuição gama, com parâmetro shape 0,4. Os QTLs e marcadores genéticos foram obtidos levando-se em conta uma MAF (minor allele frequency) de 0,01. As taxas de mutação consideradas por geração foram de 1x10-5,
tanto para loci de marcadores genéticos como para QTLs. Marcadores e QTLs foram considerados como bialélicos. Assim, as mutações geradas alteraram o alelo
1 para o 2, ou o 2 para o 1, tendo-se desconsiderado a
Seis diferentes fenótipos foram simulados, com
coeficientes de herdabilidade de QTL de 0,10, 0,25 e
0,40.
Para todas as características, levou-se em
consideração a variância fenotípica de 1,0. As simulações foram repetidas 10 vezes para cada
característica, e os resultados foram obtidos a partir
das médias e desvios-padrão das repetições, em dois cenários: um com dados de pedigree, genótipo e fenótipo (cenário 1); e outro com dados de fenótipo e genótipo, mas sem pedigree (cenário 2).
Para as análises de associação e predição dos valores genético-genômicos, o método empregado foi o ssGBLUP, e os dados foram analisados no programa BLUPF90. No caso do cenário 1, a matriz de parentesco utilizada foi a H, conforme Aguilar et al. (2010), em que são combinados dados de genótipos e pedigree. A inversa da matriz H é dada por: H A G A − − − − = + − 1 1 1 22 1 0 0 0 , em que: A é o numerador da matriz de relacionamento
para todos os animais, A22 é o numerador da matriz de
relacionamento para os animais genotipados; e G é a matriz de relacionamento genômico. Para o cenário 2, a matriz de relacionamento utilizada foi a genômica (G), que é construída apenas com as informações dos marcadores genéticos. Nos dois cenários, o modelo utilizado foi: y = m1n + Za + ε, em que: y é o vetor de
fenótipos simulados; μ é a média geral dos fenótipos; 1 é o vetor com valores um e tamanho n; Z é a matriz de incidência que relaciona os indivíduos aos fenótipos; a é o vetor de efeitos individuais dos animais; e, ε é o
vetor de resíduos.
Para as análises de associação, consideraram-se os genótipos de cinco gerações para a seguinte seleção:
na primeira selecionaram-se de forma aleatória
1.300 animais; e, na última, 500 animais.
Para as análises de validação cruzada, as gerações 1,
2, 3 e 4 formaram o grupo de treinamento, enquanto a
geração 5, para a qual os fenótipos foram zerados, foi considerada como grupo de validação, o que possibilita a avaliação da habilidade de predição nas gerações
futuras.
Após a análise de associação, calcularam-se as correlações entre valores genéticos verdadeiros e preditos, e as correlações entre valores genético-genômicos e fenótipos, que foram obtidas
em diferentes iterações, tanto para o grupo de validação quanto para o rebanho. As análises de associação genômica ampla foram representadas com gráficos Manhattan e efeitos de single nucleotide polymorphisms (SNPs) e QTLs, para cada cenário, tendo-se considerado apenas a iteração de maior correlação entre os valores genéticos preditos e verdadeiros. Para a análise de convergência, as correlações entre os valores genético-genômicos obtidos foram calculadas para toda a população, em diferentes iterações; nesta análise, utilizaram-se, em cada iteração, os pesos obtidos para os SNP da iteração
anterior, tendo-se iniciado com peso 1 para todos os
marcadores genéticos. Com os pesos dos marcadores genéticos computados a cada iteração, os marcadores com maior efeito têm seus efeitos maximizados. Assim, pode-se obter maior acurácia com uma combinação de pesos que minimize a estimação dos erros (Wang et al., 2012).
Resultados e Discussão
Os fenótipos estudados tiveram resultados
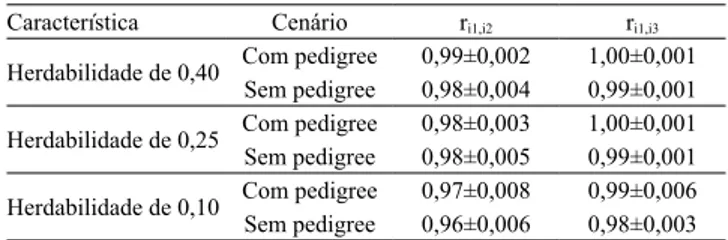
semelhantes na análise de convergência, com correlação entre os valores preditos de 0,96 a 0,99, na primeira e segunda iteração (Tabela 1), e de 0,98 a 1,00, na segunda e terceira iteração, respectivamente. Essa semelhança entre os valores genéticos indica que não há necessidade de novas iterações. Maiores correlações foram obtidas para fenótipos com maior coeficiente de herdabilidade; assim, o aumento da herdabilidade diminui o número de iterações requeridas. Os baixos valores de desvio-padrão, de 0,001 até 0,008, indicam
Tabela 1. Correlação±desvio-padrão entre os valores
genéticos preditos em diferentes iterações, no grupo de validação e em todo rebanho, tendo-se considerado características com diferentes herdabilidades e cenários de simulação.
