CENTRO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS – GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
ESTUDO GENÉTICO-CLÍNICO DE MUCOPOLISSACARIDOSES
NO ESTADO DO CEARÁ
ERLANE MARQUES RIBEIRO
ESTUDO GENÉTICO-CLÍNICO DE MUCOPOLISSACARIDOSES
NO ESTADO DO CEARÁ
Tese apresentada ao Programa de Pós-Graduação em Ciência da Saúde da Universidade Federal do Rio Grande do Norte como requisito para a obtenção do título de Doutor em Ciências da Saúde
Orientador: Prof. Dr. Carlos
Antônio Bruno da Silva
ii
CENTRO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS – GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
Coordenador do Programa de Pós-graduaçao em Ciências da Saúde
Prof. Dr.
ESTUDO GENÉTICO-CLÍNICO DE MUCOPOLISSACARIDOSES
NO ESTADO DO CEARÁ
Aprovada em: 02/ 06 / 2014 Banca Examinadora:
Presidente da banca: Prof. Dr. Carlos Antônio Bruno da Silva
Membros da banca:
Prof. Dr. José Brandão Neto Prof. Dra. Delane Maria Rêgo Prof. Dra. Ida Vanessa Schwartz
iv
Ao meu esposo Marcus Andre de Sousa e aos meus filhos, Kalina e Júlio César pelo incentivo, compreensão e paciência. Aos meus pais, Juraci e Afonso César Ribeiro, e meus familiares, pelo apoio e carinho. Aos meus amigos que me ajudaram de alguma forma nessa caminhada.
Programa de Pós-graduação do Centro de Ciências da Saúde da Universidade Federal do Rio Grande do Norte (PPGCSA-UFRN) pelo incentivo à pesquisa e oportunidade de realização deste trabalho.
Aos colegas geneticistas do Brasil pela amizade e colaboração durante o trabalho, principalmente aqueles que fazem parte da Universidade Federal do Rio Grande do Sul, em especial a Rede MPS Brasil.
Às colegas Delane Rego, Adriana Bezerra e Cristiane Fonteles pela amizade e paciência nas discussões sobre aspectos odontológicos dos pacientes com MPS.
Aos médicos Charles Marques Lourenço, Ana Elisa Kiszewski, Ida Vanessa Schwartz, Dafne Horovitz e Daniela Giovanetti, pela colaboração, paciência nas discussões e valiosas sugestões.
Ao Prof. Dr. Roberto Giugliani, por ser mestre e modelo a ser seguido, em incentivo à nossa vida profissional.
A todos os profissionais que fizeram ou fazem parte da Rede MPS Brasil no Serviço de Genética Médica do Hospital de Clínicas de Porto Alegre – Universidade Federal do Rio Grande do Sul.
Aos funcionários da secretaria do PPGCSA – UFRN pelo carinho, gentileza e colaboração.
vi trabalho.
As mucopolissacaridoses (MPS) são doenças genéticas raras decorrente da deficiência de enzimas lisossomais envolvidas no catabolismo de glicosaminoglicanos, resultando em um amplo espectro de manifestações clínicas, progressivas e multissistêmicas, exigindo tratamento por uma equipe multidisciplinar. Embora o Nordeste brasileiro seja uma região com grande taxa de consangüinidade e um efeito fundador envolvendo MPS, não há estudos caracterizando os pacientes dessa região. Nosso objetivo foi determinar o perfil epidemiológico, clínico e genético de casos não publicados com MPS provenientes do Ceará, identificando as diferenças entre outros estudos com MPS e possíveis problemas a serem enfrentados para a realização do diagnóstico precoce.
viii
MPS=mucopolissacaridoses GAG=glicosaminoglicanos AR=autossômico recessivo RLX=recessivo ligado ao X DS=dermatan sulfato HS=heparan sulfato QS=queratan sulfato CH=condroitin sulfato
TRE=terapia de reposição enzimática SUS= sistema único de saúde
x
Quadro 1. Classificação das mucopolissacaridoses Pág 70
Quadro 2: Alterações clínicas de cada tipo de
mucopolissacaridoses
Pág. 71
Quadro 3: Início dos sintomas, idade e causas de óbito de cada
tipo de mucopolissacaridoses.
Pág 72
Quadro 4: Características das medicações utilizadas para TRE em
casos de Mucopolissacaridoses
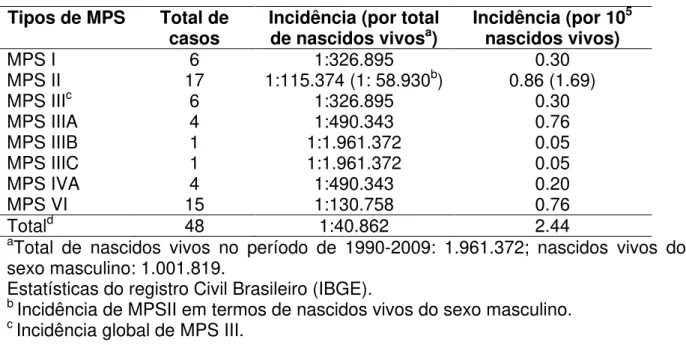
Tabela 1: Incidência dos tipos de MPS no Ceará (1990-2009) Pág. 74
Tabela 2: Incidência de MPS (por 105 nascidos vivos) em países do mundo
e no presente estudo.
Pág. 75
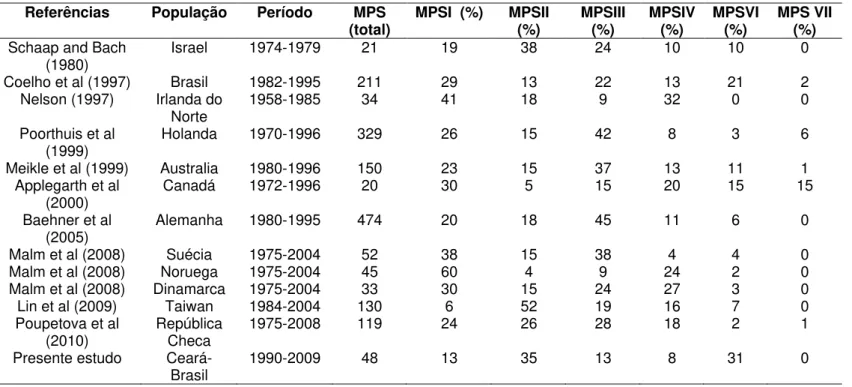
Tabela 3: Frequência dos tipos de MPS em várias populações do mundo e
no presente estudo.
Pág. 76
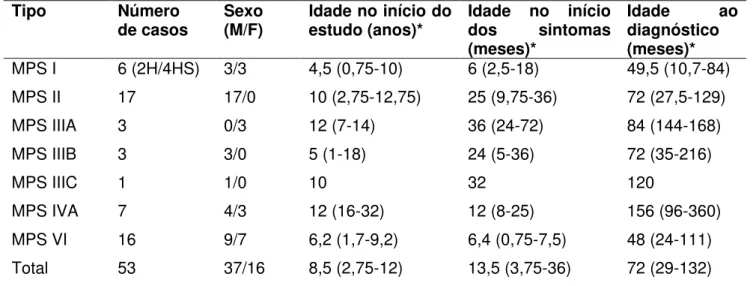
Tabela 4: Caracterização dos tipos de MPS no Ceará segundo número de
casos, sexo, idade ao início do estudo, idade do início dos sintomas e
idade ao diagnóstico.
