Cytoprotective
effect
of
1-nitro-2-phenylethane
in
mice
pancreatic
acinar
cells
subjected
to
taurocholate:
Putative
role
of
guanylyl
cyclase-derived
8-nitro-cyclic-GMP
Franc
¸
ois
Cosker
*
,
Francisco
J.B.
Lima,
Saad
Lahlou,
Pedro
J.C.
Magalha˜es
BiomedicalInstituteoftheBrazilianSemiarid(INCT-IBISAB-CNPq),DepartmentofPhysiologyandPharmacology,SchoolofMedicine,FederalUniversityof
Ceara´,Fortaleza,Ceara´ 60430-270,Brazil
1. Introduction
Acute pancreatitis (AP) is a common reason for hospital admission [1]. Its annual incidence ranges from 13 to 45 per 100,000personsworldwide[2–4].Initsnaturalhistory,theroleof reactiveoxygenspecies(ROS)productioninacinarcellshaslong
been debated and the efficacyof antioxidant regimen was not consistentlyestablished[5,6].Apartfromtheirdirectdetrimental oxidativeeffects,ROSandreactivenitrogenspecies(RNS)canalso function as second messengers in intracellular signaling [6]. Emergingevidencehasrevealedthatnitricoxide(NO)andROScan mediatesignaltransductionthatmaintainstheredoxstateofthe cell[7].Inmacrophages[8],astrocytes[9],heartmusclecells[10], and plant guard cells [11], NO and O2 react togethertoform
ONOO ,whichreactswithguanosinetriphosphate(GTP)toyield thenitrationproduct8-nitroguanosine-50-triphosphate (8-nitro-GTP), a substrate for soluble guanylate cyclase (sGC) [12] that yields 8-nitroguanosine 30,50-cyclic monophosphate (8-nitro-cGMP).Thiscompoundisauniquesecondmessengerimplicated in the regulation of ROS signaling by reacting with particular proteins that possess a thiolmoiety toform cGMP adducts (S-guanylationreaction)[13].
In the pancreas, NO promotesthe exocytosis ofvesicles and secretory granules to prevent the intracellular accumulation of activated digestive enzymes during AP-inducing stimulus. By BiochemicalPharmacology91(2014)191–201
ARTICLE INFO
Articlehistory:
Received6June2014 Accepted28July2014 Availableonline12August2014
Keywords:
Acutepancreatitis Exocrinepancreas Bilesalts Guanylylcyclase 8-Nitro-cGMP
ABSTRACT
Thenitroderivative1-nitro-2-phenylethane(NPE)wasrecentlydescribedasacompoundpossessing heme-dependentsolubleguanylylcyclase(sGC)stimulatingpropertiesinvascularsmoothmusclecells. Inthisstudy,wetestedsuchpharmacologicalpropertyofNPEinmicepancreaticacinarcellssubjectedto thebilesalttaurocholate,atypeofpathologicalstimulusthatsimulatespancreatitis.Here,isolated acinarcellsweretreatedwithNPEinordertoassesstheroleofsGConthedetrimentaleffectsinducedby taurocholate.NPEreducedtaurocholate-elicitedCa2+overload,productionofreactiveoxygenspecies
(ROS),apoptosis,necrosis,andexertedaprotectiveeffectagainstmitochondrialmembranepotential (DCm)dissipation.TheseNPE-inducedeffectswereabolishedbypretreatmentwithODQandKT5823, andaftertheblockadeofnitricoxide(NO)synthasewithL-NAME,inhibitorsofkeycomponentsofthe
sGC pathway. Contrarily to cGMP that alone increased DCm collapse and cell damage, the cytoprotectiveeffectofNPEonDCmandcellnecrosiswasalmostreproducedby8-nitro-cGMP,a secondmessengergeneratedbysGCunderoxidativestressconditions.Inconclusion,putativesGC stimulationwithNPErevealsitscytoprotectiveprofileonpancreaticcellssubjectedtotaurocholate. Moreover,ROSandNOconjunctlyappeartodrivesGCactivityinpancreaticacinarcellstoimplementan adaptivemechanisminresponsetooxidativeandCa2+stressthrough8-nitro-cGMPsynthesis.
ß2014ElsevierInc.Allrightsreserved.
Abbreviations:DCm,mitochondrialmembranepotential;AP,acutepancreatitis;
8-Br-cGMP,8-bromoguanosine3’,5’-cyclicmonophosphate;CCK,cholecystokinin; CCCP,carbonylcyanidem-chlorophenylhydrazine;FAD,flavinadenine dinucleo-tide;GTP,guanosine-50-triphosphate;LawH
2S,Lawesson’sreagent;L-NAME,N-v -nitro-L-argininemethylesterhydrochloride;mPTP,mitochondrialpermeability transitionpore;NADHandNAD+,nicotinamideadeninedinucleotide;NPE, 1-nitro-2-phenylethane;NO2-cGMP,8-nitroguanosine30,50-cyclicmonophosphate;
NO2-GTP,8-nitroguanosine-5’-triphosphate;ODQ,1H-[1,2,4]oxadiazolo[4,3-a ]quinox-alin-1-one;PI,propidiumiodide;PKG,proteinkinaseG;RNS,reactivenitrogen species;ROS,reactiveoxygenspecies;sGC,solubleguanylatecyclase;NO,nitric oxide;ATP,adenosine triphosphate;H2DCFDA,20,70-dichlorodihydrofluorescein
diacetate;DMSO,dimethylsulfoxide;NOS,nitricoxidesynthase. * Correspondingauthor.Tel.:+558533668334.
E-mailaddress:fffcosker@yahoo.fr(F.Cosker).
ContentslistsavailableatScienceDirect
Biochemical
Pharmacology
j our na l ho me p a ge : w ww . e l se v i e r . com / l oc a te / b i och e mph a rm
http://dx.doi.org/10.1016/j.bcp.2014.07.030
stimulatingsGCenzymaticactivity,NOalsoexertsacytoprotective effectsagainstotherinjuriousstimulitopancreaticacinarcells[14– 16]possibly by counteracting the intensity of pathological Ca2+
signalstowardmorenormalpatterns[17].Interestingly,cGMPitself
[18,19]doesnotreduceCa2+signalsinpancreaticacinarcells.Thus,
itishypothesizedthatthebeneficialactionsmediatedbytheNO/ sGCpathwayinahighROSproduction statecouldpreferentially involvethesynthesisof8-nitro-cGMPinsteadofcGMPalone.