Característica Cenário ri1,i2 ri1,i3 Herdabilidade de 0,40 Com pedigree 0,99±0,002 1,00±0,001
pequena variação entre as repetições, para cada fenótipo e cenário.
A acurácia dos valores genético-genômicos preditos é obtida a partir de sua correlação com os valores genéticos verdadeiros, no grupo de validação e no rebanho estudado (Tabela 2). No grupo de validação, as acurácias foram maiores para fenótipos com maior coeficiente de herdabilidade, em cada cenário. No entanto, nos fenótipos 1 e 2 – coeficientes de
herdabilidade de 0,40 e 0,25, respectivamente –, as
acurácias foram semelhantes, o que é indício de que, nesta faixa de herdabilidade, a acurácia da predição
dos valores das características varia pouco.
O uso da matriz H aumentou as acurácias obtidas, em comparação ao uso da matriz G, tanto no rebanho quanto no grupo de validação. Assim, o uso de uma matriz de relacionamento que combine informações de pedigree e genômicas parece ser mais eficiente na predição dos valores genético-genômicos do que o uso de uma matriz apenas com informações genômicas. Nos resultados obtidos para os desvios-padrão das acurácias, houve maior variação quando se utilizou a matriz G, em substituição à matriz H. Além disso, houve menor variação entre as repetições no rebanho do que no grupo de validação.
Wang et al. (2012) estudaram um fenótipo com
coeficiente de herdabilidade de 0,50, oriundo apenas dos efeitos aditivos dos QTL, em uma população que
continha cinco gerações, pedigree com 15.800 animais
e 1.500 indivíduos genotipados. Nesse caso,
para o método ssGBLUP com processo iterativo, com uso da matriz H para obtenção dos valores genético-genômicos, foram obtidas as correlações (±desvios-padrão) de 0,87 (±0,01), 0,89 (±0,01), 0,88 (±0,01), para a primeira, segunda e terceira iteração.
Os valores mais altos obtidos no presente estudo podem
estar associados ao maior coeficiente de herdabilidade utilizado; e as diferenças entre iterações, às diferentes estruturas das populações. Segundo os autores, as vantagens do uso de iterações para recalcular os valores genético-genômicos estão relacionadas ao número e à distribuição dos efeitos dos QTLs, e, quando há pequeno número de QTLs, a melhoria da acurácia na predição desses valores é imperceptível.
Ainda em relação à eficiência do método ssGBLUP iterativo, Wang et al. (2014), ao avaliar a seleção genômica do peso corporal de frangos de corte, obtiveram valores de acurácia iguais a 0,91, 0,90 e 0,88, para a primeira, segunda e terceira iterações, respectivamente. Estes resultados são corroborados
pelos obtidos no presente trabalho.
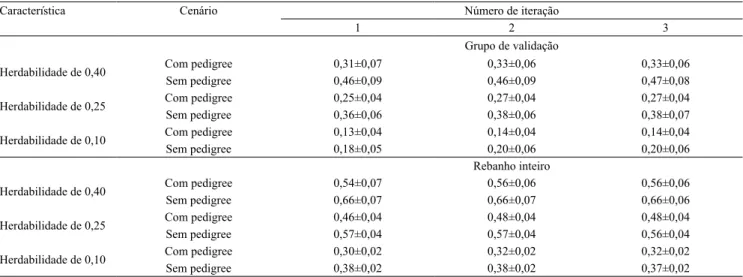
A capacidade preditiva foi avaliada a partir das
correlações entre os valores genético-genômicos
preditos e os valores fenotípicos, para o grupo de
validação e para o rebanho (Tabela 3). Para fenótipos com baixo coeficiente de herdabilidade (0,10), no
Tabela 2. Correlação±desvio-padrão entre valores genéticos preditos e verdadeiros, no grupo de validação e em todo rebanho,
tendo-se considerado características com diferentes herdabilidades, e cenários de simulação com diferentes quantidades de iterações.
