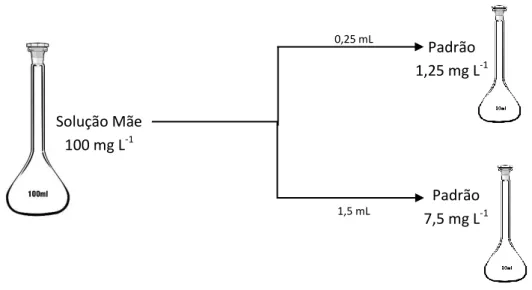
Validação e controlo da qualidade da quantificação de corantes em alimentos por HPLC-UV/Vis
113
0
0
Texto
Imagem

![Tabela 1.2 - Limites legais associados aos corantes estudados quando permitidos[3], [5- [5-9]](https://thumb-eu.123doks.com/thumbv2/123dok_br/15664974.1060965/26.892.125.807.401.669/tabela-limites-legais-associados-corantes-estudados-quando-permitidos.webp)
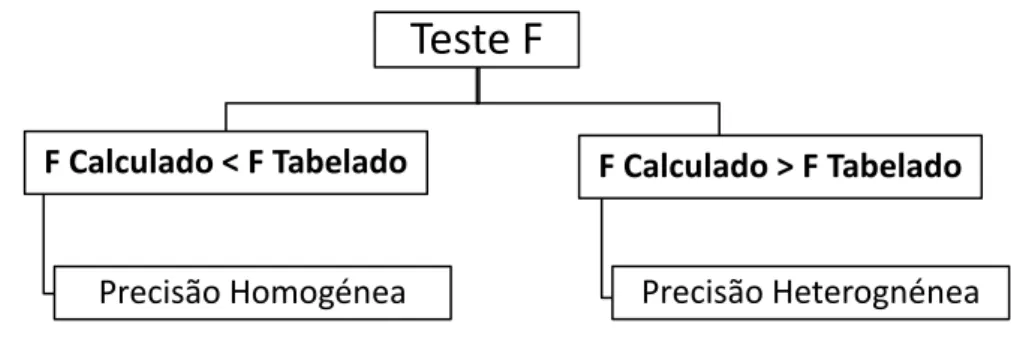
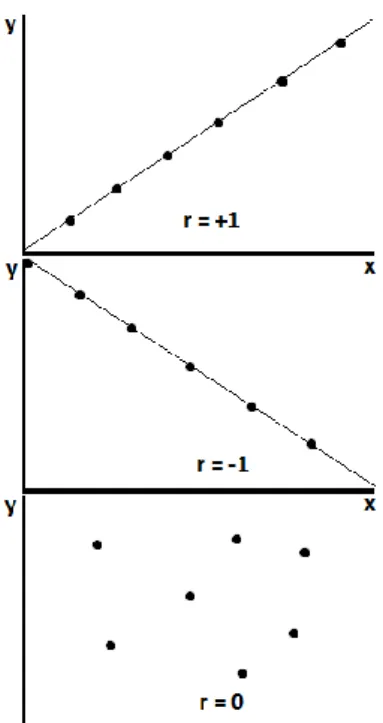
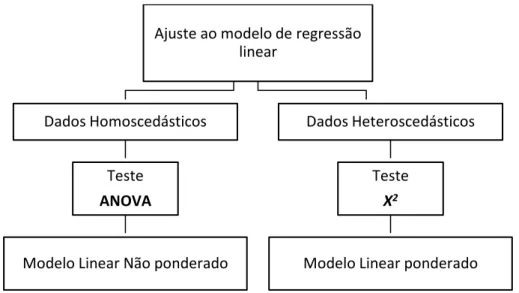
+7
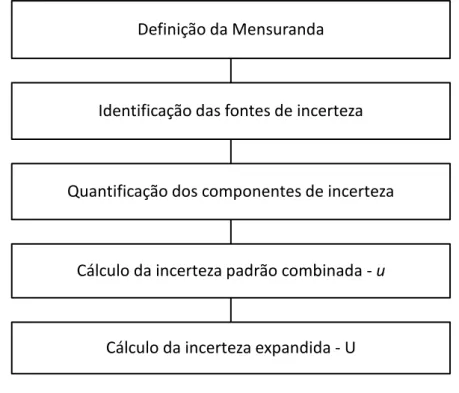


Documentos relacionados