Inthepresentstudy,wetestedsuchhypothesisbyevaluating the effects of 1-nitro-2-phenylethane (NPE), a compound that inducesNO-independentsGCstimulationinaorticsmoothmuscle
[20,21],infreshlyisolatedpancreaticacinarcellssubjectedtoanex vivomodelofAP.Ourresultsstronglyarguefortheinvolvementof sGC stimulation induced by NPE, which decreases both the intensity of Ca2+ signaling and ROS production in pancreatic
acinarcellsinresponsetothedetrimentalstimuluspromotedby thebilesalttaurocholate.Moreimportantly,NPEisrevealedasa potentcytoprotectiveagentthatmaintains
DC
manddecreases the rate of cell apoptosis and necrosis. Finally, appears to be revealedas animportant productof sGC in this cytoprotective effectofNPE.2. Materialsandmethods
2.1. Preparationofmousepancreaticacinarcells
ThestudywasapprovedbyourEthicsCommittee(protocolno. 76/2013).Pancreaticacinarcellswerefreshlyisolatedfrommale Swiss mice (25–30g). The mouse was sacrificed by cervical dislocation,and cells were isolated by mechanical dissociation afterincubationwithcollagenaseIa(8min,75UI)aspreviously described[22].Pancreaticacinar cellswereusedwithin4h. To simulateAP,cellswereincubatedwith6mMtaurocholateat378C for40–60minundervariousconditionsdetailedbelow.
2.2. ROSmeasurements
CellswereincubatedwiththeROSindicator20,70 -dichlorodi-hydrofluoresceindiacetate(H2DCFDA)(3
m
mol/l)for35minawayfrom light at room temperature and then washed to be then disposedonaconfocalmicroscope(excitation/emission:488nm/ 500–550nm)at378C.Thelaserintensitywasloweredto0.8%,and thesamplingratewasreducedto1frame/20s.Aftertaurocholate, cellexposuretoROSwasestimatedbycalculatingtheareaunder thecurve(AUC)over15min.Theresultsareexpressedastheratio offluorescence(F)dividedbytheinitialfluorescence(F0)(F/F0).
2.3. MeasurementofCa2+levels
CellswereincubatedwithFluo-4AM(5
m
mol/l)for30minat roomtemperature.After washing,cells wereseeded on poly-L -lysine-coated coverslips on the top of an inverted confocal microscope(Olympus,IX81)at 378C(excitation/emission:488/ 500–550nm;imagesamplingfrequency:1frame/5s).Theresults areexpressedastheratiooffluorescence(F)dividedbytheinitial fluorescence(F0)(F/F0).2.4. Assessmentofthemitochondrialmembranepotential(
DC
m)Cells were loaded with 10
m
mol/l tetramethylrhodamine methylester (TMRM), or with rhodamine 123 (10m
M) for 10minat378C. Attheconcentrationsapplied, thefluorescence wasquenchedwhenthedyewasaccumulatedbymitochondriaTMRM-loadedcellswereseededonpoly-L-lysine-coated cover-slipsonthetopofaninvertedconfocalmicroscope(Olympus,IX81) at378Candmonitored15minafterseeding(excitation/emission:
543/550–650nm;imagesamplingfrequency:1frame/10s).Allthe perfusion solutions contained 300nM of the dye to avoid any leakage.TheresultsareexpressedastheratioF/F0.Anincreasein
DC
mmanifestsbyadecreaseintheTMRMratioandviceversa. Rhodamine123-loadedcells werepreincubated(8min)with NPE,LawH2S,KT5823,8-Br-cGMP,NO2-cGMP,ODQ,orL-NAME. Taurocholatewasthenaddedfor1hat378Candcellimageunder aconfocalmicroscope(488/500–550nm).Mitochondrial depolar-ization induces dye diffusion throughout the cytosol. After anonymization, cells were counted and the loss of contrast of mitochondriawasinterpretedasacollapseofDC
m.2.5. Apoptosisandnecrosisquantification
Cell apoptosis activation was detected using a caspase substrate, the rhodamine 110/aspartic acid amide (10
m
mol/l), whichreleasestherhodaminedyewithinthecellduringapoptosis. Propidiumiodide(PI)(10m
g/ml)wasusedtodetectcellnecrosis. The cells were treated for 1hwith taurocholate and NPE and loaded with rhodamine 110/aspartic acid amide and PI. After washing,cellswereloaded(5min)withthenuclearstainHoechst 33342(50m
g/ml)forcellcounting.Fluorescenceevaluationwas performed sequentially under confocal microscopy (Olympus, IX81; 488/500–530nm for R110/aspartic acidamide, 488/560– 660nm for PI, and 405/425–475nm for Hoechst 33342). A minimumof200cellswascountedforeachcondition.2.6. Chemicals
Fluo-4AM,rhodamine123,rhodamine110/asparticacidamide, andHoechst 33342werefromInvitrogen(CARLSBAD,CA,USA). Poly-L-lysine, PI, Law H2S, collagenase Ia, H2DCFDA, carbonyl
cyanidem-chlorophenylhydrazone(CCCP),sodiumnitroprusside, N-
v
-nitro-L-arginine methyl ester hydrochloride (L-NAME), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one(ODQ),8-Br-cGMP,and KT 5823 were from SIGMA1 (St. Louis, MO, USA). NPE wasextracted and purified as previously described [23]. It was dissolved in DMSO (SIGMA1, finalconcentration<0.02%). The
extracellularsolutioncontained140mMNaCl,4.7mMKCl,10mM HEPES,1.13mMMgCl2,1mMCaCl2,and10mMglucose,pH7.4
withHCl(SIGMA1).8-Nitro-cGMPwaskindlygiftedbyDr.Takaaki
Akaikegroup(TohokuUniversity,Sendai,Japan).
2.7. Statisticalanalysis
Thestatisticalanalysisofresultswasperformedusingone-way analysisofvariance(ANOVA)followedbytheBonferroniposthoc testorStudent’st-testwhenappropriate.ValuesofP<0.05were
considered statisticallysignificant. Theresultsare expressed as meanSEM.