Pág. 77
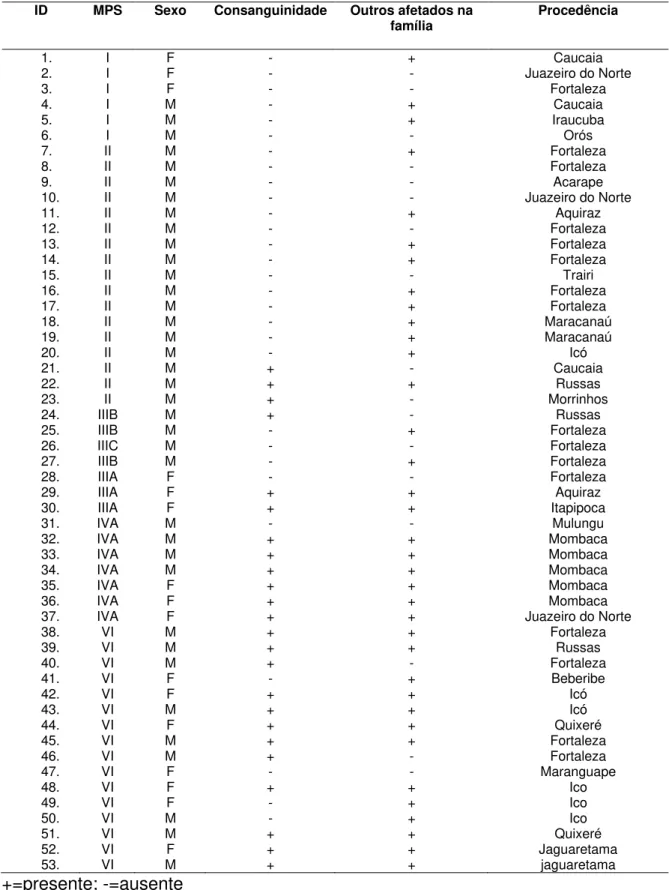
Tabela 5: Dados de peso e estatura ao nascer de 53 pacientes segundo
tipo de MPS
Pág. 78
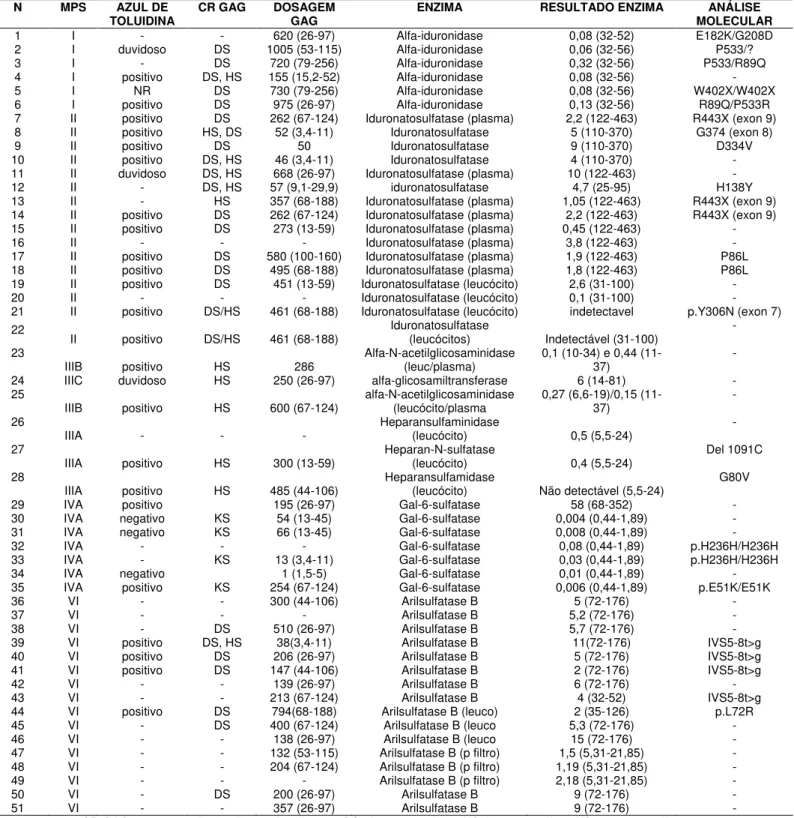
Tabela 6: Classificação dos 53 pacientes segundo tipo de MPS, sexo,
consangüinidade, presença de afetados na família e procedência.
Pág. 79
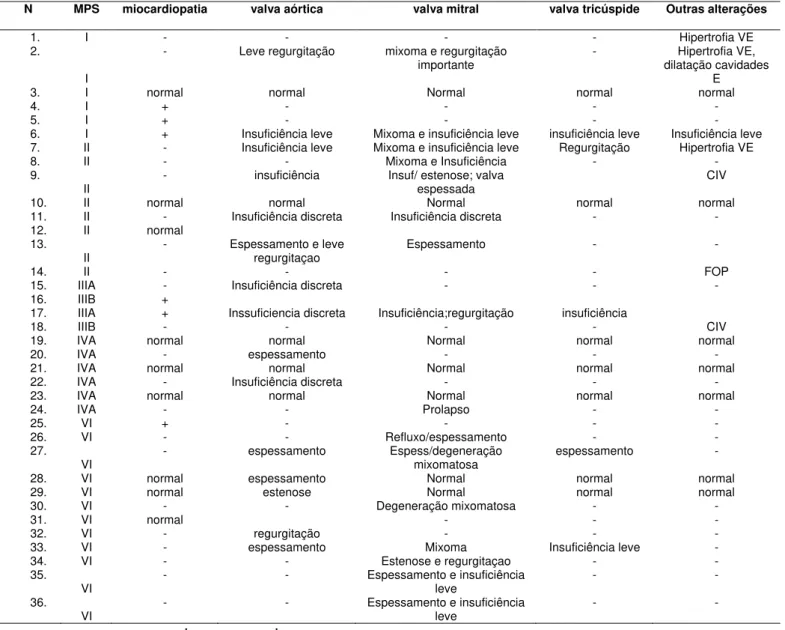
Tabela 7: Análise bioquímica e molecular de 51 pacientes com MPS no
Ceará.
Pág. 80
Tabela 8: Classificação de 36 pacientes segundo o tipo de MPS e o
resultado da avaliação de ecocardiograma.
Pág. 81
Tabela 9: Classificação de 52 pacientes segundo o tipo de MPS e as
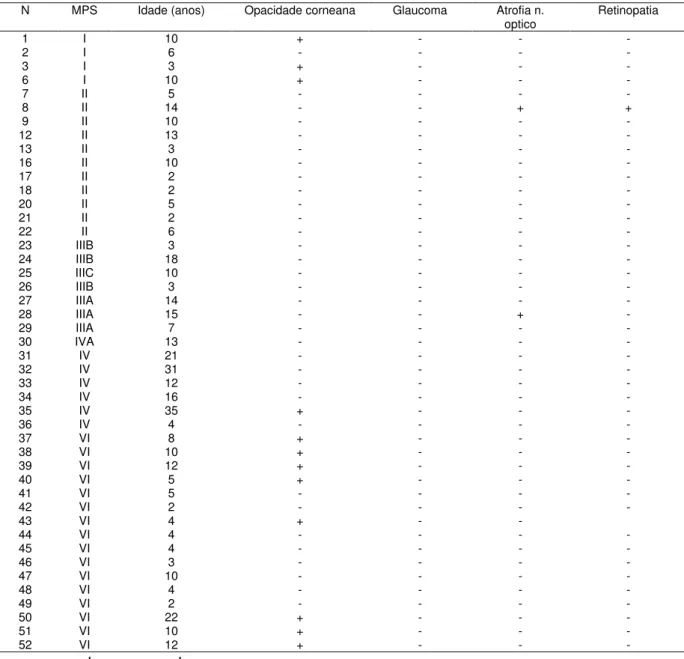
alterações oftalmológicas.
Pág. 82
Tabela 10: Classificação de 52 pacientes segundo o tipo de MPS e as
alterações gastrointestinais.
Pág. 83
Tabela 11: Classificação de 52 pacientes segundo o tipo de MPS e as
alterações otorrinolaringológicas.
xii
Tabela 13: Classificação de 52 pacientes segundo o tipo de MPS e as
alterações dermatológicas.
Pág. 86
Tabela 14: Classificação de 52 pacientes segundo o tipo de MPS e os
sinais de dismorfismos faciais.
Pág. 87
Tabela 15: Classificação de 52 pacientes segundo o tipo de MPS e as
alterações neurológicas.
Pág. 88
Tabela 16: Manifestações clínicas de 52 pacientes segundo o tipo de MPS
no Ceará.
Pág. 89
Tabela 17: Idade em que 26 pacientes iniciaram Terapia de Reposição
Enzimática (TRE) segundo o tipo de MPS no Ceará.
Pág. 90
Tabela 18: Características de 24 casos em Terapia de Reposição
Enzimática (TRE) segundo tipo de MPS, sexo, início dos sintomas, idade
ao diagnóstico, alterações clínicas, início da TRE, total de infusões,
número de interrupções da TRE e motivo das interrupções.
Pág. 91
Tabela 19: Classificação dos pacientes segundo tipo de MPS, sexo, idade
de início dos sintomas, idade ao diagnóstico, idade do óbito, se mora em
zona rural, recorrência na família, causa do óbito.
Pág. 92
Tabela 20: Características clínicas de 16 pacientes falecidos com MPS no
Ceará.
1. INTRODUÇÃO Pág. 15
2. JUSTIFICATIVA Pág. 19
3. OBJETIVOS Pág. 20
4.1. Objetivo geral Pág. 20
4.2. Objetivos específicos Pág. 20
4. MÉTODOS Pág. 21
4.1- Delineamento do estudo, local e período Pág. 21
4.2-Critérios de seleção dos casos Pág. 21
4.3-Coleta dos dados Pág. 22
4.4- Aspectos éticos Pág. 23
4.5- Análise estatística Pág. 23
5. ARTIGOS PRODUZIDOS Pág. 24
6. COMENTÁRIOS, CRÍTICAS E SUGESTÕES Pág. 56 6.1.1- O anteprojeto inicial e o estudo desenvolvido Pág. 56
6.1.2- As limitações do estudo Pág. 56
6.2 - Mérito, originalidade, e contribuição da publicação Pág. 57
6.3 - Evolução intelectual na trajetória do doutorado Pág. 58
6.4 - Metas atingidas e perspectivas de progresso Pág. 58
6.5- Inserção em grupos de pesquisa, orientações acadêmicas e
perspectivas em programas de pós-graduação
Pág. 60
6.6-Produção científica gerada a partir do projeto de pesquisa Pág. 61
7. REFERÊNCIAS Pág. 62
8. APÊNDICE Pág. 69
xiv
9. ANEXOS Pág. 96
Anexo 1- Parecer do comitê de ética do Hospital Geral César Cals
Pág. 97
Anexo 2- Parecer do comitê de ética do Hospital infantil Albert Sabin
Catalogação da Publicação na Fonte Universidade Federal do Rio Grande do Norte - UFRN
Ribeiro, Erlane Marques.
