Faculdade de Farmácia
Perfil da notificação espontânea de
suspeitas de reações adversas associadas
a Antidislipidémicos na região Sul de
Portugal, no período de 2012 a 2016
Ana Filipa Neves da Silva
Mestrado Integrado em Ciências Farmacêuticas
2017
Faculdade de Farmácia
Perfil da notificação espontânea de
suspeitas de reações adversas associadas
a Antidislipidémicos na região Sul de
Portugal, no período de 2012 a 2016
Ana Filipa Neves da Silva
Trabalho de Campo de Mestrado Integrado em Ciências Farmacêuticas
apresentada à Universidade de Lisboa através da Faculdade de Farmácia
Orientadores: Prof. Doutora Ana Paula Martins
Prof. Mestre Paula Barão
2017
Resumo
Introdução
Em Portugal, o sistema de farmacovigilância tem como base a notificação espontânea. Este método tem inúmeras vantagens que culminam no aumento do conhecimento do perfil de segurança dos fármacos. Os Antidislipidémicos, um dos grupos farmacológicos mais prescritos a nível mundial, têm uma posição fulcral no tratamento e prevenção da doença aterosclerótica, causa predominante de mortalidade e morbilidade. Dada a sua grande utilização é pertinente a monitorização da sua segurança.
Objetivo
Caracterizar o perfil de notificação espontânea de reações adversas associadas a Antidislipidémicos na Região Sul de Portugal durante o período de 2012 a 2016.
Métodos
Estudo observacional, de orientação transversal, para análise das notificações de suspeitas de reações adversas relacionadas com Antidislipidémicos, enviadas à Unidade de Farmacovigilância do Sul, diretamente por profissionais de saúde e utentes, no período 2012 a 2016.
Resultados
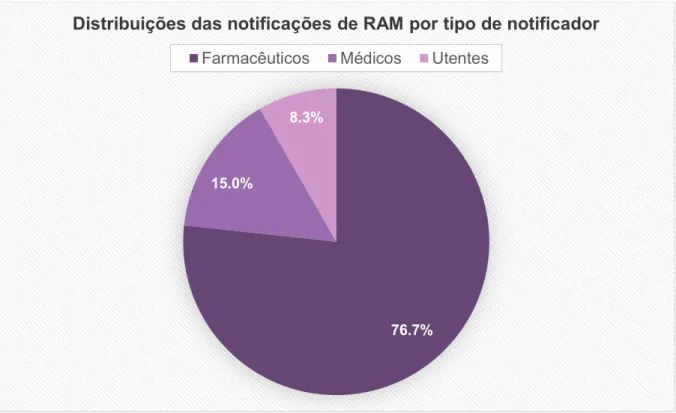
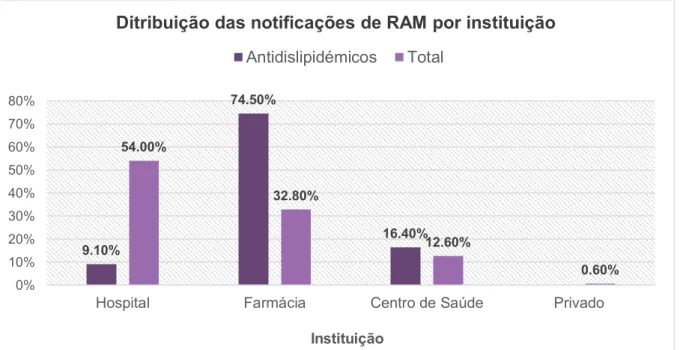
Foram recebidas 1713 suspeitas de reações adversas a medicamentos em que 60 (3,5%) eram relativas a Antidislipidémicos. O notificador mais ativo foi o farmacêutico (76,7%), a maioria a exercer funções numa Farmácia Comunitária (74,5%). A população mais afetada pertencia ao grupo etário dos idosos (60%) e ao género feminino (61,7%). As substâncias ativas mais notificadas foram a Atorvastatina (28,3%) e a Sinvastatina (25%). O SOC mais prevalente foi “Afeções musculoesqueléticas e dos tecidos conjuntivos” seguido de “Doenças gastrointestinais”, e Mialgia foi o sintoma mais notificado (20%). Grande parte dos casos suspeitos foram considerados não graves (83,3%) e 63,3% dos efeitos adversos estavam descritos no Resumo de Características do Medicamento.
Conclusões
As notificações referentes a Antidislipidémicos correspondem a apenas 3,5% do total recebido, o que demonstra uma alta taxa de subnotificação para este grupo farmacológico. Dada a limitação da não existência de trabalhos, a nível nacional e internacional, seria interessante a realização de estudos para comparação dos resultados obtidos.
Palavras-chave:
farmacovigilância, notificação espontânea, reações adversas a medicamentos, antidislipidémicosAbstract
Background
In Portugal, the pharmacovigilance system is based on spontaneous reporting. This method has numerous advantages that culminate in the increasing knowledge of the safety profile of drugs. Antidyslipidemic drugs, one of the most widely prescribed pharmacological groups, play a central role in treatment and prevention of atherosclerotic disease, a predominant cause of mortality and morbidity. Given its widespread use, it is pertinent to monitor its safety.
Objectives
To characterize spontaneous reporting of adverse reactions associated with Antidyslipidemic drugs in the Southern Region of Portugal during the period from 2012 to 2016.
Methods
Observational, cross-sectional study for the analysis of reports of suspicions of Antidyslipidemic-related adverse reactions sent to the Southern Pharmacovigilance Unit, directly by health professionals and users, between 2012 and 2016.
Results
A total of 1713 suspected adverse drug reactions were received, in which 60 (3.5%) were related to Antidyslipidemics. The most active notifier was the pharmacist (76.7%), with the majority of these working in a Community Pharmacy (74.5%). The most referenced population belonged to the age group of the elderly (60%) and to female gender (61.7%). The most commonly reported active substances were Atorvastatin (28.3%) and Simvastatin (25. The most prevalent SOC was "Musculoskeletal and connective tissue disorders" followed by "Gastrointestinal disorders", and Myalgia was the most reported symptom (20%). Most of the suspected cases were considered non-serious (83.3%) and 63.3% of adverse events were described in the Summary of Product Characteristic.
Conclusions
The spontaneous reports of Antidyslipidemic drugs correspond only to 3.5% of the total received, which demonstrates the high underreporting rate for this pharmacological group. Given the limitation of the absence of works, at national and international level, it would be interesting to carry out studies in order to compare the results obtained.
Keywords:
pharmacovigilance, spontaneous reporting, adverse drug reactions,Agradecimentos
Às minhas orientadoras, Professora Doutora Ana Paula Martins e Professora Mestre Paula Barão agradeço por me terem orientado, por todo o apoio prestado, e pelo tempo disponibilizado para a elaboração deste trabalho.
À família e amigos, em especial aos meus pais agradeço todo o amor e carinho que me têm dado ao logo da minha vida. Agradeço também por sempre acreditarem em mim e me terem proporcionado a oportunidade de realizar o meu sonho.
Ao meu namorado Rúben, agradeço o apoio incondicional em todo o meu percurso académico, a amizade, o carinho, a paciência, a confiança e força que sempre me transmitiu em todos os momentos.