3. Results
3.1. Preventionofthe
DC
mcollapsebyNPEFirstly, cells were incubated with rhodamine 123, which accumulated and remainedinside themitochondria (A; T=0s, upper-left image). The dissipation of
DC
m by theinhibitor of oxidative phosphorylation carbonyl cyanide m-chlorophenyl hydrazone(CCCP,10m
mol/l)inducedthedyediffusionintothe cytosol and loss of mitochondrial contrast (A; T=10–180s). Similarly,pancreaticacinarcellsexposedtotaurocholate(6mM), exhibitedDC
m depolarization in 70.05.4% of the cells, a percentage significantly higher than in vehicle-treated cells (Fig. 1B; 21.26.4%; n=5; P<0.001).The number of cellsthatsignificantlyreducedwhentheywerepreincubatedfor8minwitha given concentration of NPE (20–325
m
mol/l; n=5). Although nonsignificant at 20m
mol/l, the effect of NPE was maximal at 65m
mol/l(Fig.3E,34.03.2%,P<0.001)andremainedsignificantathigher concentrations (46.015.6 and 49.011.0% for 162 and 325
m
mol/lNPE,respectively).Incubation oftetramethylrhodamine(TMRM)-loaded pancre-aticacinarcellswithNPE(65
m
mol/l)resultedinaslightdecrease inF/F0,indicatingmitochondrialhyperpolarizationthat,at15minrecordingtime,didnotattainstatisticalsignificanceincomparison withcontrol(Fig.1CandD).Asapositivecontrol,CCCP(10
m
mol/l) alsoinducedunequivocaldissipationofDC
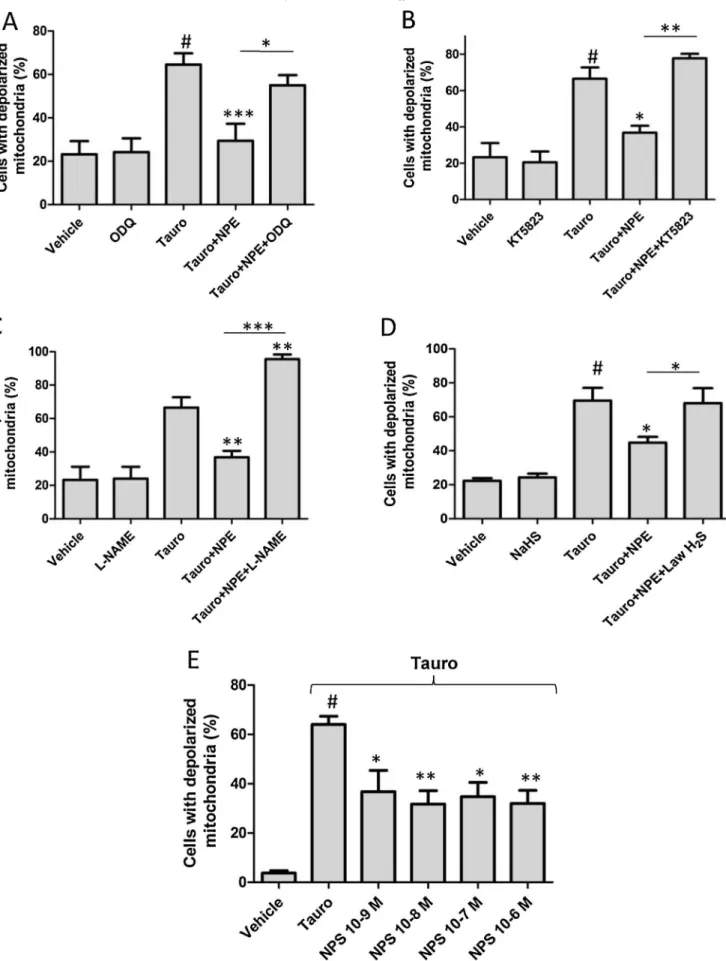
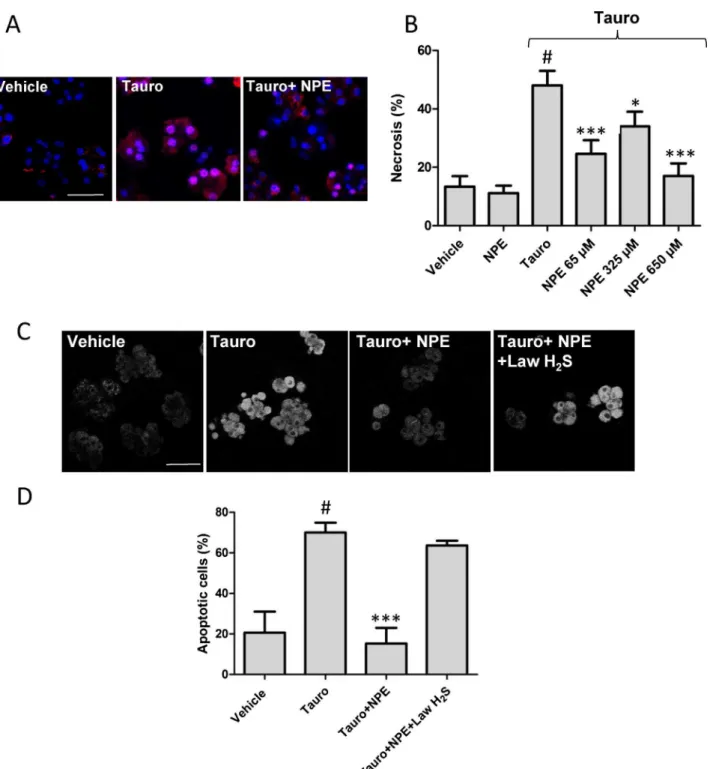
m(Fig.1C)andalostof Fig.1.ProtectiveeffectofNPEagainstmitochondrialdepolarizationcausedbytaurocholate:involvementofthesGC/PKGpathway.In(A)and(B),cellswereincubatedwith rhodamine123.LikeTMRM,rhodamine123accumulatedandremainedinsidethemitochondria(A;T=0s,upper-leftimage).Asinternalcontrol,panel(A)showsimagesof pancreaticacinarcellsincubatedwiththeinhibitorofoxidativephosphorylationCCCP(10mmol/l),whichdissipatedDCm,asindicatedbythedyediffusionintothecytosol andlossofmitochondrialcontrast(A;T=10–180s).In(B),asimilarprotocolusedtaurocholate(6mM;1hincubation)insteadofCCCP.Notice(B)thattaurocholate significantlyincreasedthepercentageofcellswithdepolarizedmitochondria,aphenomenonsignificantlydecreasedinthepresenceofarangeofNPEconcentrations(65– 325mmol/l).In(C)–(E),cellswereincubatedwithTMRM.In(C),NPE(65mmol/l)stabilizedandhyperpolarizedDCm,whiletaurocholate(6mM)inducedaslow depolarization(E).Theresultsareexpressedasmean SEM.,#P<0.001vs.vehicle;*P<0.05,**P<0.01,***P<0.001vs.Tauro(one-wayANOVAfollowedbyBonferroniposthoc test).Additionalcomparisonsbetweentreatmentsareindicatedbythehorizontallinesabovebars.
