rev bras hematol hemoter. 2017;39(1):80–83
w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Case
report
A
combination
of
the
-
␣
3.7
and
–
MEDII
alleles
causing
hemoglobin
H
disease
in
a
Brazilian
patient
Roberta
Dorta
Ferreira,
Natália
de
Oliveira
Mota,
Elza
Myiuki
Kimura,
Gisele
Audrei
Pedroso,
Maria
de
Fatima
Sonati
∗UniversidadeEstadualdeCampinas(Unicamp),Campinas,SP,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received25November2016 Accepted1December2016 Availableonline28December2016
Introduction
Alpha-thalassemiaisahereditarydiseasewithaworldwide distributioncharacterizedbyreducedorabsentsynthesisof hemoglobin␣chains.Deletionsinvolvingthe␣globingenes, whichareduplicated(␣2 and␣1)andlocatedinthe␣ clus-ter(16p13.3),arethemostcommoncausesofthediseaseand accountforover80%ofcases.Lossofafunctional␣geneinthe haploidgenomeresultsin␣+-thalassemia,whichcanoccurin aheterozygous(-␣/␣␣)orhomozygous(-␣/-␣)state,whileloss ofboth␣genesresultsin␣0-thalassemia,whichcanalsooccur inaheterozygous(–/␣␣)orhomozygous(–/–)state.Afifth␣ -thalassemicgenotypeistheresultofthecombinationofboth the␣0and ␣+ alleles (-␣/–).While thefirstthreegenotypes areassociatedwithminimalhematologicalchangesandthe fourthresultsinhemoglobin(Hb)Bart’shydropsfetaliswith intrauterineorneonataldeath,thedoubleheterozygous␣0/␣+
(-␣/–)stateleadstoHbHdisease.Thislatterischaracterized byunstablechaintetramers(4),causingchronic,moderate toseverehemolyticanemiawithmicrocytosis,hypochromia, jaundiceandhepatosplenomegaly.1,2
∗ Correspondingauthorat:DepartmentofClinicalPathology,SchoolofMedicalSciences,UniversidadeEstadualdeCampinas(Unicamp),
Campinas,SP,Brazil.
E-mailaddress:[email protected](M.deFatimaSonati).
Therearesevendeletionsthatusuallyaffectpopulations aroundtheworld:[-␣3.7,-␣4.2,-(␣)20.5,–MED,–SEA,–FIL,–THAI]. Themostcommonmethodusedtoscreenforthesedeletions ismultiplex-gappolymerasechainreaction(PCR).3Whenthe molecularbasisofthediseasecannotbeidentifiedinthisway, multiplexligation-dependentprobeamplification(MLPA)can beusedtodetectneworraredeletionsinthe␣genes,inthe wholecluster andinthe␣-majorregulatoryelement (MRE) located40kbdownstreamofthegene.1,2
WedescribethecaseofaBrazilianpatientwithHbH dis-easecausedbythecombinationofthe-␣3.7deletion,themost commoncauseof␣-thalassemiainpopulations,andararer
␣0deletionidentifiedonlybyMPLA.
Case
report
ThiscasestudywaspartofaprojectapprovedbytheResearch EthicsCommitteeoftheUniversidadeEstadualdeCampinas (Unicamp)underreferencenumber918/2007.
A31-year-oldwhiteBrazilianmaleofItaliandescentfrom Araraquara, in the state of São Paulo, with a diagnosis of
http://dx.doi.org/10.1016/j.bjhh.2016.12.001
revbrashematolhemoter.2017;39(1):80–83
81
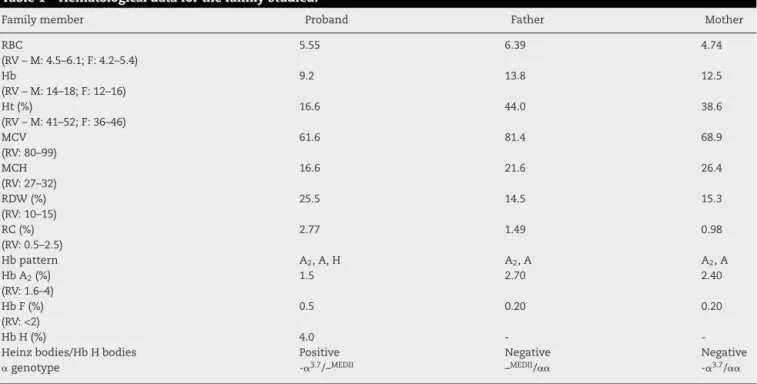
Table1–Hematologicaldataforthefamilystudied.