Estudo genético-clínico de mucopolissacaridoses no estado do Ceará / Erlane Marques Ribeiro. - Natal, 2014.
98f: il.
Orientador: Carlos Antônio Bruno da Silva.
Tese (Doutorado) - Programa de Pós-Graduação em Ciências da Saúde. Centro de Ciências da Saúde. Universidade Federal do Rio Grande do Norte.
1. Crianças com deficiência - Tese. 2. Doenças por armazenamento dos lisossomos - Tese. 3. Mucopolissacaridoses - Tese. I. Silva, Carlos Antônio Bruno da. II. Título.
1. INTRODUÇÃO
As mucopolissacaridoses (MPS) constituem um grupo de doenças genéticas relacionadas com defeitos de degradação de glicosaminoglicanos (GAG) decorrente da incapacidade funcional de enzimas lisossômicas, causando lesões progressivas em diversos órgãos e sistemas que levam a uma redução da qualidade e expectativa de vida1.
A determinação de dados epidemiológicos dessa doença e sua caracterização genético-clínica são importantes para o delineamento das necessidades dos pacientes e o planejamento das estratégias de saúde pública para atender a essa população2.
Outros trabalhos sobre MPS já foram realizados no Brasil, determinando dados de vários tipos de MPS de uma instituição que atende a esses pacientes no sudeste do país3, a frequência das MPS em uma instituição que realiza estudos laboratoriais de vários estados4 e o perfil de pacientes com um tipo de MPS5, porém nenhum deles utilizou os dados do estado do Ceará.
Para classificar o tipo de MPS é necessário identificar as alterações de GAG urinário e a deficiência enzimática específica6-11 segundo o quadro 1.
Não há estudos na literatura sobre incidência de MPS no Brasil12, mas sabe-se que em conjunto a incidência das MPS é 1,9-4,5/100.000 recém-nascidos vivos13-21.
O quadro clínico das MPS depende do substrato acumulado. O acúmulo de heparan sulfato leva a sintomas neurológicos (MPS I, II, III)22, de queratan sulfato, a alterações corneanas e esqueléticas, sem comprometimento neurológico (MPS IV). As cardiopatias são mais comuns quando há acúmulo de dermatan sulfato, como nas MPS I, II, VI 23.
comprometimento respiratório (infecções respiratórias de repetição, obstrução de vias aéreas43, apnéia do sono e alteração da função pulmonar44,45), alterações cardíacas (valvulopatia, cardiomiopatia, fibroelastose endocárdica, bloqueio cardíaco, hipertensão sistêmica e pulmonar, estenose arterial difusa, incluindo artérias coronárias46-49), abdominais (hepatoesplenomegalia, hérnia umbilical e/ou inguinal30,50,51), osteo-articulares (disostose múltipla com dolicocefalia, hipoplasia odontóide, instabilidade atlantoaxial, cifose toracolombar, vértebras em saca bucado, pedículos atargados e platisespondilia25-30, costelas em remo, clavículas pequenas e largas, escápulas alargadas e elevadas, ilíacos pequenos, ísquio e pubis alargados, acetábulo com teto oblíquo, subluxação da cabeça do fêmur, coxa valga, falanges pequenas, metacarpos cônicos, carpos pequenos e irregulares, restrição articular generalizada e indolor, dedos das mãos em gatilho e mão em garra52-54), neurológicas (hidrocefalia55,56, convulsões, atraso de desenvolvimento neurológico, retardo mental, comcomportamento autista-like, hiperatividade, agressividade22,,57, cistos aracnoideos, atrofia cortical e alterações da substância branca na ressonância magnética do crânio54,58, síndrome do túnel do carpo59, compressão da medular, instabilidade atlantoaxial, mielopatia cervical60, resposta exagerada das extremidades a mudanças de temperatura, diarréia61, perda auditiva neurossensorial e/ou condutiva. 28,30,62,63). As principais alterações clínicas em MPS estão resumidas no quadro 2.
Apesar do diagnóstico definitivo do tipo de MPS depender do estudo da atividade enzimática, as manifestações clínicas são importantes para orientar o estudo laboratorial a ser realizado64.
O estudo enzimático do sangue impregnado em papel de filtro pode permitir o diagnóstico da MPS no período neonatal, bem como na triagem de pacientes com doenças de depósito6,65,66.
Apesar do diagnóstico molecular ser mais preciso para diagnóstico pré-natal e aconselhamento genético8,63, o grande número de mutações em cada tipo de MPS resulta em alto custo do exame, impossibilitando seu uso na prática clínica, além do que não foi possível estabelecer uma correlação genótipo-fenótipo na maioria dos casos8.
O diagnóstico pré-natal pode ser realizado precocemente através do estudo da atividade enzimática nas vilosidades coriônicas ou amniócitos cultivados nos casos que a mutação da familial é conhecida8,30,63 ou tardiamente através do estudo enzimático em sangue do cordão umbilical69.
O início dos sintomas em MPS é precoce e a expectativa de vida é limitada (quadro 3). O prognóstico é dependente das complicações, principalmente para os casos com comprometimento neurológico30,70,71.
Com a determinaçao clínico-laboratorial do paciente com MPS é possível realizar o aconselhamento genético, que é um processo que envolve, além do risco de recorrência8, a compreensão pelo pacientes e familiares sobre os fatos decorrentes da doença genética em questão, incluindo a confirmação da etiologia, diagnóstico precoce, pré-natal, exames a serem realizados, probabilidade de haver outros afetados na família, indicação de grupos de apoio, prognóstico, tratamento72.
O tratamento sintomático de pacientes com MPS, que pode evitar complicações e reduzir a morbidade, deve ser realizado por uma equipe multidisciplinar8,28. As consultas com especialistas e a realização de exames complementares devem ser periódicas50.
O tratamento de suporte é indispensável, mesmo nos casos em que há terapia para doença de base a partir do transplante de células hematopoiéticas (MPS I) ou terapia de reposição enzimática (MPS I, II, IV, VI)8.
O tratamento com reposição enzimática (TRE) para MPS (quadro 4) mudou a história natural da doença, no entanto deve ser realizado antes do início das complicações para que tenha maior efetividade74,75.
Apesar das melhorias na terapêutica de MPS, ainda há a necessidade de novas modalidades de tratamento, principalmente para complicações neurológicas7,76.
O desafio atual da comunidade científica é o reconhecimento específico das bases moleculares das MPS, a correlação fenótipo-genótipo, o desenvolvimento de triagem pré-sintomática para esses casos e o tratamento precoce efetivo7.
2 – JUSTIFICATIVA
A determinação da frequência e caracterização das MPS no Ceará é desconhecida e esses dados são importantes para o delineamento das necessidades dessa população, refletindo na necessidade de treinar os profissionais de saúde para realizar diagnóstico precoce, de acessibilidade aos recursos tecnológicos para identificação do comprometimento orgânico, além de planejamento das estratégias de governo para atender a essa população.
Sendo uma doença multisisstêmica, as MPS exigem uma avaliação clínica e laboratorial em um centro de referência para avaliar o comprometimento clínico do paciente, bem como determinar o perfil bioquímico e molecular dos casos a fim de classificar essas MPS, permitindo a realização do tratamento adequado, do diagnóstico pré-natal e do aconselhamento genético, incluindo a prevenção de novos casos.
A análise molecular dos pacientes pode ainda determinar a correlação fenótipo-genótipo para prever a gravidade dos sintomas, permitir a identificação de novas mutações, caracterizando a população local, fornecendo dados inéditos sobre o perfil das MPS no Ceará, mantendo os pacientes aptos a realizar as novas terapias que surgirão em um futuro próximo.
3. OBJETIVOS
3.1 Objetivo geral
• Reconhecer o perfil genético-clínico das MPS no Estado do Ceará.
3.2 Objetivos específicos
• Determinar o perfil epidemiológicoepidemiologico da MPS no estado do Ceará, baseando-se nos dados de incidência, prevalência, morbi-mortalidade • Determinar o perfil clínico-nosológico da MPS em amostra de pacientes
estudada
4. MÉTODOS
4.1- Delineamento do estudo, local e período
Tratou-se de um estudo prospectivo de dados secundários, seccional, descritivo, observacional, contemplando todos os casos de MPS, com diagnóstico firmado até 2013 no estado do Ceará, independente do sexo e idade. Os dados primários foram provenientes do Hospital Geral Dr Cesar Cals (HGCC) e Hospital Infantil Albert Sabin (HIAS), que são as instituições/serviços de referência de diagnóstico, seguimento e terapia de doenças genéticas.
4.2-Critérios de seleção dos casos
Foram selecionados os casos com confirmação laboratorial e determinação do tipo de MPS nascidos e residentes no estado do Ceará através do estudo bioquímico (atividade enzimática no plasma e/ou leucócitos) e/ou estudo molecular. Pacientes com MPS e/ou seus representantes legais foram contatados pela investigadora para obtenção do termo de consentimento informado antes do início do estudo.