mitochondrial contrast similar to what we observed with rhodamine123(Fig.3A).Cellstreatedwithtaurocholate(6mM) showedaslowincreaseinF/F0indicativeof
DC
mdepolarization,aphenomenonnot observed when cells were preincubated with NPE (65
m
mol/l; Fig. 1E). This deleterious process ofDC
m depolarizationelicitedbytaurocholatewasslowerandobservable after40minuntil1hincubation(pleaseseefurther).3.2. InvolvementoftheNO/sGCpathwayintheeffectsinducedbyNPE
Fig.2showsthatthemitoprotectiveeffectofNPE(65
m
mol/l) was significantly lost in presence of ODQ, (25m
mol/l; n=5;Fig.2A)orKT5823(1
m
mol/l;n=5;Fig.2B),whichrespectively inhibit sGC and protein kinase G (PKG). Interestingly, in the presenceof Nv-nitro-L-arginine methyl esterhydrochloride (L -NAME;200m
mol/l;Fig.2C),aninhibitoroftheNOsynthase,NPE nomoreprotectedpancreaticcellstosufferDC
mdepolarization inresponsetothebilesalt.Indeed,almostallofthepancreatic cells(95.52.9%)showedDC
mdissipationafter6mM taurocho-lateinthepresenceofthemixtureNPEandL-NAME.Inaseparateset ofexperiments(Fig.2D),themitoprotectiveeffectofNPEwasalso not seen in pancreatic acinar cells treated with the H2S donorLawesson’s reagent (Law H2S; 12.5
m
mol/l), which revealedDC
m dissipation in a percentage similar to control cells (DC
m dissipationtauro/NPE/NaHS=68.08.9%,DC
mdissipation-tauro/NPE=44.83.4%;n=4;P<0.05).ItisnoteworthythatODQ,
KT5823,L-NAMEorLawesson’sreagentdidnotchange
DC
mwhen employedalone.Finally,weassessedthemitoprotectiveeffectofa NO-donor,sodiumnitroprusside(NPS),onpancreaticacinarcells challenged by taurocholate. As shown in Fig. 2E, NPS (1nmol/l to 1m
mol/l) significantly preventedDC
m dissipation at all concentration tested in about 50% of cells (DC
m dis-sipationtauro=64.06.7%,DC
m dissipationtauro/NPS 10–8 mol/l=31.810.8%;n=4;P<0.01).
3.3. NPEdecreasesCa2+overloadandROSproductioninducedbybile acidinjury
Both Ca2+ overloadand high ROS production trigger mPTP
opening,whichinturnfacilitate
DC
mdissipation.Weassessed successivelytheeffectofNPEonthesetwoparametersusingthe lowest protecting NPE (65m
mol/l) concentration which pre-ventsDC
mdissipation.Resemblingthephysiologicalpatternof cholecystokinin(CCK)[22],taurocholate(6mM)promotedCa2+oscillations in virtually all of the Fluo-4 AM-loaded cells (Fig.3A),whichslowlyinducedcytoplasmicCa2+accumulation
followedbyastrongCa2+spike(Fig.3A,arrow),plasmalemmal
disruptionand consequent cell death. In the presence of NPE (65
m
mol/l; Fig. 3B), taurocholate-induced Ca2+ peaks showeddecreased amplitude from 1.280.09 to 0.840.10 (Fig. 3B; n=4;P<0.01),butwithoutchangesintheirfrequency(datanot
shown). The areas under the curve of the Ca2+-elicited waves
causedbytaurocholatewerelowerinthepresenceofNPEthanin itsabsence(Fig.3panelsCandD;AUCtauro=68.09.1,AUCtauro/ NPE=48.17.9; P<0.05). The addition of ODQ (25
m
mol/l) totaurocholate+NPE-treated pancreatic cells abolished (P<0.05)
the reduction of the AUC caused by NPE (Fig. 3D; AUCtauro/ NPE=48.17.9,AUCtauro/NPE/ODQ=67.610.7).
TheindicatorofROSproductionH2DCFDArevealedaslowbut
continuousincreasein[ROS]i(n=5;Fig.3E)incellssubjectedto 6mM taurocholate. NPE (65
m
mol/l) and vehicle both slightly decreasedH2DCFDA-dependentbaselinefluorescencebyrespec-tively 7.25.3% and 7.94.7% (n=7, Fig. 2G). However, only preincubationofNPE(65
m
mol/l)decreasedsharplythe[ROS]Isignalaftertaurocholatechallenge(Fig.3FandG;
D
F/F0tauro=0.40.13,D
F/F0NPE/tauro=0.170.14).3.4. NPEprotectagainstnecrosisandapoptosis
BecauseNPE decreasedboth theintensityof theCa2+signal
inducedbytaurocholateand
DC
mdissipation,wetestedwhether italsopreventscellnecrosis.After1hexposuretotaurocholate (6mM), cells loaded with PI and Hoechst were counted (Fig.4A)and 48.05.0% (n=5) ofthe cellsrevealed necrosis,a value higher than in cells not treated with the bile salt (necrosisControl=13.43.6%;Fig.4B).Asignificantdecreaseintheamountofdeadcellsbynecrosis(24.64.7%;n=5;P<0.001)was
observedinthecellstreatedwithtaurocholateandNPE(65
m
mol/l),a remarkable cytoprotection if we take into account that NPE was added 5min after taurocholate. We also observed that NPE’s cytoprotectiveeffectdecreasedat325m
mol/l(34.011.3%;n=5), whileitalmostinhibitedtaurocholate-inducedeffectat650m
mol/l (Fig.4B)(17.011.7%;n=5).Then, we assessed the cytoprotective effect of NPE in cells challenged40minwithtaurocholate(6mM).Asrevealedbythe fluorescent dye rhodamine 110/aspartic acid amide, a caspase substrate, NPE (65
m
mol/l) completely reversed taurocholate-inducedproapoptoticeffect(Fig.4AandB).Inadifferentsetof experiment,incubationwithLawH2S(12.5m
mol/l),preventedtheprotective effect of NPE against taurocholate. The number of apoptoticcellsunderthesecircumstanceswas63.74.0%(n=3),a valuethatdidnotattaindifferenceincomparisonwithtaurocholate group(P>0.05).
3.5. NO2-cGMP,butnotcGMP,protectsagainsttaurocholate-induced collapseof
DC
mFig. 5A shows that whereascontrol cells had the indicator rhodamine 123 accumulated inside the mitochondria (left image), cells treated with taurocholate (6mM) showed dye diffusionintothecytosol(rightimage),aphenomenonindicative of
DC
mdepolarization.ThepercentageofcellswithdepolarizedDC
minresponsetotaurocholatewas61.22.6%(n=5,P<0.05)andpreincubationwithagivenconcentrationof8-bromoguanosine 30,50-cyclicmonophosphate(8-Br-cGMP;0.3–100
m
mol/l)wasnot abletodecreasethisrate.Indeed,theamountofcellsshowingDC
m depolarization was significantly higher in the presence of low concentrationof8-Br-cGMP(1m
mol/l,74.45.2%;P<0.05)thaninitsabsence(Fig.5B).Inaseparatesetofexperiments(Fig.5C), pancreaticacinarcellsshowedasignificantdecreaseinthenumber ofcellswithtaurocholate-induced
DC
mdissipationwhen incubat-edwitharangeof8-nitro-cGMPconcentrations(0.3–100m
mol/l) (DC
m dissipationTauro=76.22.5%,DC
mdissipationTauro/8-nitro-cGMP100mmol/l=52.04.2,n=5,P<0.001).