Familymember Proband Father Mother
RBC
(RV–M:4.5–6.1;F:4.2–5.4)
5.55 6.39 4.74
Hb
(RV–M:14–18;F:12–16)
9.2 13.8 12.5
Ht(%)
(RV–M:41–52;F:36–46)
16.6 44.0 38.6
MCV (RV:80–99)
61.6 81.4 68.9
MCH (RV:27–32)
16.6 21.6 26.4
RDW(%) (RV:10–15)
25.5 14.5 15.3
RC(%) (RV:0.5–2.5)
2.77 1.49 0.98
Hbpattern A2,A,H A2,A A2,A
HbA2(%)
(RV:1.6–4)
1.5 2.70 2.40
HbF(%) (RV:<2)
0.5 0.20 0.20
HbH(%) 4.0 -
-Heinzbodies/HbHbodies Positive Negative Negative
␣genotype -␣3.7/–MEDII –MEDII/␣␣ -␣3.7/␣␣
RV:referencevalues;RBC:redbloodcellcount(×109/L);Hb:hemoglobin(g/dL);Ht:hematocrit(%);MCV:meancorpuscularvolume(fL);MCH: meancorpuscularhemoglobin(pg);RDW:redcelldistributionwidth(%);RC:reticulocytecount(%).
hypochromicmicrocyticanemiawasreferredtoour labora-torytoinvestigatethecauseofhisanemia.Cellcountsand hematologicalindicesweredeterminedusinganautomated hematology analyzer (Sysmex XE5000, Sysmex, Japan) and hemoglobinanalysis wascarried out byelectrophoresison celluloseacetateinalkalineandneutralpHsandby cation-exchange high-performance liquid chromatography (HPLC) (Variant IITM, Bio-Rad Laboratories, Hercules, CA, USA). In
additiontothe hemoglobin(Hb)Aand HbA2 fractions, an
HbHfractionwasdetectedaccountingfor4%ofthetotalHb. Thepatient’sparentswereanalyzed,andalthoughtheydid nothaveanyclinicalcomplaints,bothhadminorhematologic changessimilartothosefoundin␣-thalassemia.
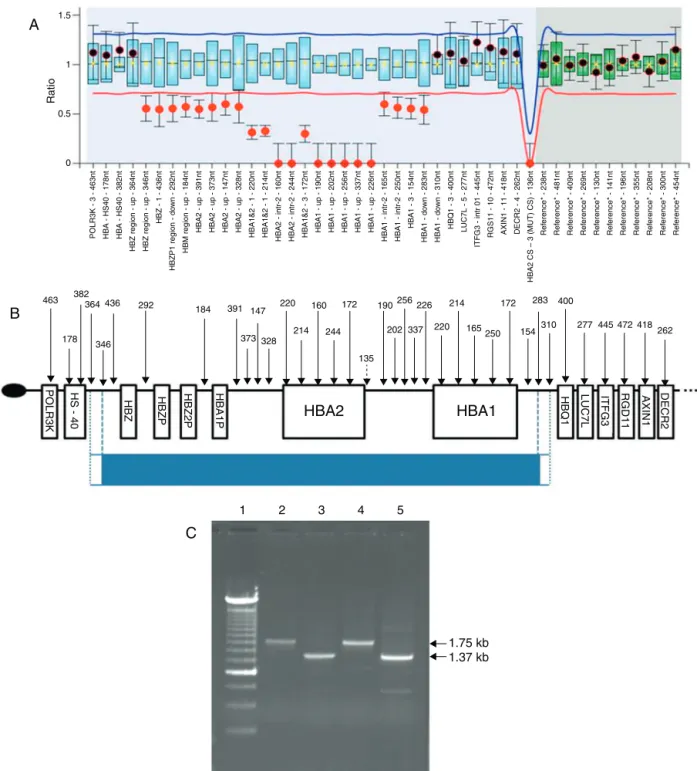
The first molecular analysis consisted of multiplex-gap PCR,3whichshowedapatterninthepatient’ssampleobserved whenthe␣3.7deletionisinahomozygousstate,aresultthat wouldnotexplaintheHbHdisease.Thesampleswerethen analyzedbyMLPAusingtheSALSAMLPAP140C1HBAkit(MRC Holland,Holland),4whichanalyzes approximately360kbof DNAextendingfrom the16ptelomericregiontotheDECR2 gene.ThefragmentswerecomparedinCoffalyser.Netto iden-tifypossiblechangesinthenumberofcopiesofthe␣alleles. In addition tothe -␣3.7 deletion, MLPAdetected a large deletionofapproximately30kbaffectingthe,,␣2,␣1,
␣2 and ␣1 genes (Figure 1A). The extent of this deletion andthe genesaffected are compatiblewithtwo previously describeddeletions:–DUTCH,describedinindividualsofDutch origin,5and–MEDII,describedinindividualsofMediterranean origin.2,6 These can bedistinguishedfrom each other by a specificPCRprotocol.5Here,thepatient’sDNAwasfirst ampli-fied with primers far from the deletion breakpoints, and the product was re-amplified by nested PCR with primers flankingthe breakpoints ofbothdeletions. With the–MEDII