4.2.1- Avaliação Bioquímica
A avaliação bioquímica dos pacientes se deu a partir de dosagem quantitativa de GAG urinários, avaliação qualitativa dos GAG na urina e medida da atividade das enzimas potencialmente deficientes em cada caso (em sangue em papel filtro, plasma, leucócitos). Os testes bioquímicos foram realizados no laboratório de Erros Inatos do Metabolismo do Serviço de Genética Médica do Hospital de Clínicas de Porto Alegre da Universidade Federal do Rio Grande do Sul através da rede MPS Brasil.
4.2.2-Análise molecular
submetido a análise por SSCP para seqüenciamento dos exons com padrões alterados de migração de acordo com o protocolo da Rede MPS Brasil, no laboratório de Erros Inatos do Metabolismo do Serviço de Genética Médica do Hospital de Clínicas de Porto Alegre da Universidade Federal do Rio Grande do Sul.
4.3-Coleta dos dados
Os dados foram obtidos a partir de fonte primária dos prontuários das instituições e naqueles obtidos diretamente de pacientes e familiares a partir de entrevista semi-estruturada. Em seguida foi realizado exame clínico de cada criança/adulto. Os exames complementares avaliados foram aqueles que faziam parte da rotina assistência aos pacientes pelo Sistema único de Saúde (SUS). Não foram solicitados exames com objetivo de pesquisa. Todas as etapas do estudo foram realizadas por um único observador, a investigadora principal.
As variáveis avaliadas foram:
Perfil epidemiológico da MPS número de casos
• tipo de MPS, Sexo, procedência, presença de consanguinidade e de outros afetados na família, caracterização epidemiológica, número de pacientes que faleceu, sexo, idade do início dos sintomas, do diagnóstico e do óbito, tipo de MPS, procedência, recorrência familiar, caracterização clínica de cada paciente, dados de mortalidade, causa de óbito
Perfil clinico-nosológico da MPS
• Dados perinatais, Idade de início de sintomas, Idade ao diagnóstico, Idade de início no estudo, caracterização dos primeiros sintomas, caracterização das manifestações clínicas (deficiência de crescimento, dismorfismo facial (face peculiar), alterações dermatológicas, oftalmológicas, odontológicas, de vias aéreas superiores, cardíacas, gastrointestinais, neurológicas e osteoarticulares).
Dados bioquímicos e Caracterização molecular
• Dosagem de GAG urinário, tipo de GAG, resultado de teste enzimático, testes moleculares.
4.4- Aspectos éticos
A pesquisa foi avaliada e aprovada pelo comitê de ética em pesquisa do Hospital Geral César Cals, Hospital Infantil Albert Sabin e pelo Conselho Nacional de Pesquisa (CONEP) tendo como número de processo CAE: 0041.1.041.602-05.
4.5- Análise estatística
5.1. – Artigo I
5.2. Artigo II
A CLINICAL STUDY OF OROFACIAL FEATURES IN 26 BRAZILIAN PATIENTS WITH DIFFERENT TYPES OF MUCOPOLYSACCHARIDOSIS.
Running Title: OROFACIAL FEATURES IN MUCOPOLYSACCHARIDOSIS
Authors:
Erlane Marques Ribeiro , Cristiane Sá Roriz Fonteles , Adriana Bezerra Freitas , Karla da Silva Alves , André Monteiro , Carlos Bruno da Silva
Abstract
Purpose: This study purpose to describe orofacial features of 26 unrelated Brazilian patients with mucopolysaccharidosis (MPS), and verify any possible associations between these findings and specific MPS-types. Methods: Patients were diagnosed with MPS, and systematically evaluated. Following consent, a clinical assessment form was completed. Facial and intraoral examination was performed, evaluating facial pattern, malocclusions, dental caries, tooth identification. Results: Midface deficiency, increased lower facial third, anterior open bite, convex profile, macroglossia, gingival enlargement and spaced arches were the most frequently observed features. These findings did not allow a differential diagnosis between different types of MPS, except for pitting enamel, which significantly associated with MPS IVA (p=.000). Open bite was statistically related to MPS types I, II, III and VI, whereas only 1 patient with MPSIVA expressed this feature (p=.043). Conclusions: Our results suggest that pitted enamel in MPS patients is most likely a feature of MPS type IVA, whereas open bite is rarely observed in these patients. Orofacial features in MPS may help pediatric dentists recognize this disorder and minimize the delay between the initial signs/symptoms and diagnosis of the disease. Future studies should focus in the longitudinal manifestations, expression and severity of MPS-associated orofacial anomalies.
Introduction
The Mucopolysaccharidoses (MPS) are a rare clinically heterogeneous group of metabolic disorders caused by a deficiency or malfunction of the lysosomal enzymes involved in the stepwise degradation of glycosaminoglycans (GAG), formerly called mucopolysaccharides. The resultant accumulation of undegraded or partly degraded GAG within lysosomes causes permanent progressive tissue and organ dysfunction. An overall incidence of 1:25.000 live births has been estimated and seven distinct clinical types and numerous subtypes of MPS have been identified. These MPS types share many clinical features with varying degrees of severity, which progress as storage of GAG affects bone, skeletal structure, connective tissues, and organs. All MPS are inherited in an autosomal recessive manner except for MPS II (Hunter syndrome) which is transmitted as an X-linked recessive disorder (Neufeld and Muenzer, 2001).
Typical features of MPS I, II and VI include coarse facies, growth impairment, deafness, cardiovascular complications, airway obstruction, hepatosplenomegaly, progressive joint stiffness and skeletal deformities (Valayannopoulos et al., 2010). Only severe forms of MPS I and MPS II manifest cognitive impairment, whereas MPS III usually does not show somatic manifestations but is marked by severe neurological symptoms (Wegrzyn et al, 2010). MPS IVA is clinically different from other forms of MPS, and is characterized by severe spondyloepiphyseal dysplasia (Montano et al, 2007). Clinical and biochemical characteristics of the mucopolysaccharidoses are summarized in Table 1.
1999), short and broad mandibular rami with narrow and flat condyles (Macleod and Macintyre, 1993; Defraia et al, 2005; Kaneyama et al, 2008). Bone marrow transplantation and enzyme replacement therapy may stabilize or reverse many aspects of MPS I, II and VI, changing the natural course of the disease(Wadenya et al, 2010). However, the impact of enzyme replacement therapy on these MPS-associated orofacial manifestations has not been demonstrated.
This study aimed to identify the most prevalent orofacial features in 26 unrelated Brazilian patients with MPS, and to verify any existent association between these findings and each of the identified types of this disease, in order to assist in clinical diagnosis.
Methods
Facial examination was performed aiming to evaluate facial pattern, proportionality and symmetry of the facial thirds, and facial convexity, whereas dental examination included the following features: occlusal pattern, eruptive disturbances, dental development, presence/absence of dental caries, identification of each individual tooth. Dental examiners were blind as to what type of MPS these patients presented. Patients that had been previously submitted to bone marrow transplantation, and those that were using medications that could potentially induce gingival overgrowth were excluded from the study. A summary of every child's medical history was provided following data collection, to be used for dental treatment planning of these patients. In order to verify association between orofacial features, gender and different MPS types and subtypes, Pearson Chi-square test was used. Statistical significance was established when p<.0.05
Results
Fifty patients were diagnosed as having MPS, out of which 39 patients fulfilled inclusion criteria (Figure 1). A total of 26 patients (9 females and 17 males) consented to participate in this study. In this final sample, distribution of MPS types were as follows: MPS I (n=4), MPS II (n=8), MPS III (n=3), MPS IVA (n=5), MPS VI (n=6). No statistical association was observed between MPS types and gender (p=.098). However, all patients who expressed MPS type II (n=8) were from the male gender.
Mucopolysaccharidosis type I (MPS I)
breathers, and presented midface deficiency, a convex profile, macroglossia, anterior open bite and spaced arches. (Table 2).
Mucopolysaccharidosis type II (MPS II)
All patients with MPS II were from the male gender. Two patients presented a mild form of the disease (ages 11 and 15 years), whereas 6 patients expressed severe MPS II (ages 4 - 9 years). These patients presented short stature (n=4), macrocephaly (n=6), respiratory abnormalities (n=7), cardiopathy (n=8), hernia (n=8), hepatosplenomegaly (n=7), joint stiffness (n=8) and skeletal abnormalities (n=8). Enzyme replacement therapy with Idursulfase, at the dose of 0.5 mg/kg weekly was implemented in 4 out of 8 patients before the beginning of this study. The patients who received treatment included 2 four year old twin males (4 infusions each), 2 unrelated six year old males (3 and 121 infusions, respectively). All patients expressed midfacial third deficiency and a convex profile. Other extra-oral features included an increased lower facial third (n=5) and enlarged lips (n=5). Intra-oral evaluation showed the presence of anterior open bite on all patients, spaced arches (n=7), macroglossia (n=6) and gingival enlargement (n=6) (Table 2). Most patients (n=7) were mouth breathers.
Mucopolysaccharidosis type III (MPS III)
We have identified 3 patients with MPS III, out of which 1 twelve year old female with MPS IIIA, 1 five year old male with MPSIIIB and 1 fifteen year old male with MPSIIIC. All patients presented macrocephaly, coarse face, developmental delay, mental retardation and behavioral disturbances, midfacial third deficiency and a convex profile and respiratory abnormalities. The most commonly observed intra-oral features were gingival enlargement (n=2) and anterior open bite (n=2) (Table 2).