3.6. NO2-cGMP,butnotcGMP,protectsagainsttaurocholate-induced necrosis
Finally, we determinedthe effect of different concentrations (1–100
m
mol/l)of8-Br-cGMP(Fig.6A)and8-nitro-cGMP(Fig.6B) on taurocholate-induced cell necrosis. Incubation of pancreatic acinar cell with 8-Br-cGMP (1–100m
mol/l) only tended to increase taurocholate-induced cell necrosis (Fig. 6A; necrosisTauro=46.51.2%, necrosisTauro/8-Br-cGMP at 1 mmol/l=52.82.2%),butthiseffectfailedtobestatisticallysignificant(P=0.053, n=4). Contrarily, 8-nitro-cGMP clearly decreased the amount of deadcells.Amongtheconcentrationstested,1
m
mol/l8-nitro-cGMP revealed thehighest protecting effect against taurocholate-elicited necrosis(Fig.6B;necrosisTauro=47.83.1%,necrosisTauro/8-nitro-cGMP1mmol/l=31.42.2%,n=5,P<0.001).Intriguingly,thisprotectiveeffect
of8-nitro-cGMPdecreasedwhenhigherconcentrationwereusedand disappearedat100
m
mol/lNO2-cGMP.TotestthehypothesisthataFig.2.ProtectiveeffectofNPEagainstmitochondrialdepolarizationcausedbytaurocholateinvolvestheNO/sGCpathway.Theeffectsoftaurocholatealoneorincombination withNPEwerecomparedinpancreaticcellstreatedwithODQ(25mmol/l)(A)orKT5823(1mmol/l)(B).NoticethateitherODQorKT-5823pretreatmentfullyreversedthe NPE(65mmol/l)-inducedmitoprotectiveeffect.PancreaticcellschallengedwithtaurocholateandNPEintheabsenceorpresenceofL-NAME(200mmol/l)(C)orLawH2S
(12.5mmol/l)(D)revealedthatbothtreatmentsreversedtheNPE-inducedmitoprotectiveeffect,especiallyinL-NAME-treatedcells.TheNO-donor,NPS(1nmol/lto1mmol/
l)(E),alsohasamitoprotectiveeffect.Theresultsin(A)–(E)areexpressedasmean SEM.#P<0.001vs.vehicle;*P<0.05,**P<0.01,***P<0.001vs.Tauro(one-wayANOVA
followedbyBonferroniposthoctest).Additionalcomparisonsbetweentreatmentsareindicatedbythehorizontallinesabovebars.
Fig.3.Taurocholate-inducedCa2+signalsandROSproductionaremodulatedbyNPE.(A–C)RepresentativetracesthatshowtherelativeFluo-4fluorescence(F/F 0)ofcells
treatedwith6mMtaurocholatealone(Tauro,A),afterincubationwith65mmol/lNPE(B),orinthepresenceof65mmol/lNPEplus25mmol/lODQ(C).Thearrow(inA) indicatesplasmamembranedisruption.Theareaundercurve(AUC;D),representingtheoverallCa2+loadinpancreaticacinarcells,wasquantifiedunderdifferentconditions.
its cytoprotective effect at high concentrations, acinar cells were incubated with DL-propargylglycine (PAG), a cystathionine
g
-lyase inhibitor, whichimpairs H2S synthesisand, consequently,8-nitro-cGMPmetabolizationincGMP[10].AsshowninFig.6C,whilePAG (3mM)didnotsignificantlymodifythenecrosisrateofacinarcells
exposed to taurocholate (6mM) and 8-nitro-cGMP (1
m
mol/l) in comparisonwithcellsnotexposedtoPAG,thecytoprotectiveeffectof thehigher8-nitro-cGMPconcentration(50m
mol/l)wassignificantly improved(necrosisTauro/8-nitro-cGMP1mmol/l=35.54.3%,necrosisTauro/8-nitro-cGMP50mmol/l=29.22.5%,n=4).
florescence(F/F0)recordedinpancreaticcellsexposedto6mMtaurocholatealone(E)orinpresenceof65mmol/lNPE(F).ROSexposurecausedbytaurocholatewas
quantifiedastheDF/F0withorwithoutpreincubationwithNPE(G).Theresultspresentedareexpressedasmean SEM.*P<0.05,#P<0.001vs.vehicle;***P<0.001vs.Tauro (one-wayANOVAfollowedbyBonferroniposthoctest).Thecomparisonbetweentreatmentsisindicatedbythehorizontallinesabovebars.
Fig.4.NecrosisandapoptosisinpancreaticcellssubjectedtotaurocholatearereducedbytreatmentwithNPE.In(A),imagesshownucleistainedwithHoechst(bluestaining) andnecrosiswasevaluatedbystainingcellswithpropidiumiodide(PI;redstaining),after1htreatmentwithtaurocholate(Tauro,6mM).Noticethedramaticreductionof necrosis(rightimageandpanelB)incellstreatedwith65mMNPEadded5minaftertheinitiationofpancreatitisbytaurocholate(Tauro+NPE).In(C),imagesshow evaluationofapoptosisinpancreaticacinarcellsstainedwithanindicatorofcaspaseactivation(fluorescentgeneralcaspasesubstrate)observed40minaftersaline(vehicle) ortaurocholate(5mM)intheabsence(Tauro)orpresenceofNPE(65mmol/l;Tauro+NPE).In(D),LawH2S(12.5mmol/l)preventedtheprotectiveeffectofNPE(65mmol/l)
againsttaurocholate.Theresultsareexpressedasmean SEM.(#P<0.001vs.vehicle;*P<0.05,***P<0.001vs.Tauro)(One-wayANOVAfollowedbyBonferronitest).Scalebars
20mmol/l.
4. Discussion
GallstonesaretheleadingcauseofAP[24],andtheinterruption ofsuchapathophysiologicalprocessisstillachallenge,especially whenconsideringthatAPcanbelife-threateningandcurrentlyhas nospecifictreatment.Invivo,ductalinfusionoftaurocholateisa widelyemployed model toinduce experimental AP because it reproducespancreaticedema,hemorrhageandnecrosisinseveral animals [25]. In the present study, taurocholate triggered Ca2+
oscillationsthatwereinitiallyverysimilartophysiological Ca2+
signals. Then, over time, we observed a progressive Ca2+
accumulationwhichprecededplasmamembranedisruptionand celldeath.Suchinvitrofindingsarecompatiblewiththeexpected cellphenomenathatareconsideredcrucialtotriggerAPdisease
[26,27].Taurocholateconcentrationsemployedhereinareclose,or evenwithin,thenaturallycriticalmicellarconcentrationrange(1– 5mM)inbileducts.Asamatteroffact,ithasbeenreportedthat taurocholate can reach concentration above 20mM in patients withgallstones[28].Effectsofbileacidswereattributedtotheir interactionswithG-protein-coupledbileacidreceptor-1(GPBAR1) inapicalsideofpancreaticacinarcells[29],butconsideringthe physiologicalroleofbileacidsindietaryfatabsorption,adetergent
action should be also considered. However, the present study showed that changes in the deleterious effects caused by taurocholatemaybeselectivelymodulatedbyNPE,thus preclud-ingtheoccurrenceofunspecificlossofthemembranepermeability inresponsetoputativedetergenteffectsoftaurocholate.