Índice
1 Introdução
... 16
1.1 Enquadramento histórico da Farmacovigilância ... 16
1.2 Impacto das RAM na Saúde Pública ... 18
1.3 Farmacovigilância e a sua importância ... 19
1.4 Reações Adversas a Medicamentos – Caracterização ... 21
1.5 Notificação espontânea e perfil dos notificadores ... 24
1.6 Sistema Nacional de Farmacovigilância ... 27
1.6.1 Enquadramento Regulamentar ... 27
1.6.2 Caracterização ... 29
1.7 Unidade de Farmacovigilância do Sul ... 30
1.8 Antidislipidémicos ... 30
1.8.1 Inibidores da HMG CoA redutase ... 31
1.8.2 Fibratos ... 32
1.8.3 Sequestradores de ácidos biliares ... 32
1.8.4 Ácido Nicotínico ... 33
1.8.5 Outros agentes modificadores de lípidos ... 34
2 Objetivos
... 35
2.1 Objetivo geral ... 35
2.2 Objetivos específicos ... 35
3 Métodos
... 36
3.1 Desenho do estudo ... 36
3.2 Recolha de informação e tratamento de dados ... 36
3.3 Variáveis em estudo ... 36
3.4 Análise estatística ... 37
4 Resultados
... 38
4.1 Caracterização da notificação de suspeita de RAM... 38
4.2 Caracterização do notificador ... 40
4.3 Caracterização do doente ... 46
4.4 Caracterização da terapêutica ... 47
4.5 Caracterização da Reação Adversa ao Medicamento ... 50
5 Discussão
... 60
5.2 Caracterização do notificador ... 61
5.3 Caracterização do doente ... 64
5.4 Caracterização da terapêutica ... 65
5.5 Caracterização da Reação Adversa ao Medicamento ... 67
6 Limitações
... 71
7 Conclusão... 72
8 Referências Bibliográficas... 74
9 Anexos... 84
9.1 Anexo I ... 84 9.2 Anexo II ... 85Índice de Figuras
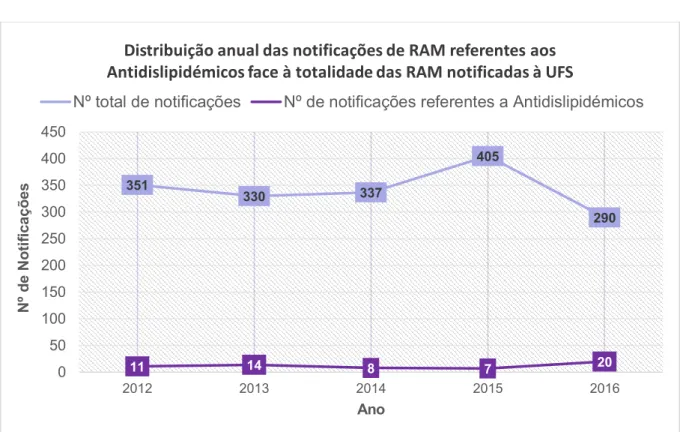
Figura 1 Distribuição anual das notificações de RAM dos Antidislipidémicos face à totalidade
de RAM, enviadas à UFS, no período de 2012 a 2016 ... 39
Figura 2 Distribuição das notificações de RAM dos Antidislipidémicos, por tipo de
notificador, enviadas à UFS no período de 2012 a 2016 ... 40
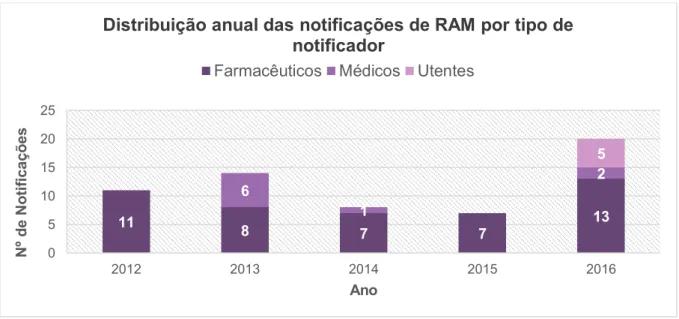
Figura 3 Distribuição anual das notificações de RAM dos Antidislipidémicos, por tipo de
notificador, enviadas à UFS no período de 2012 a 2016 ... 41
Figura 4 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
notificações, por tipo de notificador, enviadas à UFS no período de 2012 a 2016 ... 41
Figura 5 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
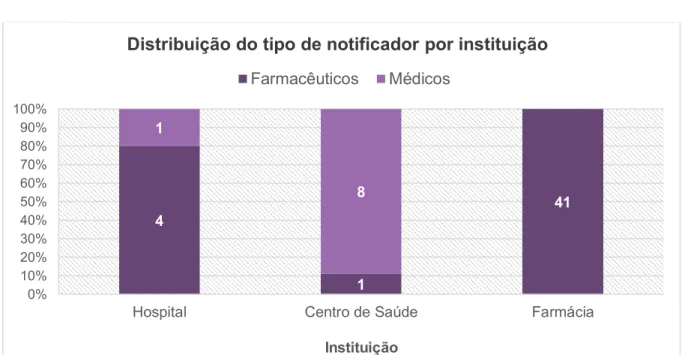
notificações, por instituição, enviadas à UFS no período de 2012 a 2016 ... 42
Figura 6 Distribuição anual das notificações de RAM dos Antidislipidémicos, por tipo de
notificador e por instituição, enviadas à UFS no período de 2012 a 2016 ... 43
Figura 7 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
notificações, por distrito, enviadas à UFS no período de 2012 a 2016 ... 43
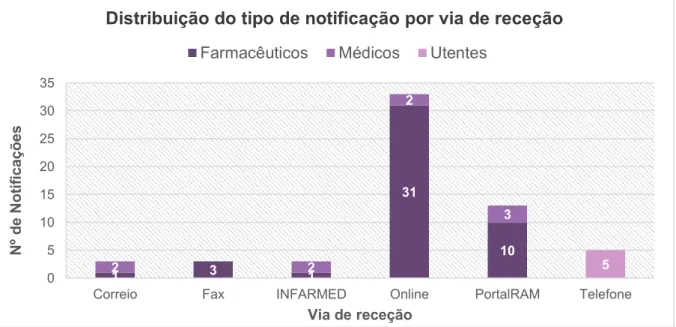
Figura 8 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
notificações, por via de receção, enviadas à UFS no período de 2012 a 2016 ... 44
Figura 9 Distribuição das notificações de RAM dos Antidislipidémicos, por tipo de notificador
e por via de receção, enviadas à UFS no período de 2012 a 2016 ... 45
Figura 10 Distribuição das notificações de RAM dos Antidislipidémicos, por ATC de 4ºnível,
enviadas à UFS no período de 2012 a 2016 ... 48
Figura 11 Distribuição das notificações de RAM dos Antidislipidémicos, por SOC, enviadas
à UFS no período de 2012 a 2016... 53
Figura 12 Distribuição das notificações de RAM totais, por SOC, enviadas à UFS no período
de 2012 a 2016 ... 53
Figura 13 Distribuição das notificações de RAM dos Antidislipidémicos, por reação adversa
mais frequente, enviadas à UFS no período de 2012 a 2016 ... 54
Figura 14 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
Figura 15 Distribuição das notificações de RAM dos Antidislipidémicos e do total de
Índice de Tabelas
Tabela 1 Classificação das RAM quanto à frequência ... 21
Tabela 2 Terapêutica lípidica segundo classificação ATC... 31
Tabela 3 Distribuição anual das notificações de RAM dos Antidislipidémicos, enviadas à
UFS, no período de 2012 a 2016 ... 38
Tabela 4 Distribuição das notificações de RAM dos Antidislipidémicos, por via de receção,
enviadas à UFS no período de 2012 a 2016 ... 44
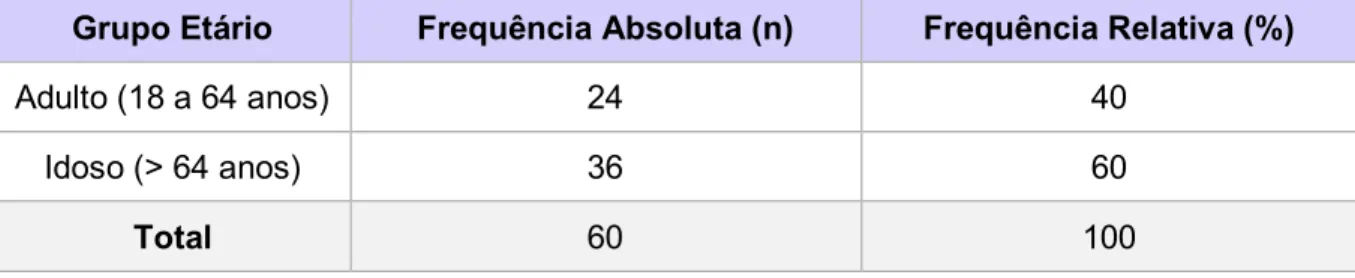
Tabela 5 Distribuição das notificações de RAM dos Antidislipidémicos, por grupo etário,
enviadas à UFS no período de 2012 a 2016 ... 46
Tabela 6 Distribuição das notificações de RAM dos Antidislipidémicos, por género e por
grupo etário, enviadas à UFS no período de 2012 a 2016 ... 46
Tabela 7 Distribuição das notificações de RAM dos Antidislipidémicos, por medicamentos
suspeitos, enviadas à UFS no período de 2012 a 2016 ... 47
Tabela 8 Distribuição das notificações de RAM dos Antidislipidémicos, por ATC de 5ªnível,
enviadas à UFS no período de 2012 a 2016 ... 49
Tabela 9 Distribuição das notificações de RAM dos Antidislipidémicos, por SOC, enviadas à
UFS no período de 2012 a 2016 ... 51
Tabela 10 Distribuição das notificações de RAM totais, por SOC, enviadas à UFS no
período de 2012 a 2016... 52
Tabela 11 Distribuição das notificações de RAM dos Antidislipidémicos, por gravidade e
descrição em RCM, enviadas à UFS no período de 2012 a 2016 ... 56
Tabela 12 Distribuição das notificações de RAM dos Antidislipidémicos, por notificador e por
gravidade, enviadas à UFS no período de 2012 a 2016... 57
Tabela 13 Distribuição das notificações de RAM dos Antidislipidémicos, por evolução da
reação, enviadas à UFS no período de 2012 a 2016 ... 57
Tabela 14 Distribuição das notificações de RAM dos Antidislipidémicos, por Critério de
Gravidade das reações Graves e por Evolução da Reação, enviadas à UFS no período de 2012 a 2016 ... 58
Tabela 15 Distribuição das notificações de RAM dos Antidislipidémicos, por imputação da
Tabela 16 Distribuição das notificações de RAM dos Antidislipidémicos, por Imputação de
Causalidade e por descrição em RCM, enviadas à UFS no período de 2012 a 2016.... 59
Tabela 17 Distribuição das notificações de RAM dos Antidislipidémicos, por Imputação de
Lista de Acrónimos
AIM – Autorização de Introdução no Mercado ALT – Alanina aminotransferase
AST – Aspartato aminotransferase
ATC – Anatomical Therapeutical Chemical CEE – Comunidade Económica Europeia
CHMP – Committee for Medicinal Products for Human Use CK – Creatina quinase
CMU – Centro de Monitorização de Uppsala CNF – Centro Nacional de Farmacovigilância
CPMP – Committee for Proprietary Medicinal Products DGRM – Direção de Gestão do Risco de Medicamentos DGTA2 – Diaglicerol acetiltransferase-2
DHA – Ácido docosahexaenóico DME – Designated Medical Events
EMA – Agência Europeia do Medicamento EPA – Ácido eicosapentaenóico
EUA – Estados Unidos da América FDA –US Food and Drug Administration HDL – Lipoproteína de alta densidade HLGT – High Level Group Term HLT – High Level Term
ICH – International Conference on Harmonisation of Technical Requirements for Registration
of Pharmaceuticals for Human Use
IME – Important Medical Events INE – Instituto Nacional de Estatística
LDL – Lipoproteína de baixa densidade LLT – Lowest Level Term
MedDRA – Medical Dictionary for Regulatory Activities NE – Notificação Espontânea
NFC – Núcleo de Farmacovigilância do Centro OMS – Organização Mundial de Saúde
PPAR – Peroxisome Proliferator-Activated Recept PRAC – Comité Europeu de Farmacovigilância PT - Preferred Term
RAM – Reação Adversa a Medicamentos
RCM – Resumo de Características do Medicamento SNF – Sistema Nacional de Farmacovigilância SOC – System Organ Class
UFC – Unidade de Farmacovigilância do Centro UFN – Unidade de Farmacovigilância do Norte
UFLVT – Unidade de Farmacovigilância de Lisboa e Vale do Tejo UFS – Unidade de Farmacovigilância do Sul