deletion, fragments of approximately 1.35 and 1.75kb are formed(Figure1C),whilewiththe–DUTCHdeletion,the frag-mentsare1.03and1.41kbinsize.Ourresultsindicatethatthe deletioninquestionconsistsofthe–MEDIIdeletion(Figure1B) in combination with the -␣3.7 deletion (-␣3.7/–MEDII) in the patientincombinationwiththenormalallele(–MEDII/␣␣)inthe patient’s father.Thepatient’smother washeterozygousfor the-␣3.7deletion(-␣3.7/␣␣).Thehematologicalandmolecular dataforthefamilyareshowninTable1.
Discussion
Alpha-thalassemiasarefrequentlycausedbydeletions.The mostcommonoftheseisthe-␣3.7deletion,whichhasa preva-lenceof20–25%inAfro-Brazilians.7,8HbHdiseaseissporadic inBrazilandisgenerallycausedbyacombinationofthe-␣3.7
deletionand–MEDI,–SEAor-(␣)20.5deletions.9MLPA,however, hasmadeitpossibletodetectrarerandevennovel␣0 dele-tionsandeventhosethatonlyaffecttheregulatoryelement. Inthefamilyanalyzedhere,HbHdiseasewastheresultofa combinationofthe-␣3.7 alleleand–MEDIIdeletion,agenetic alteration not previouslyreported inthe Brazilian or Latin American population.Thisdeletionislargerthan the–MEDI (whichisapproximately17kbofDNA)andremovesthezeta gene in addition tothe alpha genes. It hasbeen foundin Mediterranean countries, such asItaly, Turkey, Greece and Cyprus.6,10
82
revbrashematolhemoter.2017;39(1):80–831.5
1
0.5
Ratio
0
463 382364 436 292
184 391
178
POLR3K HS - 40
HBZ HBZP HBZ2P HBA1P HBQ1 LUC7L ITFG3 RGD11
HBA2
HBA1
346
1 2 3 4 5
1.75 kb 1.37 kb
POLR3K - 3 - 463nt HBA - HS40 - 178nt HBA - HS40 - 382nt
HBZ region - up - 364nt HBZ region - up - 346nt HBM region - up - 184nt
HBA2 - up - 391nt HBA2 - up - 373nt HBA2 - up - 147nt HBA2 - up - 328nt
HBA1&2 - 1 - 220nt HBA1&2 - 1 - 214nt HBA2 - intr-2 - 160nt HBA2 - intr-2 - 244nt HBA1&2 - 3 - 172nt HBA1 - up - 190nt HBA1 - up - 202nt HBA1 - up - 256nt HBA1 - up - 337nt HBA1 - up - 226nt HBA1 - intr-2 - 165nt HBA1 - intr-2 - 250nt
HBA1 - 3 - 154nt
HBA1 - do
wn - 283nt
HBZP1 region - do
wn - 292nt
HBZ - 1 - 436nt
HBA1 - do
wn - 310nt
HBQ1 - 3 - 400nt LUC7L - 5 - 277nt
ITFG3 - intr 01 - 445nt
RGS11 - 10 - 472nt AXIN1 - 11 - 418nt DECR2 - 4 - 262nt Ref
erence* - 238nt
Ref
erence* - 481nt
Ref
erence* - 409nt
Ref
erence* - 269nt
Ref
erence* - 130nt
Ref
erence* - 141nt
Ref
erence* - 196nt
Ref
erence* - 355nt
Ref
erence* - 208nt
Ref
erence* - 300nt
Ref
erence* - 454nt
HBA2 CS – 3 (MUT) CS) - 136nt
147
373 220
220 277445472418
250 214
214
244 160
165 154 310 283
172 172
135 190256
202 226
328
337
400
262
AXIN1 DECR2
A
B
C
Figure1–(A)GraphshowingtheresultfortheprobandgeneratedbytheCoffalyser.Netsoftware.Thex-axisrepresentsthe