NPEsignificantlyprotectedpancreaticacinarcellsagainstthe deleteriouseffectsoftaurocholateandstimulationofsGCappears tobethemodebywhichitinducessuchactions.Itreducedthe intensityofCa2+signalsandintracellularROSaccumulationand
stabilized
DC
m, resulting in decreases in both apoptosis and necrosis.ThemodulationofCa2+spikesappearstobeinvolvedinthecytoprotectiveeffectofNPEsinceCa2+overloadinpancreatic acinar cells has been implicated as a cardinal event in the establishmentofpancreatitis[30].Inapreviousstudy,ithasbeen demonstratedthattheactivationofsGCdecreasestheintensityof pathological Ca2+ signals induced by CCK in acinar cells and
protectsagainstcaerulein-inducedpancreatitis[17].Similarly,we observedthatNPEdecreasedtheintensityofCa2+signalsinduced
bysupraphysiologicalCCKconcentration(datanotshown). Likeinthevascularsmoothmuscle[20],theeffectsinducedby NPEwerereversedbyODQandKT5823,findingsthatsupportthe involvement of sGC and PKG on its actions. In this study, it Fig.5.ProtectiveeffectofNPEagainstmitochondrialdepolarizationcausedbytaurocholateinvolves8-nitro-cGMPratherthancGMPsynthesis.Rhodamine123accumulated andremainedinsidethemitochondria(A;leftimage,CTnegasnegativecontrol).Incubationwithtaurocholate(6mM)dissipatedDCm,allowingthedyetodiffuseintothe cytosol(A;rightimage).Taurocholate(6mM;1hincubation)significantlyincreasedthepercentageofcells(#P<0.001,vs.vehicle)thatexhibited mitochondrial
appearedlogicalthatcGMP,themostknownproductofsGC,could bedirectlyresponsibleforthesebeneficialeffectsofNPE.However, the cGMP analog 8-Br-cGMP did not reproduce any of its cytoprotective effects. Contrarily, 8-Br-cGMP revealed rather a detrimentalprofileworseningthemaintenanceof
DC
mandcell survivalat amicromolarconcentration. Thus, itisunlikely that cGMPisinvolvedinthemodulationoftaurocholate-inducedCa2+ signalinginducedbyNPE.Inlinewiththishypothesis,ithasbeen reportedthatcGMPactivatesIP3synthesisbyphospholipaseCandthecapacitiveCa2+entryinpancreaticacinarcells[18,31].These
phenomenastronglyparticipatetotheCa2+overloadinvarious
modelsofAP.Thus,thesynthesisofonlycGMPwouldfavorCa2+
overloadbutnotitsreductionperse.
During AP,the Ca2+overloadresponsiblefor cell necrosisis
increased by mitochondria dysfunction [23]. Indeed, bile acids induce an excessive ROS production and a decrease of ATP synthesis[32],whichtogether,mayinduceamarkedinhibitionof
the plasma Ca2+-ATPase [33]. Under the present experimental
conditions,NPE pretreatmentsignificantlyslowed taurocholate-inducedH2DCFDAfluorescenceincrease,reflectingadecreasein
ROS production or an increase in cell antioxidant activity.We hypothesize that NPE-dependent Ca2+modulationcouldnot be
entirely explained by the inhibition of the intracellular ROS accumulation.Indeed,theuseofantioxidantsinAPisnotsufficient topreventcelldeath,anditcanevenincreasecelldeathbynecrosis
[5,6,34].Interestingly,weobservedthatNPE,andNO2-cGMP,also
prevented
DC
mcollapse and couldeven inducemitochondrial hyperpolarization.Noteworthy,8-nitro-cGMPhasbeenreported asanovelNOderivativeinvolvedonsignaltransductioninhealth and disease [34,35].Therefore, it appearsthat NPE, through 8-nitro-cGMP synthesis, maintains mitochondrial integrity and function.Theseresultssupportthehypothesisthat8-nitro-cGMP couldbeasecondmessengersynthesizedunderstressconditions toregulatecelloxidativestate.Nonetheless,itisnoteworthythat Fig.6.8-Nitro-cGMPbutnot8-Br-cGMPreducedcellnecrosisinducedbytaurocholate.Thenumberoftaurocholate-inducednecroticcellswasevaluatedinpresenceof 8-Br-cGMP(1–100mmol/l;panelA)or8-nitro-cGMP(1–100mmol/l;panelB),butonly8-nitro-cGMPdecreasedthenumberofcellsthatenteredinnecrosis.InC,theinhibitionof 8-nitro-cGMPmetabolismbyPAGrestored8-nitro-cGMPdose-dependentcytoprotectiveeffect.Theresultsareexpressedasmean SEM.#P<0.001,vs.vehicle,*P<0.05,**P<0.01,***P<0.001(one-wayANOVAfollowedbyBonferronitest).Additionalcomparisonsbetweentreatmentsareindicatedbythehorizontallinesabovebars.
8-nitro-cGMPonlyprevented
DC
mdissipationbytaurocholateat concentrationsequalorhigherthan10m
mol/l,whileitprevented abouthalfofcellnecrosisat1m
mol/l(pleaseseeFig.6).Atthe presentmoment,wedidnotfindanyclueforsuchdiscrepancybut wecanspeculatethatitcouldbeexplainedbyastrongglycolytic ATPsynthesis tofueltheATPases anda separatestudywillbe neededtoinvestigatewhetherNPEand8-nitro-cGMPrestoreATP productionduringAPstatus.The dissipation of
DC
m is an earlymarker of the onset of apoptosis, whereas its complete collapse can be a marker of necrosis.Ourresultsalsoarguefortheinvolvementofthe sGC/8-nitro-cGMPpathwayinthepreventionofDC
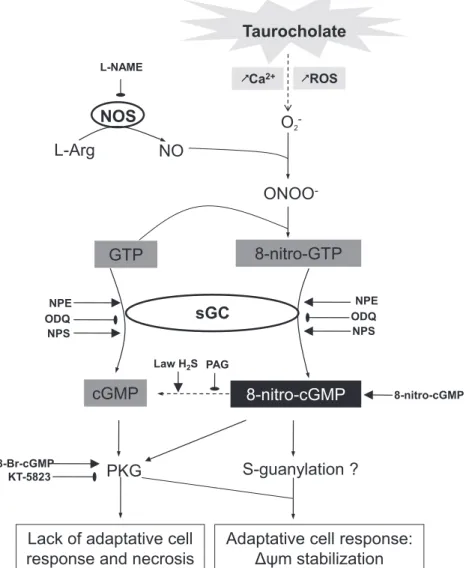
mcollapsebecause theinhibitionofthispathwayeitherbyODQorL-NAMEincreased cellulardamageinducedbythebilesalttaurocholate.Althoughthe relaxanteffectsofNPEoccurredinaNO-independentmannerin vascularsmoothmuscle[20],thepresentfindingsrevealthatNPE somehowrecruitsNOinpancreaticacinarcellstostrengthenits endogenousdefensemechanisms.Interestingly,arecentstudyby Buchwalowetal.reportedupregulationoftheentireNOsignaling machinery in pancreatitis, especially within the exocrine com-partment [35]. Therefore, under stressful NO-producing condi-tions, intracellular nitration reactions could occur to yield byproductssuchasNO2-GTP,whichisconvertedto8-nitro-cGMPbythedualactionofsGC[7].OurexperimentswithL-NAMEand ODQreinforcethehypothesisthatthismechanismactuallypasses
throughtheactivationofsGCanditisnotadirectnitrosylation reactionofcGMP.Putatively,thehighintracellularavailabilityof theelectrophilic nucleotide NO2-GTPmay beimportant tothis
enzymaticconversionandtopreventionofcelldeathinpancreatic acinarcellsduringAP(Fig.7).