Mucopolysaccharidosis type IVA (MPS IVA)
presence of pitted enamel on all patients, and the frequent expression of anterior open bite, macroglossia and spaced arches (Table 2).
Mucopolysaccharidosis type VI (MPS VI)
Six patients with MPS VI wereenrolled in this study (3 females and 3 males), with ages ranging from 1.9 to 10 years. Short stature (n=5), macrocephaly (n=5), respiratory abnormalities (n=5), cardiopathy (n=5), gibous vertebrae (n=5), hepatosplenomegaly (n=5), joint stiffness (n=5) and skeletal abnormalities (n=6) were the most commonly observed clinical features. Except one three year old male (104 infusions) and 1 seven years old male (111infusions) patient were on enzyme replacement therapy with 1 mg/kg of recombinant human N-acetylgalactosamine-4-sulphatase (rhASB), therapy was initiated prior to participation in this study. Most patients presented midfacial third deficiency (n=4) and a convex profile (n=4). Intra-orally the following features were observed: anterior open bite (n=5), spaced arches (n=5), macroglossia (n=4) and gingival enlargement (n=4).
Parafunctional habits
Non-nutritive sucking (pacifiers) was the most commonly observed parafunctional habit among MPS patients. (Table 3).
Association between Orofacial Features and Types of MPS
Although maxillary protrusion was present in most patients with MPS types II and III, most patients with MPS IV (4 out of 5 patients) and VI (5 out of 6 patients) did not express maxillary protrusion, and the number of MPSI-patients with and without maxillary protrusion were evenly distributed (2 out of 4 patients).
Discussion
In Brazil, a 4.8-year delay between the time of onset of the signs/symptoms of MPS and the establishment of a diagnosis has been previously described(Vieira et al., 2008). Specific therapies are available for MPS, and early treatment is likely to render favorable changes in the natural history of the disease(Burrow et al, 2007; Rohrbach and Clarke, 2007; Nathan and Orkin, 2007; Beck, 2010). Thus, efforts should be made to minimize this delay. We believe that orofacial characteristics of MPS in children may assist in the early recognition of this disorder. In the present study, most of the studied cases consisted of children of different ages, probably due to the high mortality rate of this disease, with the exception of MPSIVA. The observed predominance of the male gender in the present sample can be explained by the greater number of MPSII cases, known to have an X-linked recessive inheritance pattern. As for the different types of MPS, our results agreed with the findings described by Vieira et al (2008), in which a higher prevalence of MPSII and MPSVI, associated with a lower prevalence of MPS III and MPS I were reported. These results differ from the ones reported in other areas of the world(Malm et al., 2008). In addition, MPS IVB and MPS VII have also been rarely reported (Lin et al., 2009; Poupelová et al, 2010).
Pitted enamel was observed in all cases of MPS IVA, but was not noted in any of the other MPS types. This defect consists of abnormally thin enamel that is rough because of minute surface pits. The thin enamel results in altered dental shape, in addition to discolored and widely-spaced teeth. The enamel appears to be structurally weak, since it exhibits a tendency to fracture and flake off (Knirons and Nelson, 1990), predisposing these areas to dental caries. The presence of defective deciduous enamel suggests that in these patients the disease process may be active early in intrauterine life and/or immediately after birth.
The expression of orofacial features did not differ among MPS types I, II, III and VI. The most commonly observed alterations in these types of MPS were facial pattern-related alterations, in addition to macroglossia, gingival enlargement and spaced arches. The factors that may contribute with the emergence of orofacial alterations in MPS-patients are as follows: (1) MPS-associated facial alterations; (2) infiltration of GAG within the airways, leading to mouth breathing habit, macroglossia and enlargement of lips and gingiva; (3) dysostosis, which alters facial bone growth, and causes flattening of the condylar heads, (4) physiopathology of the disease may interfere prenatally in dental structure, (5) parafunctional habits. The severity of clinical alterations was also observed to increase with age, due to the progressive nature of the disease (Valayannopoulos et al, 2010).
controlled setting.
Management of MPS patients must include routine dental examinations, radiographic evaluations at a 6-month interval and regular at-home dental care(Giugliani et al., 2007), as well as preventive anticaries measures to assure oral health, avoiding further complications. Almost 50% of the presently evaluated cases had a compromised oral health, presenting a great challenge during routine dental examination. This preventive approach is of the utmost importance, since these patients may be at risk for bacterial endocarditis, as well as aspiration pneumonia due to the neurological involvement, which can greatly complicate the execution of more complex dental procedures. There are no current protocols for the evaluation and dental treatment of MPS-patients. Previously, Waldenya (2010) suggested the establishment of preventive measures for oral health maintenance in a patient with MPS I who had received bone marrow transplant (BMT). The author suggested the implementation of the same preventive measures used in patients subjected to BMT, without MPS. However, BMT guidelines are not designed to meet the specific limitations observed in most MPS-patients, such as mouth opening limitations, arm joint restrictions and claw hand deformities, which may impose difficulties in the establishment of a dental home. In addition, non-nutritive sucking habit was the most prevalent parafunctional habit observed among MPS-patients. This prevalence was greater among patients with MPSII and MPSIII, probably due to the presence of neurological alterations and behavioral disturbances, imposing a greater challenge for families to manage these alterations (Wegrzyn et al., 2007).
Acknowledgments
References
Alpoz AR, Çoker M, Çelen E, Ersin NK, Gokçen D, van Diggelenc OP, Huijmansc JG. The oral manifestation of Maroteaux-lamy syndrome (mucopolysaccharidose VI): a case report. Oral Surg. Oral Med. Oral Pathol Oral Endod 2006; 101: 632-7.
Beck M. Therapy for lysosomal Storage Disorders. Life 2010; 62 (1): 33-40.
Burrow A, Hopkin RJ, Leslie ND, Tinkle BT, Grabowski GA. Enzyme reconstitution/replacement therapy for lysosomal storage diseases T. Current Opinion in Pediatrics 2007; 19:628–35.
Cleary MA, Wraith JE. The presenting features of mucopolysaccharidosis type I (Hurler syndrome). Acta Paediatr 1995; 84: 337-9.
Defraia E, Marinelli A, Antonini A,Giuntini V. Abnormal Mandibular Growth after Craniovertebral Surgery in Morquio Syndrome Type A. Angle Orthod. 2005; 75:
461-4
Downs AT, Crisp T, Ferretti G. Hunter’s syndrome and oral manifestations: a review.
Am Academ Pediatr Dent 1995; 17 (2): 98-100.
Freitas DQ, Tempest LM, Sicoli E, Lopes-Neto FC. Bilateral dentigerous cysts: review of the literature and report of an unusual case. Dentomaxillofac Radiol 2006;
35: 464-8.
Giugliani R, Hamatz P, Wraigh JE. Management Guidelines for Mucopolysaccharidosis VI. Pediatrics 2007; 120: 405-18.
Guven G, Cehreli ZC, Altun C, Sençimen M, Ide S, Bayari SH, Karaçay S. Mucopolysaccharidosis type I (Hurler-Scheie): Oral and radiographic findings and ultrastructural / chemical features of enamel and dentin. Oral Pathol Oral Radiol Endod 2008; 105 (1): 72-8
Kaneyama K, Segami N, Hatta T. Congenital deformities and developmental abnormalities of the mandibular condyle in the temporomandibular joint. Congenital Anomalies 2008; 48: 119-25.
Keith O, Scully C, Path MRC, Weidmann GM. Orofacial features of Scheie (Hurler-Scheie) syndrome (α-L-iduronidase deficiency). Oral Surg Oral Med Oral Pathol
1990; 70: 70-4.
(Morquio’s disease type A). Oral Surg. Oral Med. Oral Pathol. 1990; 70: 176-9.
Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, Chao MC, Chiu PC, Lin SJ, Tsai LP et al. Incidence of the Mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet 2009 Part A; 149A: 960-4.
Levin LS, Jorgenson RJ, Salinas SF. Oral findings in the Morquio syndrome (mucopolysaccharidosis IV). Oral Surg. 1975; 39 (3): 390-5.
MacLeod SPR, Macintyre DR. Bilateral hypoplasia of mandibular condyles in Hurler’s syndrome. Oral Surg Oral Med Pathol 1993; 75 (5): 659-60.
Malm G, Lund AM, Mansson J, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta paediatrica 2008; 97:
1577-81.
Martin R, Beck M, Eng C, Giugliani R, Hamatz P, Muenzer J. Recognition and diagnosis of Mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008;
121:e377-86.
Montano AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: Clinical manifestation and natural course of Morquio A disease. J Inherit. Metab. Dis. 2007; 30:165–74.
Munoz-Rojas MV, Bay L, Sanchez L, van Kuijck M, Ospina S, Cabello JF, Martins AM. Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world. J Inherit. Metab. Dis 2011;
34 (5): 1029-37.
Nakamura T, Mika K, Nonaka K, Anan H, Higash S, Beppu K. Rosette formation of impacted molar teeth in mucopolysaccharidoses and related disorders.
Dentomaxillofac Radiol 1992; 21: 45-9.