Característica Cenário Número de iteração
1 2 3
Grupo de validação
Herdabilidade de 0,40 Com pedigree 0,68±0,03 0,67±0,03 0,67±0,03
Sem pedigree 0,53±0,09 0,53±0,09 0,53±0,09
Herdabilidade de 0,25 Com pedigree 0,64±0,02 0,63±0,03 0,63±0,03
Sem pedigree 0,45±0,05 0,45±0,05 0,45±0,06
Herdabilidade de 0,10 Com pedigree 0,54±0,06 0,50±0,06 0,49±0,06
Sem pedigree 0,31±0,09 0,30±0,08 0,29±0,08 Rebanho inteiro
Herdabilidade de 0,40 Com pedigree 0,87±0,01 0,85±0,02 0,85±0,02
Sem pedigree 0,68±0,07 0,67±0,07 0,67±0,07
Herdabilidade de 0,25 Com pedigree 0,81±0,01 0,79±0,02 0,79±0,02
Sem pedigree 0,61±0,04 0,60±0,04 0,59±0,04
Herdabilidade de 0,10 Com pedigree 0,72±0,04 0,66±0,04 0,65±0,04
grupo de validação, obteve-se maior correlação na segunda iteração, independentemente do cenário
avaliado. Quando o rebanho foi avaliado, obtiveram-se
as maiores correlações entre valor genético-genômico e fenótipo também na segunda iteração, consideradas as informações de pedigree; e, quando a matriz de relacionamento G foi utilizada, a primeira e a segunda iteração foram iguais. Esses resultados são indicativos de que, para fenótipos com baixo coeficiente de herdabilidade, o uso dos resultados da segunda iteração
pode ser mais adequado do que o uso dos da primeira
iteração.
Ao se considerar o rebanho, obtiveram-se
correlações entre os valores genéticos verdadeiros e fenótipos mais elevadas na segunda iteração, com a matriz H, para todos os fenótipos estudados. Porém, quando utilizada a matriz G, observaram-se duas situações quanto às habilidades preditivas: 1, maiores valores na primeira iteração; ou 2, resultados iguais entre primeira e segunda iteração. Esses resultados
foram semelhantes aos obtidos no grupo de
validação, em que as maiores correlações na segunda iteração – exceto para o fenótipo 2 – foram obtidas quando a matriz G foi utilizada. Além disso, obteve-se maior habilidade preditiva com uso da matriz G; assim, as informações de marcadores foram suficientes para substituir as informações de parentesco.Com a
diminuição do coeficiente de herdabilidade, houve diminuição também da habilidade preditiva.
Em geral, esses resultados concordam com os de
Wang et al. (2014), que estimaram habilidade preditiva de 0,44, 0,52 e 0,52, para a primeira, segunda e terceira
iterações, respectivamente.
Os gráficos de soluções dos efeitos dos SNPs foram obtidos para a primeira iteração, em todos os fenótipos (Figuras 1, 2 e 3), em razão da maior acurácia na predição dos valores genético-genômicos obtidos nesta iteração, independentemente do fenótipo e do cenário. Os gráficos, para cada fenótipo, foram semelhantes nos dois cenários; indício de que a capacidade de identificação de QTL é igual, independentemente da ausência ou presença de informações de pedigree. Portanto, essas informações não foram determinantes para a detecção de QTLs. Além disso, o uso do método ssGBLUP foi eficiente no estudo da arquitetura genética dos fenótipos
estudados, tendo mostrado muitos loci com pequeno efeito e poucos com grande efeito.
Para a característica com coeficiente de herdabilidade de 0,40, as regiões que continham QTLs na simulação foram identificadas por meio dos gráficos de soluções dos efeitos dos SNPs. Identificou-se a presença de um QTL no cromossomo cinco, além de vários outros de pequeno efeito. Além disso, foram encontrados indícios da presença de QTL no final do cromossomo
Tabela 3. Correlação±desvio-padrão entre valores genéticos preditos e fenótipos, no grupo de validação e em todo rebanho,
tendo-se considerado características com diferentes herdabilidades, e cenários de simulação com diferentes quantidades de iterações.
Característica Cenário Número de iteração
1 2 3
Grupo de validação
Herdabilidade de 0,40 Com pedigree 0,31±0,07 0,33±0,06 0,33±0,06
Sem pedigree 0,46±0,09 0,46±0,09 0,47±0,08
Herdabilidade de 0,25 Com pedigree 0,25±0,04 0,27±0,04 0,27±0,04
Sem pedigree 0,36±0,06 0,38±0,06 0,38±0,07
Herdabilidade de 0,10 Com pedigree 0,13±0,04 0,14±0,04 0,14±0,04
Sem pedigree 0,18±0,05 0,20±0,06 0,20±0,06 Rebanho inteiro
Herdabilidade de 0,40 Com pedigree 0,54±0,07 0,56±0,06 0,56±0,06
Sem pedigree 0,66±0,07 0,66±0,07 0,66±0,06
Herdabilidade de 0,25 Com pedigree 0,46±0,04 0,48±0,04 0,48±0,04
Sem pedigree 0,57±0,04 0,57±0,04 0,56±0,04
Herdabilidade de 0,10 Com pedigree 0,30±0,02 0,32±0,02 0,32±0,02
Figura 1. Soluções dos SNP – ±(●), para cromossomos 1, 3 e 5, e ±(●),para cromossomos 2 e 4 – e efeitos dos QTLs – ±(*), para o fenótipo com herdabilidade 0,40 –, com uso de informações de pedigree, genótipo e fenótipo (A), ou apenas de genótipo e fenótipo (B).