Usingapharmacologicalapproach,wefurtherelucidatedhow 8-nitro-cGMPtriggersitsadaptivemechanisminacinarcells.Like cGMP,8-nitro-cGMPactsthroughPKGactivation.Additionally,it actsthroughposttranslationalmodificationsofproteinthiolsvia cGMPadduction(proteinS-guanylation)[13].Indeed,8-Br-cGMP was unable toreproduce the cytoprotective effectof NPE. The experimentwithKT5823showedthatPKGwasnecessaryforthe protective effectof NPE. Such resultssoundimportant because theysupportthehypothesisthatthemitoprotectiveeffectofNPE cannotbeexplainedbycGMP,especiallywithinthephysiological range of concentrations used herein. At low concentration (1
m
mol/l),8-Br-cGMPincreasedDC
mdissipation.Ifthe activa-tion of PKG was necessary but insufficient by itself to confer complete protection for pancreatic cells, thus S-guanylation appearedtohave moreof aprotective influencein thepresent protective effects of NPE. Such reasoning is supported by the reversalofthesemitoprotectiveeffectsafterthesulfhydrationof 8-nitro-cGMPwithLawH2S,confirmingthatthismoleculeensurestheimplementationofanadaptiveresponsetooxidativestressin different cell types [13,36]. Moreover, we showed that the
L-Arg
NO
NOS
O
2-ONOO
-GTP
8-nitro-cGMP
cGMP
sGC
PKG
Tauro
cholate
S-guanylatio
n ?
Adaptativ
e ce
ll res
ponse:
Δψm s
tabiliz
ation
8-nitro-GTP
ODQ NPE
ODQ NPE L-NAME
ROS
KT-5823 8-Br-cGMP
Lack of
adaptative ce
ll
response
and necros
is
LawH2S PAG
8-nitro-cGMP
&
Ca
2+&
NPS NPS
progressivelossofcytoprotectiveeffectsusinghighconcentrations of 8-nitro-cGMP could be reverted be the inhibition of its metabolismincGMP.Theseresultsareinagreementwithprevious resultsincardiactissue[10].
ThepresentstudyrevealsthatthesGCstimulatingpropertiesof NPEispotentiallyofconsiderablepathophysiologicalimportance. NPE prevented necrosis in pancreatic acinar cells exposed to taurocholate,mostprobablythrough8-nitro-cGMPsynthesisby thesGC. Altogether, thesetwo biological products of thesame enzyme, namely sCG, appear to have opposite effects on cell survival in AP (Fig. 7). Supporting this possibility, the total dissipationof
DC
mandcompletelossoftheprotectiveeffectof NPEobservedaftertheblockadeofNOsynthesisbyL-NAMEare highlyrevealing.Indeed,intheabsenceofNO,8-nitro-cGMPcould notbefurther synthesized, andsubsequentsCG stimulationby NPEonly ledtocGMP production and celldeath. Thus, likean adaptationtostresspreviouslydescribedinplantguardcellsin whichcGMP and8-nitro-cGMPwereshowntoplay differential rolesinthesignalingpathwaysandcontrolstomataopeningand closing[11],thepresentstudydemonstratesthatoxidativestress andNOtogethermodulatealsoinvertebratestheeffectsofcGMP andinduceanadaptivemechanisminresponsetooxidativeand Ca2+stressinpancreaticacinarcells.Acknowledgements
TheauthorsthankDr.J.G.Maia,Dr.P.J.Sousa,Dr.M.H.Souza, andDr.L.V.Costa-Lotufoforkindlyprovidinguswith 1-nitro-2-phenylethane,LawH2S,Rhodamine123andPI.Theauthorsalso
expressmanythankstoDr.TakaakiAkaikeforthesynthesisand kind donation of NO2-cGMP. This work was supported by the
ConselhoNacional deDesenvolvimentoCientı´fico eTecnolo´gico (CNPq; grant no. 573928/2008-8 INCT) and Fundac¸a˜o Cearense paraoDesenvolvimentoCientı´ficoeTecnolo´gico(FUNCAP).
References
[1]PeeryAF,DellonES,LundJ,CrockettSD,McGowanCE,BulsiewiczWJ,etal. BurdenofgastrointestinaldiseaseintheUnitedStates:2012update. Gastro-enterology2012;143:1179–87[e1–3].
[2]YadavD, LowenfelsAB.The epidemiologyof pancreatitisand pancreatic cancer.Gastroenterology2013;144:1252–61.
[3]SatohK,ShimosegawaT,MasamuneA,HirotaM,KikutaK,KiharaY,etal. NationwideepidemiologicalsurveyofacutepancreatitisinJapan.Pancreas 2011;40:503–7.
[4]ShenHN,LuCL,LiCY.Epidemiologyoffirst-attackacutepancreatitisinTaiwan from2000through2009: anationwidepopulation-basedstudy.Pancreas 2012;41:696–702.
[5]SiriwardenaAK,MasonJM,BalachandraS,BagulA,GallowayS,FormelaL,etal. Randomised,doubleblind,placebocontrolledtrialofintravenousantioxidant (n-acetylcysteine,selenium,vitaminC)therapyinsevereacutepancreatitis. Gut2007;56:1439–44.
[6]LeungPS, ChanYC. Roleof oxidative stress inpancreatic inflammation. AntioxidRedoxSignal2009;11:135–65.
[7]FujiiS,AkaikeT.Redoxsignalingby8-nitro-cyclicguanosinemonophosphate: nitricoxide-andreactiveoxygenspecies-derivedelectrophilicmessenger. AntioxidRedoxSignal2013;19:1236–46.
[8]SaitoY,SawaT,YoshitakeJ,ItoC,FujiiS,AkaikeT,etal.Nitricoxidepromotes recyclingof8-nitro-cGMP,acytoprotectivemediator,intointactcGMPincells. MolBiosyst2012;8:2909–15.
[9]IharaH,AhteshamAK,IdaT,KasamatsuS,KuniedaK,OkamotoT,etal. Methodologicalproofofimmunochemistryforspecificidentificationof 8-nitroguanosine30,50-cyclicmonophosphateformedingliacells.NitricOxide
2011;25:169–75.