Nathan DG and Orkin SH. Musing on genome medicine: enzyme-replacement therapy of the lysosomal storage diseases. Genome medicine 2009; 1: 1141-3.
Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill, 2001: 3421–52
data in different populations. J Inherit Metab Dis, 2010; 33: 387-96.
Roberts MW, Barton NW, Constantopoulous G, Butler DP, Donahue AH. Occurrence of multiple dentigerous cysts ina patient with Maroteaux-Lamy syndrome (mucoplysaccharidosis, type VI). Oral Surg. 1984; 50: 169-75.
Rohrbach M, Clarke JTR. Treatment of Lysosomal Storage Disorders Progress with Enzyme Replacement. Therapy Drugs. 2007; 67 (18): 2697-2716.
Rolling I, Clausen N, Nyvad B, Sindet-Pedersen S. Dental findings in three siblings with Morquio’s syndrome. Int. J. Ped. Dent. 1999; 9: 219-24.
Smith KS, Hallett KB, Hall RK, Wardrop RW, Firth N. Mucopolysaccharidosis: MPS VI and associated delayed tooth eruption. Int. J. Oral Maxillofac. Surg. 1995; 24:
176-80.
Wadenya RO; Stout AM; Gupta A; Monge J. Hurler syndrome: a case report of a 5-year follow-up of dental findings after bone marrow transplantation. Spec. Care Dentist 2010; 30 (1): 14-17.
Wegrzyn G, Jakobkiewick-Banecka J, Narajczyk M, Wisniewski A, Piotrowska E, Gabig-Ciminska M, Kloska A, Slominska- Wojewodzka M,Korzon-Burakowska A, Wegrzyn A. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Medical Hypoteses 2010; 75: 605-9.
Valayannopoulos V, Nicely H, Harmatz P , Turbeville S. Mucoplysaccharidosis VI.
Orphanet Journal of Rare Dis 2010;5 (5): 1-20.
Figure 1: Patient recruitment flow chart.
Table 1: Clinical and biochemical characteristics of the mucopolysaccharidoses. Table 2: Distribution of orofacial features of 26 patients with different types of MPS. Table 3: Distribution of parafunctional habits among MPS patients
Table 1
Type OMIM Clinical
Dysmorphis m
Disostose
Multiplex Corneal Opacity Mental Retardatio n
Urinary GAG Deficient Enzyme
IH (Hurler) #607014 ++++ ++++ Y Y DS/HS L-iduronidase IHS
(Hurler-Scheie) #607015 +++ +++ Y N DS/HS L-iduronidase
IS (Scheie) #607016 ++ ++ Y N DS/HS L-iduronidase
IIA (Hunter –
attenuated) #309900 +++ +++ N N DS/HS Iduronate 2 sulfatase IIB (Hunter –
severe) #309900 +++ +++ N Y DS/HS Iduronate 2 sulfatase IIIA (Sanfillipo A) #252900 ++ + N Y HS Heparin sulfamidase
IIIB (Sanfillipo B) #252920 ++ + N Y HS
N-Acetylglucosaminidase
IIIC (Sanfillipo C) #252930 ++ + N Y HS Heparan
acetyl-
CoA:alpha-glucosaminide N-acetyltransferase IIID (Sanfillipo D) #252940 ++ + N Y HS N-Acetyl-Alpha-D-Glucosaminidase
IVA (Morquio A) #253000 ++++ ++++ Y N KS Galactosamine 6
sulfate sulfatase IVB (Morquio B) #253010 ++++ ++++ Y N KS Beta galactosidase VI
Table 2
MPS Type I§ I I I II§ II II II II II II II III§ III III IVA§ IVA IVA IVA IVA VI§ VI VI VI VI VI
Age (years) 4 5 11 10 6 6 7 4 11 15 4 4 5 12 15 35 17 13 33 10 12 5 1 11 2 8 Extraoral features*
Enlarged Lips + - + - + + + - + + + - - + + - + + - + + - - + + +
Lip Competence + + - - + - - + + - - + + - + + + + + - - + + - +
-Mouth Breather + + + + + + + - + + + - + + - - - + - + + + - + - +
Midface Deficiency
+ + + + + + + + + + + - + + + + + + + + + + + + - +
Increased Lower Third
+ + + - + + + - + + - - - + + + + + + + + - - + - +
Convex Profile + + + + + + + + + + + + + + + + + + + + + + + + + +
Intraoral feature*
Deciduous
Dentition + + - - - - - + - - + + + - - - - - - - - + + - +
-Mixed Dentition - - + + + + + - + - - - - + + - - - - - + - - + - +
Permanent
Dentition - - - - - - - - - + - - - - - + + + + + - - - - -
-Gingival
Enlargement - - + + + - + + + + - - - + + + - + - - + + + + - +
Biprotrusion - - + - + - + - - + - - - + - - - - - + - - - + -
-Macroglossia + - + + + + + + - + + - + - - + + - - + + + - + - +
Cross-bite + + - - + + - - + - - + - + - + - + - - + - - - - +
* Where “+” means presence, and “- “ means absence.
§ MPS I, n=4; MPS II, n=8; MPS III, n=3; MPS IVA, n=5; MPS VI, n=6.
bite
Conoid Incisors - - + - - - - - + + - - - + - - - - - - - - - - -
-Crowding - - - + - - - - - + - - - + - - - - + + - + - - -
-Pitted enamel - - - - - - - - - - - - - - - + + + + + - - - - -
-Spaced arches + + + + + + + + - + + + + - - + - - + - + + + - + +
Table 3
Parafunctional
habits MPS In=4 MPS IIn=8 MPS IIIn=3 MPS IVAn=5 MPS VIn=6 Totaln=26
Pacifier 2 3 1 2 1 9
Finger sucking 0 2 1 0 1 4
Bottle-feeding 1 3 0 0 0 4
Bruxism 1 1 0 0 0 2
Biting objects 0 2 1 0 0 3
Nail biting 0 1 0 0 0 1
Table 4.
*
Data expressed as the number of patients with a specific type of MPS affected by a feature or habit in relation to the total number of patients affected by this type of MPS. ** Chi-square
test. Significance established at p < .05.
MPS types I* II III IV VI p-value**
COMENTÁRIOS, CRÍTICAS E SUGESTÕES
6.1.1- O anteprojeto inicial e o estudo desenvolvido
O anteprojeto inicial previa o estudo genético-clínico dos pacientes com MPS do Ceará. No período em que o estudo foi desenvolvido aconteceram mudanças no conhecimento dos pesquisadores sobre MPS, com a incorporação de novos tratamentos. Essas mudanças fizeram com que os estudos que relatavam resultado de terapia fossem mais atrativos dos que os estudos clínicos descritivos para publicação. As publicações científicas sobre quadro clínico dos pacientes com MPS passaram a exigir um número maior de sujeitos, o que é difícil de ser obtido para uma doença genética rara, sendo necessário aumentar o tempo do estudo, além do determinado no cronograma.
6.1.2- As limitações do estudo 6.1.2.1- Modelo do estudo
Inicialmente escolhido foi um estudo quantitativo, descritivo, observacional limitado no tempo, sem a realização de um estudo piloto e apenas no estado do Ceará. Apesar de que o modelo correspondeu às expectativas dos investigadores, gerando dados com grande validação interna, a publicação internacional do trabalho foi difícil com esse modelo.
6.1.2.2.- Número de pacientes
Por se tratar de uma doença genética rara, o estudo teve um pequeno número de casos se for considerado cada tipo de MPS em separado. Se o estudo fosse multicêntrico, envolvendo os casos de outros estados do país, reduziria essa limitação.
6.1.2.3- Falta de reconhecimento precoce dos casos de MPS
No Brasil em média há um intervalo de 4,8 anos entre os primeiros sintomas de MPS e o diagnóstico etiológico12. A falta de reconhecimento precoce dos casos pode resultar na sub-estimativa da freqüência das MPS no Ceará, mesmo com a estratégia de educação em saúde realizada a partir da distribuição de folder, cartazes, artigos e palestras.
6.1.2.4- A carência do sistema de transporte
serviços de saúde terciários para consultas e exames, foi responsável pela ausência da avaliação clínica-laboratorial para muitos casos de MPS.
6.1.2.5- A demora na realização dos exames
A realização dos exames para avaliação do comprometimento orgânico dos pacientes com MPS pelo SUS foi limitante para a avaliação adequada de muitos casos de MPS, devido a morosidade da realização dos exames. Como esse trabalho foi observacional sem patrocínio para realização de exames e custeio de avaliações, os pacientes realizaram exames/consultas pela rotina de assistência clínica.
6.1.2.6- A alta morbi-mortalidade dos pacientes
A redução da expectativa de vida dos pacientes com MPS impediu a inclusão de alguns sujeitos da pesquisa e a realização da avaliação clínico-genética completa em alguns casos.
6.1.2.7-Estudo molecular
Para alguns casos não foi possível a determinação da mutação em questão, apesar de ter sido procuradas as mutações mais frequentes para esses tipos de MPS (tabela 6). Seria necessário realizar o sequenciamento do DNA para alguns casos, o que não foi possível nesse estudo.
6.1.2.8-Relação genótipo-fenótipo
Não foi possível determinar a relação genótipo-fenótipo para a maioria dos casos de MPS em que a mutação foi identificada, exceto para os casos de MPS I.