URF – Unidades Regionais de Farmacovigilância VLDL - Lipoproteína de muito baixa densidade
16
1 Introdução
1.1 Enquadramento histórico da Farmacovigilância
Os medicamentos, essenciais para a prevenção, tratamento e diagnóstico de muitas patologias, têm como fundamento primordial a promoção de bem-estar e de qualidade de vida.(1) Porém existem riscos inerentes aos quais os seus utilizadores estão expostos e que devem ser ponderados, quando conhecidos, face aos benefícios que são esperados.
O conhecimento da ocorrência destes possíveis riscos data de tempos antigos como o referenciam algumas fontes bibliográficas. São exemplos o código de Hammurabi (2200 a.C.) em que vem descrito que “quem infligir dano ao seu doente deverá ter as suas mãos
amputadas”, os ensinamentos de Hipócrates (460-370 a.C) “primum non nocere" e de
Galeno (131-201) que alertava contra os perigos dos enganos advindos de prescrições mal escritas ou até Paracelso (1634) que dizia que “a toxicidade das substâncias é condicionada
pela sua dose”. (2)
A partir do séc. XX, com a evolução técnica e científica, o desenvolvimento e comercialização de novos fármacos permitiu reduzir acentuadamente a morbilidade e mortalidade, levando a um aumento da esperança média de vida. O maior número de utilizadores de medicamentos bem como o crescimento da população idosa e portadores de doenças crónicas e degenerativas, levaram a uma maior incidência de reações adversas ao medicamento (RAM). (1)
São vários os casos descritos de grandes incidentes com fármacos, que de certa forma permitiram a evolução da vigilância de medicamentos ao longo dos tempos até à forma como a conhecemos atualmente.
Em 1936, nos Estados Unidos da América (EUA), a utilização de uma formulação contendo sulfanilamida, para infeções por Streptococcus, dissolvida em etilenoglicol provocou a morte de 107 pessoas por insuficiência renal. (3,4) A toxicidade da utilização do solvente neste incidente atraiu a atenção da comunidade cientifica o que originou a promulgação da primeira lei de farmacovigilância, “Federal Food, Drug and Cosmetic Act", aprovada pelo Congresso dos EUA em 1938. Esta lei exigia que a toxicidade dos medicamentos fosse avaliada antes da obtenção da licença para a sua comercialização. (4)
Em 1952, é publicado o primeiro tratado sobre reações adversas intitulado “Side
effects of drugs, Meyer L”. (2)
Em 1960, a US Food and Drug Administration (FDA) determina que nos EUA passem a ser registadas as reações adversas aos medicamentos e em 1962 é feita uma alteração à lei “Federal Food, Drug and Cosmetic Act", com o “Kafauver-Harris Amendments” sendo que
17
para além do estudo da toxicidade do fármaco é exigida a comprovação da eficácia terapêutica dos medicamentos a fim de garantir a sua segurança. (1) Esta exigência foi consequência de uma das maiores tragédias iatrogénicas medicamentosas, a “tragédia da Talidomida”, contribuindo assim para a existência de sistemas de farmacovigilância a nível mundial na maioria dos países. (5)
A Talidomina, utilizada em gestantes para o tratamento de náuseas e vómitos, foi responsável pelo nascimento de cerca de 10.000 crianças em todo o mundo com anomalias congénitas (nomeadamente focomélia), por centenas de mortes neonatais e por um número não determinável de abortos espontâneos em cerca de 49 países, entre 1958 e 1962. (1,6– 8)
Em 1968 a Organização Mundial de Saúde (OMS) inicia o Programa de Monitorização Internacional de Reações Adversas. (7) Este programa tinha como objetivo detetar reações adversas não verificadas nos respetivos ensaios clínicos sendo no inicio constituído por 10 países: Austrália, Canadá, Checoslováquia, EUA, Irlanda, Nova-Zelândia, Países Baixos, Reino Unido, República Federal da Alemanha e Suécia.(1,2,9) Em 1978 o Centro Internacional de Monitorização de Medicamentos foi transferido da sede da OMS, na Suíça, para Uppsala, na Suécia, dando origem ao Centro de Monitorização de Uppsala (CMU). (10) Este centro baseia-se na troca de informações entre sistemas nacionais de farmacovigilância de mais de 150 países, os quais fornecem relatórios de suspeitas de RAM, contabilizando até ao momento na sua base de dados, a VigiBase®, mais de 14
milhões de processos. (11,12) Com o conjunto destas notificações recebidas pelo CMU e após a sua análise por peritos selecionados para o efeito, sempre que necessário geram-se alertas e implementam-se ações em função do fenómeno verificado. (2)
A nível europeu os acidentes com medicamentos desencadearam a criação de leis e normas para a comercialização segura e eficaz de fármacos, começando pela Diretiva nº 65/65/CEE. (2)
Em 1975 é publicada a Diretiva nº75/319/CEE, instituiu-se um Comité de Especialidades Farmacêuticas (Committee for Proprietary Medicinal Products - CPMP) composto por representantes dos Estados-membros e da Comissão e são renovados os princípios da Diretiva nº65/65/CEE. (2)
Em 1993 é publicada a diretiva nº93/39/CEE na qual surge a designação de farmacovigilância e se definem conceitos relativos a reações adversas. (2)
Em 1995, o Conselho da Europa institucionaliza a Agência Europeia do Medicamento (European Medicines Agency - EMA) que definiu os procedimentos necessários para autorização de introdução do medicamento no mercado (AIM) e recebe e regula todas as informações provenientes dos Estados-membros, da OMS e dos titulares de AIM. (1,2)
18
Para consagrar a informação recebida a EMA desenvolveu a base de dados EudraVigilance, sistema para notificação de suspeitas de efeitos secundários em fases de pré e pós-autorização de introdução do medicamento no mercado. Esta base permite que sejam detetados sinais de suspeitas de reações adversas ouobternovas informações sobre reações previamente conhecidas. Estas serão avaliados pelo Comité dos Medicamentos para Uso Humano (CHMP) (que substituiu em Maio de 2004 o CPMP) e pelo Comité Europeu de Farmacovigilância (PRAC), com inicio em 2012, podendo estes recomendar uma ação regulamentar. (13–15)
É no contexto da criação da EMA que a partir de 1995 se torna obrigatória a criação de um Sistema Nacional de Farmacovigilância para cada Estado-Membro. (2)
1.2 Impacto das RAM na Saúde Pública
A partir do séc. XX, com o desenvolvimento da indústria farmacêutica, a iatrogenia medicamentosa assume-se como um problema de saúde pública devido à morbilidade e mortalidade que lhe está associada. (3)
São vários os estudos realizados que pretendem caracterizar as taxas destes dois parâmetros referidos anteriormente. Um dos estudos que mais destaque teve foi o de Lazarou e equipa, uma meta-análise que envolveu 39 estudos prospetivos realizados em hospitais americanos. (1) Neste estudo a taxa de incidência de RAM foi de 15,1% (10,9% durante o internamento) das quais 6,7% foram graves e 0,32% fatais. Extrapolando estes dados percentuais para números absolutos verificou-se que em 1994, nos hospitais americanos, ocorreram 2 216 000 reações adversas graves (702 000 ocorridas durante o internamento) das quais 106 000 se revelaram fatais. O estudo de Lazarou mostrou ainda que as reações adversas foram a 4ª causa de mortalidade nos EUA, atrás de causas como doença cardíaca (743 460), doença oncológica (529 904) e acidente vascular cerebral (150 108). (2)