probesandthey-axistheratiooftheintensityoftheprobandsampletothemeanintensityofreferencesamples.Aratioof
1indicatesthepresenceofbothalleles,aratioof0.5thelossofonealleleand0thelossofthatregioninbothalleles.(B)
Schematicrepresentationofchromosome16p13.3.Theovalrepresentsthetelomericregion,thearrowsshowthelocations
oftheprobesandtheboxesthegenes.Thebluelinecorrespondstothedeletedfragment,thedottedlinesdenotethefirst
andlastdeletedprobeandtheregionsbetweenthedottedanddashedlinesshowwherethebreakpointsmaybe(adapted
fromMRC-Holland,2014).(C)Agarosegelwiththenested-PCRamplifiedproduct.The1.75kband1.35kbbandscorrespond
tothe–MEDIIdeletion.5Sample1isthemolecularweightmarker(240bpladder);samples2and3arefromtheproband,and
revbrashematolhemoter.2017;39(1):80–83
83
Financial
support
Thisstudy wascarriedout withfinancialsupportfromthe Fundac¸ão de Amparo à Pesquisa do Estado de São Paulo (FAPESP)(Grantno.2014/00984-3;fellowshipno.2015/21184-8), theConselhoNacionaldeDesenvolvimentoCientíficoe Tec-nológico(CNPq)andtheCoordenac¸ãodeAperfeic¸oamentode PessoaldeNívelSuperior(CAPES)oftheBrazilianMinistryof Education.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
r
e
f
e
r
e
n
c
e
s
1. BunnHF,ForgetBG.Hemoglobin:molecular,geneticand clinicalaspects.Philadelphia,London,Toronto:W.B.Saunders Company;1986.
2. HarteveldCL,HiggsDR.Alpha-thalassaemia.OrphanetJRare Dis.2010;5:13.
3. ChongSS,BoehmCD,CuttingGR,HiggsDR.Simplified multiplex-PCRdiagnosisofcommonsoutheastAsian
deletionaldeterminantsofalpha-thalassemia.ClinChem. 2000;46(10):1692–5.
4.HarteveldCL,VoskampA,PhylipsenM,AkkermansN,den DunnenJT,WhiteSJ,etal.Nineunknownrearrangementsin 16p13.3and11p15.4causing␣andthalassaemia
characterizedbyhighresolutionmultiplexligationdependent probeamplification.JMedGenet.2005;42(12):922–31.
5.HarteveldKL,LosekootM,FoddeR,GiordanoPC,BerniniLF. TheinvolvementofAlurepeatsinrecombinationeventsat the␣-globingenecluster:characterizationoftwo
␣◦-thalassaemiadeletionbreakpoints.HumGenet. 1997;99(4):528–34.
6.KutlarF,Gonzalez-RedondoJM,KutlarA,GurgeyA,AltayC¸, EfremovGD,etal.Thelevelsofzeta,gammaanddeltachains inpatientswithHbHdisease.HumGenet.1989;82(2):179–86.
7.SonatiMF,FarahSB,RamalhoAS,CostaFF.Highprevalenceof alpha-thalassemiainaBlackpopulationofBrazil.
Hemoglobin.1991;15(4):309–11.
8.SonatiMF,CostaFF.HemoglobinBart’sinaBrazilianblack population.BrazJMedBiolRes.1990;23(5):395–6.
9.WenningMR,KimuraEM,CostaFF,SaadST,GervásioS,de JorgeSB,etal.Alpha-globingenes:thalassemicandstructural alterationsinaBrazilianpopulation.BrazJMedBiolRes. 2000;33(9):1041–5.