6.2- Mérito, originalidade, e contribuição da publicação
O artigo “A Clinical Study Of Orofacial Features In 26 Brazilian Patients With Different Types Of Mucopolysaccharidosis” incluiu a avaliação
Cals e Departamento de Odontologia da Universidade Federal do Ceará), envolvendo profissionais de diversas áreas de saúde.
O artigo “ Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI” foi uma colaboração de vários serviços de genética do país, que reuniu o maior número de casos do mundo com MPS VI com idade inferior a 5 anos com resultado de avaliação clínica multiprofissional relacionada com a terapia de reposição enzimática (TRE), contribuindo para aumentar o conhecimento da comunidade científica sobre tratamento precoce de MPS VI. Existe a necessidade de acompanhamento a longo prazo dessas crianças para posteriormente realizar uma nova publicaçãoo que mostre os resultados de TRE a longo prazo.
6.3- Evolução intelectual na trajetória do doutorado
Durante o período do doutorado foram adquiridos conhecimentos sobre bioestatística, metodologia científica e redação do artigo científico que contribuíram para o enriquecimento intelectual e científico da doutoranda. A revisão de literatura sobre MPS foi realizada periodicamente, fazendo com que a doutoranda estivesse em constante atualizaçãoo científica sobre MPS.
Além disso, outros estudantes e profissionais de saúde tiveram um enriquecimento de seus conhecimentos sobre MPS a partir do estabelecimento da linha de pesquisa em MPS no Ceará.
6.4- Metas atingidas e perspectivas de progresso 6.4.1-Delineamento do perfil das MPS no Ceará
recebidas, faltas ao tratamento, motivos para as faltas (tabela 17) e mortalidade (tabelas 18 e 19).
6.4.2- Benefícios aos pacientes com MPS
A partir da realização desse trabalho houve uma melhor qualidade de atendimento, acompanhamento e tratamento dos pacientes com MPS no Ceará e a orientação das famílias quanto aos fatos médicos da doença a partir do aconselhamento genético.
6.4.3- Repercussão para profissionais de saúde da região
Realizou-se um trabalho de educação em saúde com orientação aos profissionais de saúde quanto ao diagnóstico precoce dos casos de MPS e necessidade de encaminhamento para centros que permitam o diagnóstico etiológico, principalmente para profissionais das equipes de saúde da família em vários municípios do interior do Ceará. Esse trabalho terá continuidade mesmo depois da defesa da tese.
6.4.4- Repercussão para tomadas de decisão do governo
Apesar da geração de dados que permitam o planejamento estratégico governamental para garantir melhor acesso desses pacientes aos recursos tecnológicos para diagnóstico e acompanhamento do comprometimento multisistêmico de pacientes com MPS, reduzindo as complicações, o custo do tratamento e a morbimortalidade desses casos, não houve interesse do governo sobre esse assunto. O trabalho proporcionou a doutoranda o título de coordenadora do grupo técnico da Secretaria de Saúde do Estado do Ceará para tratamento de doenças metabólicas e a realização de consultorias nesse assunto.
6.4.5- Repercussão para a sociedade
Com a divulgação dos dados sobre MPS foi possível o apoio da sociedade civil para o diagnóstico e tratamento precoce, principalmente das associações de apoio a pacientes com doenças genéticas como a ACDG (Associação Cearense de Doenças Genéticas). Com a educação em saúde continuada combatemos o preconceito quanto aos casos de MPS, já que em alguns tipos a inteligência está preservada e os pacientes podem ser indivíduos economicamente ativos na sociedade.
A partir dos estudos em MPS foi gerada a necessidade de trabalhar também em estudos qualitativos, o que deverá ser efetivado após a defesa do doutorado com esse tema. Essa linha de pesquisa será continuada a partir de estudos multicêntricos e pesquisa com colaboração da equipe multidisciplinar que foi estimulada a trabalhar com MPS a partir desse estudo.
6.5- Inserção em grupos de pesquisa, orientações acadêmicas e perspectivas em programas de pós-graduação
Esse trabalho permitiu que a doutoranda fosse selecionada como a única brasileira a participar do grupo de estudos internacionais em alterações respiratórias de pacientes com MPS II, além da participação como palestrante em vários eventos científicos.
A partir desse trabalho foi possível participar de projetos multicêntricos dos seguintes grupos de pesquisa:
(1) Serviço de Genética Médica da Universidade Federal do Rio Grande do Sul;
(2) Serviço de Genética Médica da UFBA coordenado pela Dra Angelina Acosta em Salvador
(3) Grupo de pesquisa multicêntrico internacional Registry MPS I
(4) Grupo de pesquisa multicêntrico internacional HOS (Hunter Outcome Survey).
A partir do anteprojeto envolvendo as MPS no Ceará foi estimulada a produção de trabalhos de término de curso na graduação na Estácio FMJ e pós-graduação na UNIFOR (Universidade de Fortaleza), além da colaboração no trabalho de pós-graduação da Dra. Raquel Boy na Universidade do Rio de Janeiro. Outros trabalhos de pós-graduação serão desenvolvidos na Universidade Federal do Ceará e no Hospital Geral de Fortaleza envolvendo o tema desse trabalho. Foi construída uma base de pesquisa em MPS no Hospital Infantil Albert Sabin, que hoje é centro de referência para tratamento de doenças metabólicas no estado do Ceará. Esta base de pesquisa é reconhecida nacional e internacionalmente e continuará gerando dados observacionais após a defesa da tese de doutorado.
formação acadêmica a partir das novas perspectivas de estudos e publicações com grupos fortes do país e exterior que trabalham nessa mesma linha de pesquisa.
6.6-Produção científica gerada a partir do projeto de pesquisa
• Participação em 24 eventos científicos nacionais e internacionais • 7 artigos completos publicados em periódicos
6. REFERÊNCIAS
1. Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW et al. eds. The metabolic and molecular basis of inherited disease, vol 3, 8th ed. New York: McGraw-Hill, 2001. p. 3421–37. 2. Giugliani R. Inborn errors of metabolism in Latin America: challenges and
opportunities. J Inherit Metabol Dis 2010; 33 (2): 315-320.
3. Albano LM, Sugayama SS, Bertola DR, Andrade CE, Utagawa CY, Puppi F et al. Clinical and laboratorial study of 19 cases of mucopolysaccharidoses. Rev Hosp Clin Fac Med Sao Paulo 2000; 55: 213-218
4. Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr 1997; 156: 650-654.
5. Azevedo ACMMM, Schwartz IV, Kalakun L, Brustolin S, Burin MG, Beheregaray APC et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin. Gent. 2004; 66:208-213.
6. Coutinho MF, Lacerda L, Alves S. Glycosaminoglycan storage disorders: a review. [internet] Biochem Res Int 2012 [cited 2014 may 20] Avaliable from: http://www.hindawi.com/journals/bri/2012/471325/
7. Giugliani R. Mucopolysaccharidoses: From understanding to treatment, a century of discoveries. Genet Mol Biol 2012; 35 (4 suppl): 924-931 (a).
8. Giugliani R, Federhen A, Rojas MVM, Vieira T, Artigalás O, Pinto LL et al. Mucopolysaccharidosis I, II and VI: Brief review and guidelines for treatment. Genet Mol Biol 2010; 33 (4): 589-604.
9. Sheth J, Patel P, Sheth F, Shah R. Lysosomal storage disorders. Indian Pediatr. 2004; 41 (3): 260-265.
10. Fuller M, Meikle PJ, Hopwood JJ. Glycosaminoglycan degradation fragments in mucopolysaccharidosis I. Glycobiology 2004; 14 (5): 443-450.
11. OMIM-Online Mendelian Inheritance in Man. [internet]. Johns Hopkins University. [cited 2014 may 20]. Avaliable from:
12. Vieira T, Schwartz I, Muñoz V, Pinto L, Steiner C, Ribeiro M et al. Mucopolysaccharidoses in Brazil: What Happens From Birth to Biochemical Diagnosis? Am J Med Genet 2008; 146A:1741–1747.
13. Nelson J. Incidence of the Mucopolysaccharidoses in Northern Ireland. Hum Genet 1997; 101:355–358.
14. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 1999; 105:151–156.
15. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders.JAMA 1999; 281:249–254.
16. Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 2000; 105:e10.
17. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H et al. Prevalence
of lysosomal storage diseases in Portugal. Eur J Hum Genet 2004; 12(2):87–92
18. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlsch€utter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 2005; 28:1011–1017. 19. Malm G, Lund AM, Mansson J, Heiberg A. Mucopolysaccharidoses in the
Scandinavian countries: incidence and prevalence. Actapaediatrica2008; 97: 1577-1581.
20. Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am J Med Genet 2009; 149A: 960– 964.
21. Poupětová H, Ledvinová J, Berná L, Dvořáková L, Kožich V, Elleder M. The
birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33: 387–396.
22. Wegrzyn G, Jakobkiewicz-Banecka J, Narajczyka M, Wisniewski A, Piotrowska E, Gabig-Ciminska M et al. Why are behaviors of children suffering from various neuronopathic types of mucopolyssacharidoses different? Med Hypotheses 2010; 75: 605-609.