Entre 2001 e 2002 Pirmohamed e os seus colaboradores verificaram que em Inglaterra as reações adversas foram a causa de cerca de 6,5% das intervenções hospitalares, o que representou cerca de 1225 intervenções, e pelo menos 5700 mortes. (16) De entre os doentes afetados, verifica-se que os idosos apresentam a taxa de incidência mais elevada. Numa revisão sistemática de 25 estudos observacionais, a percentagem de internamentos devido a reações adversas foi de 4% para crianças, 6% para adultos e 11% para idosos. Esta última percentagem pode ser explicada pelo fato de a partir dos 65 anos a farmacocinética do organismo alterar-se, existindo assim comorbilidades e, portanto, a necessidade de polimedicação que pode, por si só, induzir reações por interação
19
medicamentosa. (17)
Associado aos problemas de saúde do doente devido às reações adversas está também o aumento dos gastos com os serviços de saúde utilizados para o seu tratamento e assistência. (18) O impacto económico relacionado com efeitos secundários aos medicamentos pode ser maior que o custo total de cuidados cardiovasculares ou de diabetes. (19) Foi realizada uma dissertação de mestrado, em Portugal, subordinada ao tema dos custos devidos à iatrogenia medicamentosa relativos à região Centro e segundo o autor a estimativa de gastos relacionados com a morbilidade, para os três anos em estudo (2012-2014), foi de cerca de 321.387,96€. (20) Pode verificar-se que as reações adversas aos medicamentos têm um peso enorme para os recursos limitados destinados à saúde. Estima-se que nos países ocidentais estas reações ocorram em cerca de 6,7% dos doentes hospitalizados totalizando cerca de 5 a 9% dos gastos hospitalares. (18)
A iatrogenia por medicamentos demonstra ter um enorme impacto na saúde pública, diminui a eficácia do tratamento, reduz a qualidade de vida do doente, e provoca um encargo a nível financeiro nos sistemas de saúde numa altura em que se pretende, através do uso racional do medicamento e de outras medidas, diminuir a pressão financeira que se verifica nos dias de hoje. (21)
1.3 Farmacovigilância e a sua importância
Para que um novo fármaco seja aprovado pela entidade reguladora do medicamento é necessário que demonstre evidência sobre qualidade, eficácia e segurança, ou seja, que apresente uma relação benefício/risco favorável para o doente. Dada a possibilidade da ocorrência de efeitos adversos raros e graves, que podem não ser detetados no desenvolvimento do medicamento, a vigilância na fase IV ou pós-comercialização é essencial. (22) Fazem parte desses efeitos as RAM raras, tardias, resultantes de exposição crónica ao medicamento, resultantes de interações medicamentosas ou mesmo por utilização off-label do fármaco. (23)
São vários os fatores que contribuem para que estes efeitos sejam dificilmente detetados nas fases anteriores. Exemplo disso é o número limitado de pessoas expostas ao medicamento nos ensaios clínicos face à posterior utilização na vida real, bem como, o fato de serem seguidos em locais e sob condições extremamente controladas. O fato de os medicamentos na fase experimental não serem testados em populações especiais como são as grávidas, crianças ou idosos constitui outro fator. Existem ainda outros aspetos de segurança que só são verificados na fase IV, nomeadamente, o abuso e efeitos de sobredosagem ou mesmo erros de medicação que só são minimizados após a utilização
20
mais habitual do medicamento. (24) Existe, por isso, a necessidade de existência de um sistema que monitorize e avalie a relação beneficio-risco do medicamento no contexto real– o sistema de Farmacovigilância.
A Farmacovigilância, palavra derivada de “pharmakon” (fármaco em Grego) e “vigilare” (manter um olho sobre/monitor em Latim), visa de forma generalizada a segurança do medicamento com todos os aspetos que lhe estão implícitos. (25) Segundo a OMS, esta compreende a ciência e os processos inerentes à deteção, avaliação, compreensão, bem como, prevenção de reações adversas e outras questões relacionadas com fármacos. (22)
Tendo por base a informação recolhida e analisada, um dos objetivos gerais da farmacovigilância é a deteção precoce de sinais/alertas de segurança que podem ser definidos como, “informação que surge de uma ou múltiplas fontes (incluindo observações ou experiências), em que há sugestão de uma nova associação potencialmente causal, ou de um novo aspeto sobre uma associação conhecida entre uma intervenção (ex.: administração de um fármaco) e um evento ou conjunto de eventos relacionados, quer sejam benéficos ou adversos, que é considerada de probabilidade suficiente para justificar uma ação verificatória”. (26) Assim um sinal poderá ser por exemplo um efeito adverso ainda não descrito, o aumento da gravidade ou frequência de um efeito adverso conhecido, interações novas entre medicamentos ou ainda uma informação nova sobre contraindicações, precauções de utilização ou mesmo sobredosagem. (27,28)
De forma a concretizar o que é pretendido, há varias etapas na farmacovigilância que devem ser verificadas e que tornam a vigilância do medicamento um processo sistemático. Numa primeira fase é fundamental proceder à recolha de informação sobre a reação adversa tendo em conta a sua natureza, gravidade, características clinicas e consequências. Posteriormente procede-se à documentação e análise da informação recolhida com o objetivo de verificar a relação causal entre o fármaco e os efeitos adversos. Quando essa relação é observada torna-se essencial tomar medidas corretivas para eliminar ou minimizar os perigos que resultaram dessas mesmas reações, medidas essas que requerem monitorização para ser verificado o seu impacto. (24)
Quanto aos objetivos específicos da farmacovigilância fazem parte: a intenção de melhorar o atendimento, as intervenções médicas e a segurança dos utilizadores do medicamento; melhorar a saúde e segurança pública; contribuir para a utilização segura, eficaz e racional dos fármacos; avaliar benefícios e riscos associados ao uso do medicamento, bem como, promover a educação e formação em farmacovigilância e a comunicação eficaz com o público. (22)
Por vezes as questões de segurança relacionam-se com problemas de outra ordem, designadamente, com a qualidade do produto. São exemplos destas questões alterações
21
das características organoléticas, problemas ao nível da embalagem (embalagem secundária sem rótulo, sem lote ou datas de validade expiradas) ou medicamentos falsificados. Quando estes inconvenientes ocorrem as autoridades reguladoras solicitam aos titulares de AIM que tomem medidas no sentido de corrigir estas falhas, o que muitas vezes passa pela retirada de circulação do produto ou lote em questão. (24)
1.4 Reações Adversas a Medicamentos – Caracterização
Em 1975 a OMS definiu a reação adversa a medicamentos como “resposta a uma droga que é nociva e não intencional e ocorre em doses normalmente usadas no homem para a profilaxia, diagnóstico ou terapia da doença, ou para modificação da função fisiológica”. (7,29,30) No entanto a partir de 2012, alterada pela Diretiva 2010/84/EU do Parlamento Europeu e do Conselho, a definição de RAM passa a ser definida como “reação nociva e não intencional a um medicamento”. (6) Esta definição contempla RAM resultantes da utilização do fármaco nos termos da AIM ou casos de sobredosagem, erros de medicação e uso off-label, bem como, as reações resultantes de exposição ocupacional.
Em 1991 Rawlins e Thompson classificaram as reações em tipo A e B. (30) Posteriormente esta classificação foi alvo de atualizações, tendo sido acrescentadas novas categorias que foram nomeadas de forma mnemónica de A a F: (31)
A- Aumentadas: estas reações são relacionadas com o mecanismo de ação do fármaco e
são dependentes da dose. São comuns de ocorrerem, podem ser previstas e têm baixa mortalidade. Um exemplo são os efeitos anticolinérgicos provocados pelos antidepressivos tricíclicos.
