De Novo Assembly of the Donkey White Blood Cell Transcriptome and a Comparative Analysis of Phenotype-Associated Genes between Donkeys and Horses.
Texto
Imagem
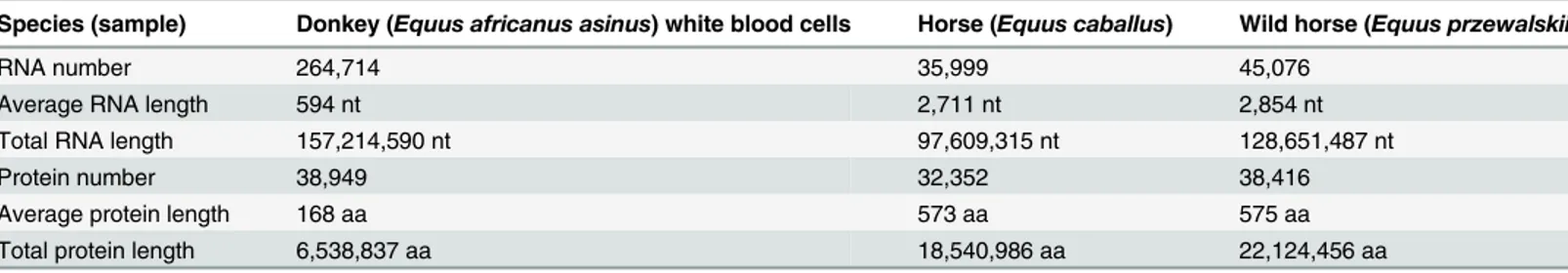
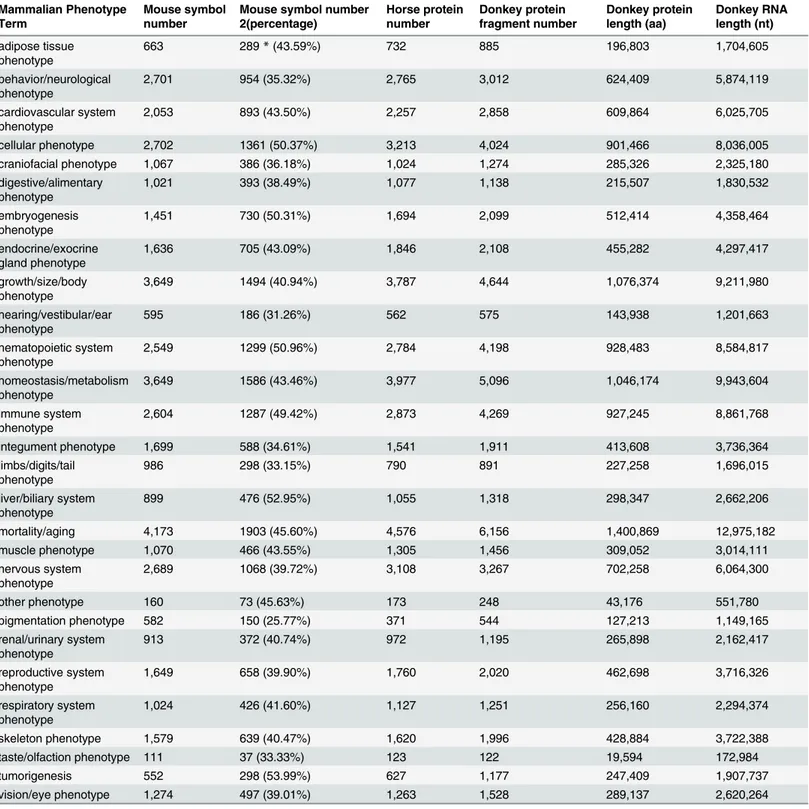
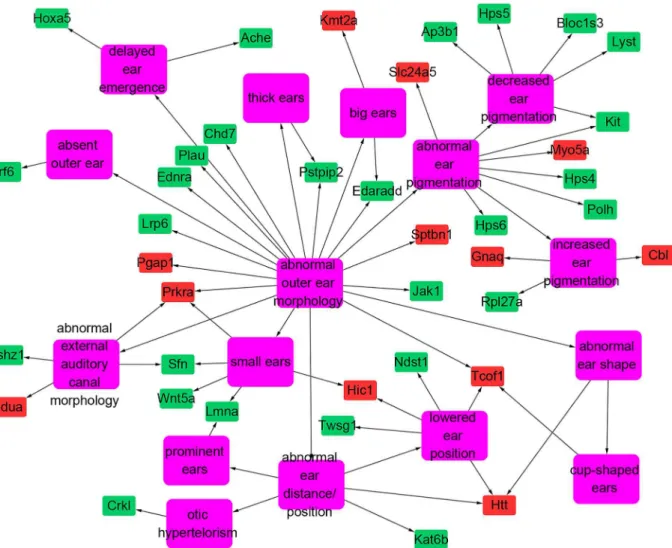

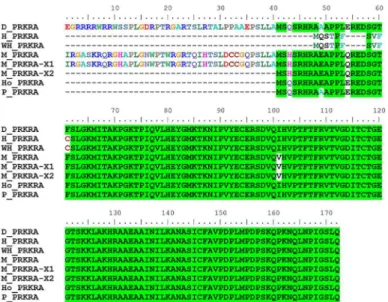

Documentos relacionados
Este relatório relata as vivências experimentadas durante o estágio curricular, realizado na Farmácia S.Miguel, bem como todas as atividades/formações realizadas
É nesta mudança, abruptamente solicitada e muitas das vezes legislada, que nos vão impondo, neste contexto de sociedades sem emprego; a ordem para a flexibilização como
Os dados foram obtidos por meio da intervenção direta da educadora com os educandos, no transcurso de um processo de (re)alfabetização, utilizando como instrumento de ação
The table reports multivariate OLS regressions explaining the size of the equity mutual funds across countries using the fund level variables, excluding the fund asset dummies and
Tomando essa constatação como ponto de partida, neste artigo, através do relato sucinto de investigação longitudinal, respondemos à questão de pesquisa (que conhe- cimentos para
Após a realização deste estudo concluiu-se que não existia nenhum sistema de informação que se pudesse utilizar com resolução ao problema existente, isto porque os sistemas
Deste modo, concluímos que é necessário estarmos atentos ao tripé básico da formação do ser humano: afetividade, cognição e motricidade, apresentado neste estudo
A contribuição de autores como Veyne, Certeau e White, e as futuras apropriações por Spiegel e Munslow, dentre outros que abordam a questão da escrita da história não pode