24. Schwartz IVD, Ribeiro M, Mota JG, Toralles MBP, Correia P, Horovitz D. et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta paediatr 2007; 96: 63-70.
25. Wood TC, Harvey K, Beck M, Burin MG, Chien Y, Church H et al. Diagnosing mucopolysaccharidosis IV A. J. Inherit Metab Dis 2013; 36: 293-307.
26. Ruijter J, Broere L, Mulder MF, Van der Ploeg AT, Rubio-Gozalbo ME, Wortmann SB, et al. Growth in patients with mucopolysaccharidosis type III (Sanfilippo disease). J Inherit Metab Dis out 2013 (epub ahead of print )
27. Deckera C, Yub Z, Giugliani R, Schwartz IV, Guffond N, Telese EL et al. Enzyme replacement therapy for mucopolysaccharidosis VI: growth and pubertal development in patients treated with recombinant human N-acetylgalactosamine 4 sulfatase. J Pediatr Rehabil Med 2010; 3 (2): 89-100.
28. Martins AM, Dualibi AP, Norato D, Takata ET, Santos ES, Valadares ER et al. Guidelines for the Mucopolysaccharidosis Type I. J Pediatr 2009; 155 (4): S32-46. 29. Burton B, Giugliani R. Diagnosing Hunter syndrome in pediatric practice:
practical considerations and common pitfalls. Eur J Pediatr 2012; 171 (4): 631-639. 30. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S.
Mucopolysaccharidosis VI. Orphanet J Rare Dis2010; 5 (5): 1-20.
31. Ashworth JL, Biswas S, Wraith E, Lloyd C. Mucopolysaccharidoses and the Eye. Surv Ophthalmol 2006; 51: 1-17.
32. Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol 2013; 24 (5): 379-388.
33. Keith O, Scully C, Path MRC, Weidmann GM. Orofacial features of Scheie (Hurler-Scheie) syndrome (α-L-iduronidase deficiency). Oral Surg Oral Med Oral Pathol 1990; 70: 70-74.
34. Antunes LA, Nogueira AP, Castro GF, Ribeiro MG, de Souza IP. Dental findings and oral health status in patients with mucopolysaccharidosi: a case series. Acta Odontol Scand 2013; 71 (1): 157-167.
35. Alpoz AR, Çoker M, Çelen E, Ersin NK, Gokçen D, van Diggelenc OP et al. The oral manifestation of Maroteaux-lamy syndrome (mucopolysaccharidose VI): a case report. Oral Surg. Oral Med. Oral Pathol Oral Endod2006; 101: 632-637. 36. Downs AT, Crisp T, Ferretti G. Hunter’s syndrome and oral manifestations: a
review. Am Academ Pediatr Dent 1995; 17 (2): 98-100.
et al. A. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis 2014; 37 (2): 263-268.
38. Freitas DQ, Tempest LM, Sicoli E, Lopes-Neto FC. Bilateral dentigerous cysts: review of the literature and report of an unusual case. Dentomaxillofac Radiol 2006; 35: 464-468.
39. Roberts MW, Barton NW, Constantopoulous G, Butler DP, Donahue AH. Occurrence of multiple dentigerous cysts in a patient with Maroteaux-Lamy syndrome (mucoplysaccharidosis, type VI). Oral Surg. 1984; 50: 169-175.
40. Cleary MA, Wraith JE. The presenting features of mucopolysaccharidosis type I (Hurler syndrome). Acta Paediatr 1995; 84: 337-339.
41. Guven G, Cehreli ZC, Altun C, Sençimen M, Ide S, Bayari SH et al. Mucopolysaccharidosis type I (Hurler-Scheie): Oral and radiographic findings and ultrastructural / chemical features of enamel and dentin. Oral Pathol Oral Radiol Endod 2008; 105 (1): 72-78
42. Kinirons MJ, Nelson J. Dental findings in mucopolysaccharidosis type IV A (Morquio’s disease type A). Oral Surg Oral Med Oral Pathol 1990; 70: 176-9.
43. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in Mucopolysaccharidoses. Paediatr Respir Rev 2011; 12: 133-138.
44. Lin H, Chen M, Lin C, Chen C, Lin D, Chuang C et al. Polysomnographic Characterisitics in Patients With Mucopolysaccharidoses. Pediatr Pulmonol 2010; 45:1205-1212.
45. Berger KI, Fagondes SC, Giugliani R, Hardy KA, Lee KS, Mc Ardle C, Scarpa
M, Tobin MJ, Ward SA, Rapoport DM. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metabol Dis 2013; 36: 201-210.
46. Van den Broek L, Backx AP, Coolen H, Wijburg FA, Wevers R, Morava E et al. Fatal coronary artery disease in an infant with severe mucopolysaccharidosis type I. Pediatr 2011; 127(5):e1343-1346.
47. Dangel J. H. Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders – clinical and echocardiographic findings in 64 patients. Eur J Pediatr 1998; 157: 534-538.
49. Brands MMMG, Frohn-Mulder IM, Hagemans MIC, Hop WCJ, Oussoren E, Helbing WA et al. Mucopolysaccharidosis: Cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI. J Inherit Metabol Dis 2013; 36: 227-234.
50. Muenzer J, Wraith JE, Clarke LA and the International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatr 2009; 123: 19-29.
51. Beck M. Mucopolysaccharidosis Type II (Hunter Syndrome): clinical picture and treatment. Curr Pharm Biotechnol 2011; 12 (6):861-866.
52. Palmucci S, Attina G, Lanza ML, Belfiore G, Capello G, Foti PV et al. Imaging findings od mucopolysaccharidoses: a pictoral review. Insights Imaging 2013; 4: 443-459.
53. Lachman R, Martin KW, Castro S, Basto MA, Adams A, Teles EL. Radiologic and neurorradiologic findings in the mucopolysaccharidoses. J Ped Rehabil Med: An Interdisciplinary Approach 2010; 3: 109-118.
54. Rasalkar DD, Chu WCW, Hui J, Chu C-M, Paunipagar BK, Li C-K. Pictorial review od mucopolysaccharidosis with emphasis on MRI features of brain and spine. The British J Radiol 2011; 84: 469-477.
55. Aliabadi H, Reynolds R, Powers CJ, Grant G, Fuchs H, Kurtzberg J. Clinical outcome of cerebrospinal fluid shunting for communicating hydrocephalus in mucopolysaccharioses I, II and III: a retrospective analyses of 13 patients. Neurosurg 2010; 67 (6): 1476-1481.
56. Seto T, Kono K, Morimoto K, Inuone Y, Shintaku H, Hattori H et al. Brain Magnetic Resonance Imaging in 23 Patients with Mucopolysaccharidoses and the Effect of Bone Marrow Transplantation. Ann Neurol 2001; 50: 79-92.
57. Verity C, Winstone AM, Stellitano L, Will R, Nicoll A. The epidemiology of progressive intellectual and neurological deterioration in childhood Arch Dis Child 2010;95:361–364
59. White K, Kimb T, Neufeld JA. Clinical assessment and treatment of carpal tunnel syndrome in the mucopolysaccharidoses. J Pediatr Rehabil Med 2010: 3: 57–62.
60. .Castilhos RM, Blank D, Netto COB, Souza CFM, Fernandes LNT, Schwartz IVD et al. Severity score system for progressive myelopathy: development and validation of a new clinical scale. Braz J Med Biol Res 2012; 45: 565-572.
61. Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Rujter GJG, Wevers RA et al. Mucoplysaccharidosis Type IIIA: Clinical Spectrum and Genotype-Phenotype Correlations. Ann Neurol 2010; 68: 876-887.
62. Kariya S, Schachern PA, Nishizaki K, Paparella MM, Cureoglu S. Inner Ear Changes in Mucopolysaccharidosis Type I/Hurler Syndrome. Otol Neurotol 2012; 33: 1323-1327.
63. Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Munoz V et al. Recognition and Diagnosis of Mucopolysaccharidosis II (Hunter Syndrome). Pediatrics 2008; 121: e377-e386.
64. Leister S, Giugliani R. A useful routine for biochemical detection and diagnosis of mucopolysaccharidoses. Genet. Mol. Biol 1998; 21 (1):163-167.
65. Muller KB, Rodrigues MDB, Pereira VG, Martins AM, DÁlmeida V. Reference values for lysosomal enzymes activies using dried blood spots samples – a Brazilian experience. Diagn Pathol [internet] 2010; 5: 65.[cited 2014 may 20]. Avaliable from: http://www.diagnosticpathology.org/content/5/1/65
66. Giugliani R. Newborn screening for lysosomal diseases: current status and
potential interface with population medical genetics in Latin America. J Inherit Metab Dis 2012; 5 (5): 871-877 (b).
67. Lopez-Marin L, Gutierrez-Solana LG, Azuara LA, de Las Heras RS, Rodriguez AD, Extremera VC. Detection by Urinary GAG Testing of Mucopolysaccharidosis Type II in an At-Risk Spanish Population. JIMD Rep 2013; 10: 61-68.
68. Lage S, Prieto JA, Andrade F, Sojo A, Sanjurjo P, Aldamiz-Echevarria L J.
Reability of a Visual Test for the Rapid Detection of Mucopolysaccharidoses: GAG-Tests. J Clin Lab Anal 2011; 25: 179-184.