B- Bizarras: estas reações, não relacionadas com a ação farmacológica do medicamento,
não são dependentes da dose. Não são comuns, a sua ocorrência não é previsível sendo que ao ocorrerem têm uma taxa de mortalidade elevada. São exemplos reações imunológicas como hipersensibilidade à Penicilina ou reações idiossincráticas como porfiria aguda ou hipertermia maligna.
C- Crónicas: estas reações estão associadas ao efeito cumulativo do medicamento sendo
que os efeitos secundários são relacionados com dose e tempo de exposição ao fármaco. São reações pouco comuns e um exemplo é a supressão do eixo hipotalâmico-hipofisário-adrenal por corticosteroides.
D- Retardadas: do inglês Delayed, estas reações ocorrem ou aparecem algum tempo após
o uso do medicamento. O seu surgimento está relacionado com a dose e o tempo de utilização sendo que são pouco comuns de ocorrerem. É exemplo de reação o aparecimento de adenocarcinoma vaginal em filhas de mulheres tratadas com
22
Dietilestilbestrol na gravidez.
E- Fim de Tratamento: do inglês End of use, estas reações, farmacologicamente
previsíveis, ocorrem aquando da suspensão do medicamento. Normalmente a condição do doente melhora quando o fármaco é reintroduzido. Um exemplo é a ocorreria de isquemia miocárdica devida à suspensão de β-bloqueadores.
F- Falência terapêutica: estas reações estão relacionadas com a falha na eficácia do
medicamento. São reações comuns que podem estar relacionadas com a dose, interações medicamentosas ou mesmo problemas de qualidade. Um exemplo é gravidez indesejada por dosagem inadequada de anticoncecional oral quando utilizados concomitantemente indutores enzimáticos.
Relativamente à gravidade das RAM, a OMS considera uma reação adversa grave qualquer ocorrência médica indesejável que, independentemente da dose, resulte em morte, coloque a vida em risco, requeira internamento hospitalar ou prolongue a hospitalização pré-existente, resulte em incapacidade persistente ou significativa ou seja uma anomalia/defeito congénito. (32)
Com o objetivo de facilitar a classificação de reações adversas suspeitas e apoiar a análise de dados e avaliação de casos nas atividades diárias de farmacovigilância, um grupo de trabalho da EudraVigilance coordenou o desenvolvimento de uma lista de termos médicos importantes denominada Important Medical Events (IME). Esta lista, destinada apenas para orientação, foi desenvolvida durante a revisão da lista atual do MedDRA versão 12.1, sendo que os critérios de inclusão foram baseados na definição oficial de gravidade de RAM supracitada. (33) Para além desta lista existe outra denominada Designated Medical
Events (DME) desenvolvida pela EMA. Esta lista tem como finalidade ajudar a identificar
quais os relatórios de suspeita de RAM que devem ter uma avaliação prioritária servindo assim como uma rede de segurança que garante que sinais importantes não sejam perdidos. Desta lista fazem parte termos do MedDRA que representam eventos adversos raros e graves e que são tipicamente devidos a certos medicamentos o que auxilia numa revisão do beneficio/risco do medicamento. Assim a identificação de um pequeno número de um determinado DME, aquando de uma revisão de casos levará provavelmente à geração de um sinal. (34,35)
No que diz respeito à frequência de ocorrência de reações adversas, consideram-se cinco categorias como indicado abaixo, resultantes do trabalho desenvolvido pelo CIOMS Working Group V. (36)
23
Tabela 1 Classificação das RAM quanto à frequência
Muito frequente ≥1/10 > 10%
Frequente ≥1/100 to <1/10 > 1% e < 10% Pouco frequente ≥1/1,000 to <1/100 > 0.1% e < 1 % Rara ≥1/10,000 to <1/1,000 > 0.01% e < 0.1% Muito rara <1/10,000 < 0.01%
As reações adversas são consideradas um diagnóstico clinico que pode ser baseado em sintomas apresentados, em resultados de exames de diagnóstico ou ainda suportado pela identificação de fatores de risco do indivíduo. (37) Através de uma revisão da literatura é possível determinar exemplos de fatores que podem influenciar o aparecimento de reações adversas abrangendo estes: características do doente, do medicamento suspeito, de outras terapêuticas concomitantes, ou da existência de doenças de base. (38–40)
Relativamente ao doente verifica-se que o género feminino é maioritariamente afetado, e que a faixa etária mais preocupante é a superior a 65 anos. Estes são considerados um grupo de risco uma vez que nesta faixa etária aumentam as alterações farmacocinéticas e farmacodinâmicas. Relativamente às mulheres, são um grupo de risco porque, além de apresentarem uma diferença hormonal e corporal em relação aos homens, têm uma metabolização diferente em alguns fármacos. (38–40) A polimedicação também pode aumentar o risco de aparecimento de efeitos secundários, devido à possibilidade de interação entre medicamentos, bem como a existência de patologias de base como insuficiência renal ou hepática, uma vez que estas vias de eliminação e metabolização do medicamento ficam afetadas. (38,39) Outro fator referenciado é a variação genética do metabolismo do fármaco que afeta certas populações. Por exemplo a população asiática, tais como os chineses e os japoneses, têm menor prevalência de metabolizadores da família do citocromo P450 (como o CYP2C18, CYP2C19 e CYP2D6) o que torna estes doentes mais suscetíveis a RAM de medicamentos metabolizados por estas enzimas. (41) Existem ainda outros fatores que estão relacionados com hábitos sociais, como o consumo de bebidas alcoólicas ou tabaco. O álcool afeta o metabolismo de muitos fármacos, podendo torná-los mais tóxicos, favorecendo o desenvolvimento de efeitos indesejados. Relativamente ao tabaco está provado alterar o processo metabólico de certas enzimas hepáticas funcionado como indutor enzimático. Foram estudadas e comprovadas interações fármaco-tabaco com teofilina, insulina, contracetivos orais entre outros. (42)
24
1.5 Notificação espontânea e perfil dos notificadores
Existem vários métodos que podem ser utilizados no âmbito dos Sistemas de Farmacovigilância. São estes os métodos geradores de hipóteses, que contemplam a notificação espontânea (NE) e a publicação de casos como os case reports; os métodos verificadores de hipóteses dos quais fazem parte os estudos de coorte, estudos de caso-controlo e ensaios clínicos controlados e aleatorizados; e por fim os métodos que englobam a geração e a verificação de hipóteses como a monitorização prescrição-evento. (6)
O método mais disseminado a nível mundial é a notificação espontânea, cuja informação pode posteriormente ser investigada através de estudos farmacoepidemiológicos. (2,43) O objetivo das notificações, em conjunto com outras atividades de pós-autorização, é identificar reações adversas graves ou raras, cujo conhecimento melhora a compreensão do perfil de segurança de novos medicamentos, permitindo assim a geração de sinais. (44)
As suspeitas de reações adversas podem ser reportadas por profissionais de saúde, titulares de AIM e pelo próprio doente desde 2012 com a introdução da nova legislação europeia de farmacovigilância, Diretiva 2010/84/EU. (1,45) Para que essa suspeita reportada seja válida é necessário no mínimo a verificação de quatro requisitos: um notificador, um doente, pelo menos uma reação adversa suspeita e pelo menos um medicamento suspeito. (46)
Para a análise da notificação existem outros campos que podem ser avaliados e quando necessário o notificador é contactado de forma a prestar outras informações complementares que se considerem pertinentes. (46) No que diz respeito à via de notificação pode ser utilizada a via online (através de preenchimento de um formulário no portal RAM), telefone ou envio de uma ficha de notificação por correio, fax ou e-mail. (6) A ficha de notificação tem um formato em papel de tamanho A4 e é necessário o preenchimento dos seguintes campos: a) reação adversa ao medicamento; b) medicamento(s) suspeito(s); c) medicamento(s) concomitante(s); d) doente; e e) profissional de saúde. (Anexo I) (2)
Uma vez reportada a notificação, há todo um processo sequencial que deve ser respeitado do qual fazem partes as seguintes fases: a) receção; b) validação; c) verificação de duplicações; d) codificação; e) registo na base de dados; f) análise técnico-científica com imputação de causalidade e g) deteção de problemas que quando necessário convergem na geração de sinais. (2)
25
causalidade que, segundo a OMS, é “um método pelo qual se estima a relação relativa entre agente (que é o fármaco) e as reações adversas”. (47) Por outras palavras a imputação de causalidade é definida como a probabilidade de um medicamento especifico ser a causa de um efeito secundário observado, sendo que o que se pretende é avaliar a conexão causal entre estes dois fatores. (48,49)
A imputação de causalidade é realizada recorrendo à introspeção global com base na escala de avaliação da causalidade da OMS, e com utilização pontual de algoritmos como é o caso do Algoritmo de Naranjo. (2,49) A escala da OMS, utilizada pelo Sistema Nacional de Farmacovigilância (SNF), divide a causalidade de uma reação adversa ao medicamento em seis categorias: (31)
1. Definitiva: acontecimento clinico ou alteração laboratorial que ocorre com uma relação temporal plausível e que não pode ser explicado por doenças concomitantes ou outros fármacos;
2. Provável: acontecimento clinico ou alteração laboratorial que ocorre com uma relação temporal aceitável, em que o nexo de causalidade com doenças concomitantes ou outros fármacos é pouco provável e em que a evolução após a suspensão do fármaco é aceitável do ponto de vista clinico.