três, no início do cromossomo quatro, na parte central do cromossomo quatro e na parte final do cromossomo cinco. Nestes locais, há presença de QTLs simulados próximos. Os resultados indicam que o método proposto é capaz de identificar regiões com maior relevância para o fenótipo estudado.
Para a característica com coeficiente de herdabilidade 0,25, os QTLs simulados tiveram efeito menor do que com herdabilidade 0,40. Porém, a eficiência na identificação de QTLs foi semelhante
entre os fenótipos com diferentes herdabilidades, o que
é indício de ocorrência de um QTL de grande efeito na parte central, no quinto cromossomo, e muitos QTLs de pequeno efeito. Além disso, foram encontrados indícios da presença de QTLs no final do cromossomo três, no início e na parte central do cromossomo quatro, e na parte final do cromossomo cinco, que são locais com presença de QTLs simulados próximos.
Para a característica com herdabilidade 0,10, foram
identificados QTLs nas regiões inicial e intermediária dos cromossomos dois e três. No primeiro cromossomo, foi simulado um QTL que não foi identificado pela análise de associação, possivelmente por não haver
outros QTL próximos, o que diminuiu seu poder de detecção.
Os coeficientes de herdabilidade tiveram influência na identificação dos QTLs, pois quanto maior a
herdabilidade, maiores foram os efeitos estimados
para os marcadores genéticos presentes nas regiões próximas aos QTLs simulados.
Conclusões
1. As informações de parentesco melhoram a predição dos valores genético-genômicos em uma população em seleção.
2. Quanto à habilidade preditiva, as informações de marcadores genéticos são suficientes para substituir as informações de parentesco.
3. O método ssGBLUP iterativo é eficiente para análise de associação genômica ampla e evidencia regiões de QTLs associadas a características de interesse econômico.
4. Maiores herdabilidades favorecem a identificação de potenciais reprodutores para as próximas gerações e de regiões associadas a características de interesse.
Agradecimentos
À Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp), ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), e à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), pelo apoio financeiro.
Referências
AGUILAR, I.; MISZTAL, I.; JOHNSON, D.L.; LEGARRA, A.; TSURUTA, S.; LAWLOR, T.J. Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. Journal of Dairy Science, v.93, p.743-752, 2010. DOI: 10.3168/jds.2009-2730. BRITO, F.V.; BRACCINI NETO, J.; SARGOLZAEI, M.; COBUCI, J.A.; SCHENKEL, F.S. Accuracy of genomic selection in simulated populations mimicking the extent of linkage disequilibrium in beef cattle. BMC Genetics, v.12, p.80-89, 2011. DOI: 10.1186/1471-2156-12-80.
CALAFELL, F.; GRIGORENKO, E.L.; CHIKANIAN, A.A.; KIDD, K.K. Haplotype evolution and linkage disequilibrium: a simulation study. Human Heredity, v.51, p.85-96, 2001. DOI: 10.1159/000022963.
LEGARRA, A.; AGUILAR, I.; MISZTAL, I. A relationship matrix including full pedigree and genomic information. Journal of Dairy Science, v.92, p.4656-4663, 2009. DOI: 10.3168/jds.2009-2061. SARGOLZAEI, M.; SCHENKEL, F.S. QMSim: a large-scale genome simulator for livestock. Bioinformatics, v.25, p.680-681, 2009. DOI: 10.1093/bioinformatics/btp045.
WANG, H.; MISZTAL, I.; AGUILAR, I.; LEGARRA, A; MUIR, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genetic Research, v.94, p.73-83, 2012. DOI: 10.1017/S0016672312000274.
WANG, H.; MISZTAL, I.; AGUILAR, I.; LEGARRA, A; FERNANDO, R.L.; VITEZICA, Z.; OKIMOTO, R.; WING, T.; HAWKEN, R.; MUIR, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Frontiers in Genetics, v.5, article 134, 2014. DOI: 10.3389/ fgene.2014.00134.