[10]NishidaM,SawaT,KitajimaN,OnoK,InoueH,IharaH,etal.Hydrogensulfide anionregulatesredoxsignalingviaelectrophilesulfhydration.NatChemBiol 2012;8:714–24.
[11]JoudoiT,ShichiriY,KamizonoN,AkaikeT,SawaT,YoshitakeJ,etal.Nitrated cyclic GMP modulates guard cell signaling in Arabidopsis. Plant Cell 2013;25:558–71.
[12]SawaT,Ihara H,IdaT,FujiiS,NishidaM,AkaikeT.Formation,signaling functions, and metabolisms of nitrated cyclic nucleotide. Nitric Oxide 2013;34:8–10.
[13]SawaT,ZakiMH,OkamotoT,AkutaT,TokutomiY,Kim-MitsuyamaS,etal. ProteinS-guanylationbythebiologicalsignal8-nitroguanosine30,50-cyclic
monophosphate.NatChemBiol2007;3:727–35.
[14]JaworekJ,JachimczakB,TomaszewskaR,KonturekPC,PawlikWW,SendurR, etal.Protectiveactionoflipopolysaccharidesinratcaerulein-induced pancre-atitis:roleofnitricoxide.Digestion2000;62:1–13.
[15]KawabataA,KurodaR,NishidaM,NagataN,SakaguchiY,KawaoN,etal. Protease-activatedreceptor-2(PAR-2) inthepancreasandparotid gland: immunolocalizationandinvolvementofnitricoxideintheevokedamylase secretion.LifeSci2002;71:2435–46.
[16]KonturekPC,JaworekJ,ManiatoglouA,BoniorJ,MeixnerH,KonturekSJ,etal. Leptinmodulatestheinflammatoryresponseinacutepancreatitis.Digestion 2002;65:149–60.
[17]YangF,WangY,SternfeldL,RodriguezJA,RossC,HaydenMR,etal.Theroleof freefattyacids,pancreaticlipaseandCa+signallingininjuryofisolatedacinar cellsandpancreatitismodelinlipoproteinlipase-deficientmice.ActaPhysiol (Oxf)2009;195:13–28.
[18]MoustafaA,SakamotoKQ,HabaraY.AfundamentalroleforNO-PLCsignaling pathwayinmediatingintracellularCa2+oscillationinpancreaticacini.Nitric Oxide2011;24:139–50.
[19]PandolSJ,Schoeffield-PayneMS.CyclicGMPmediatestheagonist-stimulated increaseinplasmamembranecalciumentryinthepancreaticacinarcell.JBiol Chem1990;265:12846–53.
[20]BritoTS,LimaFJ,Araga˜oKS,deSiqueiraRJ,SousaPJ,MaiaJG,etal.The vasorelaxant effectsof1-nitro-2-phenylethaneinvolvestimulationofthe soluble guanylate cyclase-cGMP pathway. Biochem Pharmacol 2013;85:780–8.
[21]deSiqueiraRJ,MacedoFI,InteraminenseLeF,DuarteGP,Magalha˜esPJ,Brito TS,etal.1-Nitro-2-phenylethane,themainconstituentoftheessentialoilof Anibacanelilla,elicitsa vago-vagalbradycardiacand depressorreflexin normotensiverats.EurJPharmacol2010;638:90–8.
[22]CoskerF,ChevironN,YamasakiM,MenteyneA,LundFE,MoutinMJ,etal. The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate(NAADP)synthasethatcouplesreceptoractivationtoCa2+ mobi-lizationfromlysosomesinpancreaticacinarcells.JBiolChem2010;285: 38251–59.
[23]LahlouS,Magalha˜esPJ,deSiqueiraRJ,FigueiredoAF,InteraminenseLF,Maia JG,etal.CardiovasculareffectsoftheessentialoilofAnibacanelillabarkin normotensiverats.JCardiovascPharmacol2005;46:412–21.
[24]YadavD,LowenfelsAB.Trendsintheepidemiologyofthefirstattackofacute pancreatitis:asystematicreview.Pancreas2006;33:323–30.
[25]WanMH,HuangW,LatawiecD,JiangK,BoothDM,ElliottV,etal.Reviewof experimentalanimalmodelsofbiliaryacutepancreatitisandrecentadvances inbasicresearch.HPB(Oxf)2012;14:73–81.
[26]MuiliKA,WangD,OrabiAI,SarwarS,LuoY,JavedTA,etal.Bileacidsinduce pancreaticacinarcellinjuryandpancreatitisbyactivatingcalcineurin.JBiol Chem2013;288:570–80.
[27]KimJY,KimKH,LeeJA,NamkungW,SunAQ,AnanthanarayananM,etal. Transporter-mediatedbileaciduptakecausesCa2+-dependentcelldeathinrat pancreaticacinarcells.Gastroenterology2002;122:1941–53.
[28]HayDW,CahalaneMJ,TimofeyevaN,CareyMC.Molecularspeciesoflecithins inhumangallbladderbile.JLipidRes1993;34:759–68.
[29]PeridesG,LaukkarinenJM,VassilevaG,SteerML.Biliaryacutepancreatitisin miceismediatedbytheG-protein-coupledcellsurfacebileacidreceptor Gpbar1.Gastroenterology2010;138:715–25.
[30]PetersenOH,TepikinAV,GerasimenkoJV,GerasimenkoOV,SuttonR,Criddle DN.Fattyacids,alcoholandfattyacidethylesters:toxicCa2+signalgeneration andpancreatitis.CellCalcium2009;45:634–42.
[31]XuX,StarRA,TortoriciG,MuallemS.DepletionofintracellularCa2+stores activatesnitric-oxidesynthasetogeneratecGMPandregulateCa2+influx.J BiolChem1994;269:12645–53.
[32]VoroninaSG,BarrowSL,SimpsonAW,GerasimenkoOV,daSilvaXavierG, RutterGA,etal.DynamicchangesincytosolicandmitochondrialATPlevelsin pancreaticacinarcells.Gastroenterology2010;138:1976–87.
[33]BruceJI,ElliottAC.Oxidant-impairedintracellularCa2+signalinginpancreatic acinarcells:roleoftheplasmamembraneCa2+-ATPase.AmJPhysiolCell Physiol2007;293:C938–50.
[34]BoothDM,MurphyJA,MukherjeeR,AwaisM,NeoptolemosJP,Gerasimenko OV,etal.Reactiveoxygenspeciesinducedbybileacidinduceapoptosisand protect against necrosis in pancreatic acinar cells. Gastroenterology 2011;140:2116–25.
[35]BuchwalowI,SchnekenburgerJ,TiemannK,SamoilovaV,BankfalviA, Por-embaC,etal.L-Arginine-NO-cGMPsignallingpathwayinpancreatitis.SciRep 2013;3:1899.
[36]SawaT,IharaH,AkaikeT.Antioxidanteffectofanitratedcyclicnucleotide functioning as an endogenous electrophile. Curr Top Med Chem 2011;11:1854–60.