3. Possível: acontecimento clinico ou alteração laboratorial que ocorre com uma relação temporal aceitável mas que pode também ser explicada por doenças concomitantes ou outros fármacos.
4. Impossível: acontecimento clinico ou alteração laboratorial com uma relação temporal que torna improvável o nexo de causalidade com o fármaco e em que a associação com outros fármacos ou doenças concomitantes constitui uma explicação plausível.
5. Condicional/ Não classificada: acontecimento clinico, incluindo uma alteração de um parâmetro laboratorial, reportado como uma reação adversa, sobre a qual são necessários mais dados para uma avaliação adequada ou então esses dados ainda estão a ser analisados.
6. Inacessível/ Não Classificável: acontecimento ou reação adversa que não pode ser avaliada devido a informação insuficiente ou contraditória que não pode ser complementada ou verificada.
Posteriormente as notificações são enviadas para a base de dados europeia, EudraVigilance, permitindo o acesso à consulta pela EMA e pelos Estados-Membros. (2) Se
26
a reação for grave deverá ser enviada para a base no período de 15 dias e se for não grave deverá ser enviada num período de 90 dias. (50)
A notificação espontânea tem um contributo bastante significativo para o conhecimento do perfil de segurança do medicamento, e como qualquer método tem vantagens e desvantagens. São vantagens o facto de não interferir com a prescrição médica, utilizar várias fontes de informação, permitir identificar fatores de risco, permitir gerar sinais e detetar RAM raras precocemente, ser um método pouco dispendioso, abranger todos os medicamentos durante o seu ciclo de vida e permitir uma monitorização em larga escala, uma vez que envolve toda a população utilizadora de medicamentos. As principais desvantagens são a impossibilidade de determinação de taxas de incidência e a elevada taxa de subnotificação, estimando-se assim que apenas 6% das reações adversas sejam reportadas aos Sistemas de Farmacovigilância. (2,44)
Em Portugal, em 2008, verificou-se serem reportadas 175 notificações/milhão de habitantes em contraste com o proposto pela OMS de 250 notificações/milhão de habitantes. (51) Segundo a Autoridade Nacional de Medicamentos e Produtos de Saúde, I.P. (INFARMED), em 2014, o número de notificações espontâneas total foi de 4618, mas as reportadas por profissionais de saúde e doentes foi de apenas 2464 perfazendo um índice de 237 notificações/milhão de habitantes, continuando abaixo do recomendado pela OMS. (52,53)São vários os fatores estudados que demonstram contribuir para a baixa notificação de reações. Em 1976, Inman identificou sete atitudes que contribuíam para tal, sendo elas a complacência (crença que as RAM graves estão bem documentadas aquando da comercialização do medicamento); a ignorância (crença de que apenas as RAM graves e inesperadas devem ser notificadas); a desconfiança (crença de que só se deve notificar uma RAM quando se tem a certeza sobre o nexo de causalidade com o medicamento); a insegurança (crença em ser quase impossível determinar se um medicamento é ou não responsável por uma RAM); a indiferença (crença de que um único caso não é suficiente para contribuir para o conhecimento médico); a letargia (por falta de interesse ou tempo para notificar); e o medo (da responsabilização e de consequências legais após a notificação). (54,55)
É importante implementar estratégias para aumentar o número de notificações espontâneas, mas também é fundamental não descurar a qualidade da informação reportada. (25)
O médico, através das suas decisões terapêuticas e do seu conhecimento sobre o doente e a doença, desempenha um papel relevante para o conhecimento do perfil de segurança dos medicamentos. Quanto maior for o investimento na sua formação e sensibilização, maior será a compreensão acerca da necessidade de reportar suspeitas de
27
RAM. (2)
O enfermeiro, dado ao seu contacto primário com os doentes e devido à administração do fármaco, é o primeiro a observar possíveis sinais de RAM. (56)
Relativamente ao farmacêutico, o seu envolvimento nos sistemas de farmacovigilância é crucial e tem sofrido uma evolução graças a uma mudança no seu papel passando de “dispensador de medicamentos” para prestador de cuidados farmacêuticos. Deste modo o farmacêutico tornou-se essencial na gestão de doenças crónicas, onde pode detetar problemas relacionados com medicamentos como: dosagens incorretas, reações adversas, interações medicamentosas ou até mesmo ineficácia terapêutica. Tudo isto permite melhorar a segurança do medicamento, a produtividade e diminuir tempo e custos ao sistema de saúde. (25)
A inclusão dos doentes, como possíveis notificadores, permitiu aumentar a notificação espontânea e ajudar na deteção precoce de RAM graves ou inesperadas. Através de perspetivas diferentes, os doentes podem fornecer informações relativas a sintomas e RAM que poderiam passar despercebidas aos prestadores de cuidados de saúde. Os doentes podem ainda contribuir reportando situações associadas a medicamentos não sujeitos a receita médica. (57)
1.6 Sistema Nacional de Farmacovigilância
1.6.1 Enquadramento Regulamentar
O SNF foi implementado em Portugal em 1992, após entrada do país para a Comunidade Económica Europeia (CEE). (2) É formalizado pelo Despacho Normativo 107/92 de 27/06/92 que define a organização inicial do sistema, determina o estudo e análise da informação relativa a reações adversas notificadas quer por profissionais de saúde quer pela indústria farmacêutica e anuncia a criação do Centro Nacional de Farmacovigilância (CNF) (2,23)
Em 1993, pelo Decreto-Lei nº 10/93 de 15/01/93, emerge o Instituto Nacional da Farmácia e do Medicamento (INFARMED), hoje nomeado Autoridade Nacional do Medicamento e Produtos de Saúde I.P., no qual é integrado o SNF. (2,6)
Com o objetivo da descentralização do sistema, assente na portaria nª605/99, em 2000 foram criadas quatro Unidades Regionais de Farmacovigilância (URF): Unidade de Farmacovigilância do Norte (UFN), Núcleo de Farmacovigilância do Centro (NFC), Unidade de Farmacovigilância do Sul (UFS) e Unidade de Farmacovigilância dos Açores. No ano de 2002, foi aprovado o Decreto-Lei nº 242/2002 de 05/11/02 que revoga a portaria nº605/99
28
com o fim de regularizar em Portugal as normas de acordo com descrito na Diretiva nº 2001/83/CE de 06/11/01 da Comunidade Europeia. (6)
Em 2003 verifica-se a entrada numa nova fase do sistema com a publicação do novo regulamento interno do INFARMED na portaria nº 271/2003 passando a existir a Unidade de Farmacovigilância de Lisboa e Vale do Tejo (UFLVT) e a Unidade na Região Autónoma da Madeira. (6) O Sistema passa a ser constituído pelo Departamento de Farmacovigilância do INFARMED, que coordena as atividades de farmacovigilância através da Direção de Gestão do Risco de Medicamentos (DGRM). (1)
Em 2006, com o objetivo de agregar toda a legislação existente sobre o medicamento, é publicado o Decreto-Lei 176/2006 que funciona como base de regulamentação do Sistema Nacional de Farmacovigilância de Medicamentos para Uso Humano. (1)
Em 2012 entra em vigor a nova legislação europeia de farmacovigilância, a Diretiva 2010/84/EU e a Diretiva nª 2012/26/UE de 25/10/12, que alteram a Diretiva 2001/83/CE no que diz respeito à Farmacovigilância. Foram aprovados os Decretos-Lei nº 20/2013 de 14/02/13 e Decreto-Lei nº 128/2013, de 5/09/13 para transpor para o ordenamento jurídico nacional as Diretivas anteriores e que introduziram grandes alterações ao nível do SNF. (23) Fazem parte das alterações implementadas, o início da tomada de posição do doente como notificador, a maior abrangência do conceito de reação adversa, o processo de notificações mais simplificado e ainda a AIM do medicamento condicionada pela apresentação de um plano de gestão de risco, no qual deve constar as atividades e medidas de farmacovigilância necessárias à identificação, caracterização, prevenção e/ou minimização de riscos. Ficou ainda designado nesta Diretiva a possibilidade das autoridades requererem estudos de pós-comercialização bem como monitorização adicional para medicamentos específicos. Ficou também definida a necessidade da criação de portais nacionais de medicamentos com ligação ao portal europeu, que permitam a divulgação de informação como os Resumo de Características do Medicamento (RCM) e folhetos informativos, relatórios de avaliação, resumos de planos de gestão de risco e formas de notificação de suspeitas de RAM. (1)
No inicio de 2017 o INFARMED procedeu ao alargamento da rede de Unidades de Farmacovigilância, passando a existir oito Unidades: Porto, Coimbra, Lisboa, Setúbal e Santarém, Guimarães, Beira Interior, Algarve e Alentejo e ainda a Unidade do INFARMED, com responsabilidades sobre as regiões autónomas dos Açores e Madeira e alguns concelhos dos distritos de Lisboa e Leiria. O objetivo deste reforço, para além do aumento dos recursos humanos, é o de promover maior envolvimento dos profissionais de saúde na vigilância dos medicamentos. (58)
29
1.6.2 Caracterização
No que diz respeito às características do SNF este tem como base a notificação espontânea sendo que a recomendação é para que todas as reações adversas sejam reportadas independentemente de serem graves ou não graves, descritas ou não descritas no RCM. No que se refere aos titulares de AIM estes são obrigados a enviar todas as notificações graves recebidas, quer dos utentes quer dos profissionais de saúde, ocorridas em território nacional, e as graves e inesperadas ocorridas em território internacional. (1,2)
As notificações recebidas são processadas, analisadas e registadas numa base de dados central. Após a atribuição da causalidade pelo parecer dos peritos, o notificador recebe informação sobre o nexo de causalidade entre a reação adversa e o medicamento suspeito. Posteriormente o SNF comunica aos profissionais de saúde e aos utilizadores dos medicamentos as medidas de segurança adotadas ou a adotar para prevenção de RAM. (1,2)
Relativamente à base de dados central utilizada em Portugal, esta foi desenvolvida em 2004, é denominada SVIG, e é nesta que são introduzidas as notificações de RAM enviadas às Unidades ou notificadas diretamente ao INFARMED. (1)
Na base de dados SVIG as reações adversas são classificadas segundo o Dicionário Médico para Atividades Regulamentares, MedDRA (Medical Dictionary for Regulatory
Activities) desenvolvido pela Conferência Internacional sobre Harmonização de Requisitos
Técnicos para o Registo de Medicamentos de Uso Humano, ICH (International Conference
on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Este é composto por terminologia médica especifica de forma harmonizada para
facilitar (a nível internacional) a partilha de informação. (1,2,59) A codificação é apenas qualitativa uma vez que não inclui níveis de gravidade ou intensidade de cada termo. (2) Relativamente à classificação MedDRA esta foi desenvolvida com base numa estrutura hierárquica que proporciona graus ou níveis de supra-ordenação e subordinação. O termo supra-ordenado diz respeito a um termo vasto incluído num grupo geral. Assim, hierarquicamente, existem os seguintes níveis: SOC (System Organ Class); HLGT (High Level Group Term); HLT (High Level Term); PT (Preferred Term) e LLT (Lowest Level Term). (60)
Ainda sobre a base de dados SVIG, os medicamentos inscritos são classificados segundo a Classificação ATC (Anatomic Therapeutical Chemical), utilizada para codificar as moléculas com ação terapêutica, sendo este sistema adotado pela OMS. (1)
30
1.7 Unidade de Farmacovigilância do Sul
Antes de realizada a recente reestruturação do SNF, uma das unidades existentes era a Unidade de Farmacovigilância do Sul, nascida em 2004 através de um protocolo entre o INFARMED e a Faculdade de Farmácia da Universidade de Lisboa. Integrada no SNF, a Unidade pretendia colaborar na proteção da saúde pública pela monitorização do perfil de segurança dos medicamentos de uso humano comercializados em Portugal. (61) Era da sua competência a receção, análise, avaliação e validação das notificações de suspeita de RAM determinando no final o nexo de causalidade. (23)
A Unidade do Sul abrangia as áreas geográficas do Alentejo e Algarve, totalizando uma área de 32.327 Km2 e representando um total de 939 445 habitantes, segundo dados do
Instituto Nacional de Estatística (INE). (61) No que diz respeito à densidade populacional, o Alentejo é caracterizado por ter 23,1 habitantes por Km2 enquanto que o Algarve tem 88,4
habitantes por Km2. (62)
Podiam reportar para a UFS profissionais de saúde a exercer funções em Hospitais, Centros de Saúde ou Farmácias comunitárias. Fazem parte da zona Sul 5 Hospitais Distritais Gerais públicos: Hospital José Maria Grande (Portalegre); Hospital do Espirito Santo (Évora); Hospital José Joaquim Fernandes (Beja); Hospital de Faro e Hospital do Barlavento Algarvio (Portimão) e 3 Hospitais de Nível I: Hospital de Santa Luzia de Elvas; Hospital de S. Paulo (Serpa) e Hospital Distrital de Lagos. Relativamente aos Centros de Saúde, o Algarve conta com 44 unidades e 248 extensões e o Alentejo tem 16 centros de Saúde com 68 extensões. Em relação às Farmácias comunitárias, a região está coberta por 274 estabelecimentos. (61)
Com o objetivo de divulgar a atividade da farmacovigilância, promover a formação, elaborar e participar em projetos científicos, além de uma equipa multidisciplinar, a UFS contava também com o apoio de 10 delegados de farmacovigilância para promover a notificação espontânea na área abrangente. (61)
1.8 Antidislipidémicos
A doença cardiovascular, uma das principais causas de morte, está fortemente relacionada com a arteriosclerose, estando a esta associados vários fatores de risco como é o caso da dislipidemia. (63,64) A dislipidemia refere-se a um conjunto de distúrbios metabólicos caracterizados por um excesso ou deficiência de partículas lipoproteicas. Como resultado verifica-se o aumento de concentrações plasmáticas da lipoproteína de baixa densidade (colesterol-LDL) e/ou triglicéridos e a deficiência em lipoproteína de alta densidade (colesterol-HDL). (65)
31
Segundo a classificação ATC, os Antidislipidémicos pertencem ao grupo C10. (66) Na tabela 2 é possível observar de forma sumária as classes e subclasses integradas neste grupo. Em anexo é possível analisar a tabela completa com as substâncias ativas pertencentes a cada subclasse. (Anexo II)
Tabela 2 Terapêutica lipídica segundo classificação ATC
C10 - Agentes modificadores de lípidos
C10A
Agentes modificadores de lípidos em Monoterapia
C10AA - Inibidores da HMG CoA redutase C10AB - Fibratos
C10AC - Sequestradores de ácidos biliares C10AD - Ácido Nicotínico e derivados
C10AX - Outros agentes modificadores de lípidos C10B
Agentes modificadores de lípidos em Combinação
C10BA – Combinação de inibidores da HMG CoA redutase com outros agentes modificadores de lípidos
C10BX – Inibidores da HMG CoA redutase com outros medicamentos
1.8.1 Inibidores da HMG CoA redutase
Os Inibidores de HMG-CoA redutase, também denominados Estatinas, são considerados segundo as atuais Guidelines como a primeira linha farmacoterapêutica para a redução do colesterol-LDL. (67)
As Estatinas, descobertas em 1970, atuam inibindo competitiva e reversivelmente a enzima 3-hidroxi-3-metilglutamil-coenzima A redutase, o que inibe a conversão de HMG-CoA em mevalonato na via de síntese do colesterol. (68–70) Esta inibição resulta num aumento da expressão dos recetores de colesterol-LDL nas membranas dos hepatócitos o que acelera a depuração das partículas desta fração. (71,72)
Embora as Estatinas sejam geralmente bem toleradas pelos utilizadores existem algumas reações adversas documentadas sendo clinicamente relevantes a miopatia, a hepatotoxicidade e o aumento da glucose no sangue. (73,74) Os efeitos adversos relacionados com o músculo esquelético mais frequentes são a mialgia, a miosite ou a rabdomiólise. (68) A sintomatologia inclui dores musculares, fraqueza, rigidez e caibras sendo a rabdomiólise a complicação mais grave podendo levar a insuficiência renal. (67,68,75) A hepatotoxicidade pode ser acompanhada pelo aumento sérico da aspartato aminotransferase (AST) e da alanina aminotransferase (ALT), porém a inflamação do fígado é rara. (76,77) As alterações ao nível do metabolismo da glucose podem contribuir para o aparecimento de Diabetes Mellitus do tipo 2 ou afetar o controlo em doentes já com a
32
patologia. (68,73,75) Outras possíveis RAM incluem toxicidade renal, alterações gastrointestinais nomeadamente diarreia, dor abdominal e náuseas e ainda erupção cutânea e rubor. (68,73,76) Foram ainda documentadas alterações ao nível do sistema nervoso central resultando em diminuição cognitiva associada a perda aguda de memória bem como sintomas de irritabilidade. Fazem parte também das RAM documentadas casos de doença pulmonar intersticial, neuropatia periférica, disfunções pancreáticas e sexuais. (76,78,79)
1.8.2 Fibratos
Os Fibratos são uma classe de antidislipidémicos cujo mecanismo de ação é baseado na ativação da subunidade alfa dos recetores intracelulares PPAR (Peroxisome
Proliferator-Activated Receptors) que aumentam positivamente a transcrição de múltiplos
genes que facilitam o metabolismo lipídico. (80,81)A ativação da PPAR-alfa reduz os níveis sérios de triglicéridos e aumenta a lipoproteína lípase, a oxidação de ácidos gordos e os níveis plasmáticos de colesterol-HDL. (82–85)
Os compostos de ácido fíbrico são geralmente bem tolerados podendo, no entanto, ocorrer determinados efeitos secundários. (86) Entre as reações adversas descritas fazem parte alterações gastrointestinais cuja sintomatologia passa por náuseas e cólicas acompanhadas de dor de cabeça. (86,87) Outros efeitos mais frequentes são fadiga, cefaleias, erupção cutânea, urticária, formação de cálculos biliares, anemia, aumento do risco de trombose e ansiedade. (65,82,85,86) Algumas reações adversas têm uma frequência dependente da dose como é exemplo o aumento dos valores séricos das transaminases hepáticas, da homocisteína e da creatinina, apesar da utilização de Fibratos não estar associada a um risco aumentado de insuficiência renal. (82,87–89) É conhecido também o aumento do risco de miopatia com fraqueza muscular e dor associadas tendo sido relatados casos de rabdomiólise. (86,87,89) Esta, é uma RAM grave associada ao uso de Fibratos em monoterapia ou em combinação com outras substâncias ativas (especialmente Estatinas) que influenciam o metabolismo dos mesmos.(86)
1.8.3 Sequestradores de ácidos biliares
Os Sequestradores de ácidos biliares são resinas que atuam sobre os ácidos biliares, moléculas anfipáticas, sintetizadas a partir do colesterol e que facilitam a absorção de gordura ao nível do intestino. (90) As resinas, carregadas positivamente, ligam-se aos ácidos biliares, carregados negativamente, formando um complexo insolúvel que interfere com a reabsorção intestinal dos ácidos biliares e que sofre uma excreção fecal. (90–92) O
33
efeito direto resulta numa regulação positiva de síntese de novo ácido biliar a partir de colesterol hepático. A quantidade de colesterol intracelular diminui à medida que os sais biliares são sintetizados, o que desencadeia uma produção aumentada de recetores de colesterol-LDL e uma maior depuração desta fração lipídica. (93,94)
No que diz respeito às reações adversas e a interações medicamentosas estas ocorrem maioritariamente nas resinas mais antigas e limitam o uso desta classe de antidislipidémicos. (95) No entanto os efeitos secundários predominantes em todos os Sequestradores de ácidos biliares estão relacionados com alterações gastrointestinais com sintomatologia associada de dor abdominal, inchaço, flatulência, obstipação, diarreia, náuseas, vómitos e dispepsia. (96–98)
1.8.4 Ácido Nicotínico
A Niacina, também designada ácido nicotínico, uma vitamina do complexo B, é um dos fármacos mais antigos utilizados no tratamento da dislipidemia e afetam praticamente todas as frações lipídicas. (99,100) O mecanismo de ação tem como base a inibição da enzima hepática DGTA2 (Diaglicerol acetiltransferase-2), principal enzima responsável pela síntese de triglicéridos. (99,100)Para além desta inibição, a Niacina aumenta a degradação da apolipoproteína B o que resulta numa secreção diminuída de partículas de colesterol-LDL e VLDL (lipoproteina de muito baixa densidade). (101) Subjacente a este mecanismo o ácido nicotínico pode ainda diminuir a lipólise dos triglicéridos no tecido adiposo, diminuindo o transporte de ácidos gordos livres para o fígado. (102)
No que diz respeito às reações adversas provocadas pela Niacina o efeito predominante, mesmo em doses baixas, é o rubor ou vermelhidão da pele causado por uma vasodilatação ao nível da face e do tronco superior e que pode vir acompanhado de prurido e sensação de queimadura. (95,100,103–106)Ainda ao nível de alterações cutâneas foram ainda verificados casos de Acantose nigiricans e pele seca. (85,103) São também efeitos secundários possíveis da Niacina alterações gastrointestinais com sintomatologia associada de náuseas, vómitos, dor abdominal, diarreia e dispepsia. (103) Foram verificados ainda casos de hepatotoxicidade, hiperuricemia com ativação da gota e hiperglicemia com redução da sensibilidade à insulina o que representa uma limitação ao uso do ácido nicotínico em doentes com Diabetes Mellitus. (95,103,107) Efeitos músculo-esqueléticos com miopatia foram reportados bem como congestão nasal, arritmias e fibrilhação, mais comum em doentes idosos. (85,103,104,108) O ácido nicotínico é responsável igualmente por provocar alterações oculares nomeadamente visão turva, edema das pálpebras, proptose, perda de cílios e edema macular, sintomas estes reversíveis e relacionados com a
34
dose. A bibliografia consultada reporta ainda casos de conjuntivite e ambliopia tóxica. (85,103,108) Devido aos seus possíveis efeitos teratogénicos, a utilização da Niacina está contraindicada em mulheres grávidas.(85)
1.8.5 Outros agentes modificadores de lípidos
• Ácido Gordos Ómega-3
Atualmente as Guidelines sugerem a utilização de ácidos gordos Ómega-3 como complemento de outras terapias caso a redução de triglicéridos não esteja a ser satisfatória ou em caso de hipertrigliceridemia grave. (109,110) Os ómega-3 prescritos pelos médicos contêm principalmente ácido eicosapentaenóico (EPA) e ácido docosahexaenóico (DHA) e reduzem em cerca de 30% os níveis plasmáticos de triglicéridos. (111,112) Relativamente ao mecanismo de ação este ainda não está comprovado, porém acredita-se que seja multifatorial. (112) Estudos recentes mostraram que os ómega-3 estão implícitos na diminuição da lipogénese hepática, no aumento da oxidação de ácidos gordos, na inibição de enzimas chave envolvidas na síntese de triglicéridos e no aumento da expressão da lipoproteína lípase. (111,112)
Relativamente aos efeitos adversos dos ácidos gordos ómega-3 as principais reações verificadas são referentes a alterações gastrointestinais nomeadamente náuseas, vómitos e diarreia. (113,114) Alguns estudos com estes Antidislipidémicos demonstraram também um aumento no tempo de sangramento após a sua utilização, porém sem ultrapassar os valores normais de referência.(115)
35
2 Objetivos
2.1 Objetivo geral
Caracterizar o perfil de notificação espontânea de reações adversas associadas a Antidislipidémicos na Região Sul de Portugal durante o período de 2012 a 2016.
2.2 Objetivos específicos
Relativamente às notificações espontâneas associadas aos Antidislipidémicos reportadas no período supracitado pretendeu-se:
• Quantificar a amostra face às restantes notificações recebidas no mesmo período; • Quantificar as notificações recebidas por ano, por instituição e por via de receção;
• Caracterizar o notificador responsável por reportar a RAM (se profissional de saúde e respetiva categoria ou se doente);
• Caracterizar o doente segundo género e idade;
• Caracterizar o medicamento suspeito da RAM segundo o número total de medicamentos associados, por tipo de medicamento (genérico ou não) e por classificação ATC (até ao 5º nível);
• Caracterizar as RAM reportadas segundo gravidade, número e classificação dos SOC (System Organ Class), evolução do estado e imputação de causalidade
36
3 Métodos
3.1 Desenho do estudo
Estudo observacional, de orientação transversal, para análise das notificações de suspeitas de reações adversas relacionadas com Antidislipidémicos, enviadas à Unidade de Farmacovigilância do Sul (UFS), diretamente por profissionais de saúde e utentes, no período 2012 a 2016.
3.2 Recolha de informação e tratamento de dados
A informação alvo de tratamento e análise foi recolhida a partir da base de dados das notificações de RAM reportadas à UFS, no período de 1 de janeiro de 2012 a 31 de dezembro de 2016. O registo de cada notificação diz respeito a uma única pessoa afetada que poderá estar associada a um ou mais medicamentos suspeitos e a uma ou mais RAM.
As RAM reportadas foram classificadas com recurso ao dicionário MedDRA (versões 15.0 à 19.1) por SOC/PT/LLT e os medicamentos suspeitos foram classificados segundo o sistema ATC (Anatomical Therapeutical Chemical).
Os dados existentes na base de dados da UFS, devidamente autorizados, foram exportados para um ficheiro Excel e alvo de análise.
3.3 Variáveis em estudo
As variáveis do estudo, presentes na base de dados que foram alvo de tratamento foram as seguintes:
A. Caracterização da Notificação
• Distribuição anual das RAM no período de 2012 a 2016;
• Via de receção (INFARMED, Portal RAM, site online da UFS, correio para a UFS, telefone UFS e e-mail UFS).
B. Caracterização do Notificador
• Categoria profissional (médico, farmacêutico, enfermeiro, delegado de farmacovigilância) ou utente;