UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Characterization of CFTR nonsense mutations
using novel CFTR minigenes
João Pedro Pacheco Conde de Amorim
DISSERTAÇÃO
MESTRADO EM BIOQUÍMICA Especialização em Bioquímica Médica
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Characterization of CFTR nonsense mutations
using novel CFTR minigenes
João Pedro Pacheco Conde de Amorim
DISSERTAÇÃO
MESTRADO EM BIOQUÍMICA Especialização em Bioquímica Médica Orientadores: Doutora Anabela S. Ramalho
Professora Doutora Margarida D. Amaral 2013
3
Index
I Agradecimentos ... 5 II Resumo ... 7 III Abstract ... 9 IV Abbreviations ...10 V Notes ...13 1 Introduction ...141.1 Cystic Fibrosis – Overview ...14
1.2 Clinical Features of Cystic Fibrosis ...15
1.3 CFTR structure and function ...16
1.4 Classification of CFTR mutations ...18
1.4.1- Class I – Mutations that lead to no protein production ...19
1.4.2- Class II – Mutants that prevent intracellular traffic ...19
1.4.3- Class III - Mutations affecting the regulation of the chloride channel ...19
1.4.4- Class IV - Mutations that lead to defective chloride transport ...20
1.4.5 – Class V – Mutations that lead reduced levels of protein ...20
1.4.6– Class VI – Reduced stability or altered regulation of separate ion channels ...20
1.5 Current and upcoming Cystic Fibrosis treatments ...20
1.6 Nonsense-mediated mRNA decay and CF nonsense mutations ...22
1.7 PTC-containing transcripts resistant to NMD ...24
1.9 Premature termination codon read-through ...25
2 Objectives ...27
3 Materials and Methods ...28
3.1 Production of vectors to study the susceptibility of CFTR mutant transcripts to NMD ...28
3.1.1 Bacterial strain ...28
3.1.2 Plasmid vectors ...28
3.1.3 Production of competent bacteria ...29
3.1.4 Transformation of competent bacteria ...29
3.1.5 DNA extraction and quantification ...30
4
3.1.7 Site-directed Mutagenesis ...31
3.1.8 DNA sequencing ...32
3.2 Production of transient and stable cell lines ...33
3.2.1 Characterization, culture and maintenance of cell lines ...33
3.2.2 Transient transfections ...34
3.2.3 Flp-In system for the establishment of stable cell lines ...34
3.3 RT-PCR analysis of CFTR PTC-containing transcripts ...36
3.3.1 RNA extraction ...36
3.3.2 cDNA synthesis ...38
3.3.3 Polymerase chain reaction ...37
3.3.3.1 Semi-quantitative analysis ...40
3.4 Biochemical and functional characterization of CFTR nonsense variants ...38
3.4.1 Preparation of total protein extracts ...38
3.4.2 Western blot ...40
3.4.3 Immunofluorescence ...39
3.4.4 Iodide Efflux ...40
3.5 Pharmacological treatments ...41
3.5.1 Pharmacological indirect inhibition of Nonsense-mediated mRNA decay ...41
3.5.2 Pharmacological induction of PTC read-through ...41
4 Results ...42
4.1 Production of plasmid vectors ...42
4.2 Evaluation of the effects of nonsense mutations on CFTR expression ...45
4.3 Assessment of NMD susceptibility of CFTR nonsense mutants ...48
4.4 Assessment of intracellular localization and function of CFTR nonsense mutants ...51
4.5 PTC read-through and NMD inhibition in CFTR nonsense mutants by chemical compounds ...54
5 Discussion ...55
6 Final remarks and future perspectives ...60
7 Bibliography ...62
8 Appendices ...70
Appendix I – pcDNA5/FRT plasmid map ...70
Appendix II – pOG44 plasmid map ...71
5
I
Agradecimentos
Gostaria de aproveitar este espaço, para a todos aqueles que de alguma forma contribuíram para a sua realização, e que me apoiaram ao longo deste último ano.
Agradeço em primeiro às minhas orientadoras, à Doutora Anabela S. Rama-lho por me ter aceitado no seu projeto, por toda a sua ajuda, disponibilidade e con-fiança, que foram essenciais para o desenvolvimento deste trabalho, e por estar sem-pre pronta a discutir atá as ideias mais absurdas. À Professora Margarida Amaral, agradeço a forma como me acolheu no seu grupo, e por providenciar um ambiente de trabalho altamente estimulante que sempre nos desafia a trabalhar e a pensar melhor, e a encarar os erros e tentativas falhadas como oportunidades que de al-guma forma podem ser aproveitadas.
Agradeço a todos os meus colegas de laboratório, que muitas vezes foram bem mais que isso, por toda ajuda, até nos momentos mais difíceis, e por fazerem com que o meu dia-a-dia nunca se tornasse rotineiro. Um enorme obrigado à Veró-nica, a quem devo muito do que aprendi e por sempre me dizer aquilo que precisava de ouvir, quer eu quisesse quer não. Agradeço à Sara Canato pela sua boa disposição e por nunca levar a mal nenhuma das minhas piadas, mesmo aquelas de gosto mais duvidoso, à Susana “Suz&Tina” Igreja e à sua cadela Lola por nunca deixarem que os meus dias se tornassem entediantes, à Ana Marta “Chica Bacana” Romão toda a ajuda nos stainnings, e por me apresentar a todo o universo de vídeos recônditos do Youtube, e por introduzir todo um novo padrão do que é a indumentária própria para o local de trabalho. À Ana Cachaço agradeço todas as lições de vida que retirei dos seus eloquentes monólogos e ainda toda ajuda em todo o trabalho de micrósco-pia. Agradeço ainda ao Prof. Carlos, à Marta, ao José, à Sara Afonso, Ines, Inna, Onófrio e Nikhil por todo o apoio.
Agradeço aos meus três mosqueteiros, Cátia, Carlos e Sid, por estarem sem-pre comigo nos bons e nos maus momentos durante o meu percurso nesta faculdade, com quem cresci e aprendi imenso, com quem sei que posso sempre contar para julgar os meus comportamentos mais infames.
Agradeço ainda aos meus amigos de longa data, Carlos e Graciano e por esta-rem sempre disponíveis quando preciso mesmo quando não lhes digo nada durante
6
meses, e à minha grande amiga Tânia, que mesmo do outro lado do Oceano Atlântico não se importa de me ouvir reclamar sobre tudo e mais alguma coisa e por encontrar sempre uma forma de me pôr a sorrir.
Por fim gostaria de agradecer à minha família. À minha tia Guida, ao meu tio Lico e ao Lima por se preocuparem sempre comigo por em várias instâncias terem-me dado o apoio necessário para concluir este trajeto. Ao terem-meu primo Francisco, por ser um autêntico irmão, com quem posso contar para tudo. E especialmente aos meus Avós, Luís e Isabel, que me criaram e educaram melhor do que se fossem meus pais, que fizeram todos os esforços e sacrifícios possíveis e imagináveis para que eu pudesse fazer sempre aquilo que ambicionava, e a quem devo tudo o que sou hoje.
7
II
Resumo
A Fibrose Quística (FQ) é a doença recessiva autossómica letal mais comum na população caucasiana e apresenta, na Europa, uma taxa de incidência de 1 em 3500 recém nascidos, enquanto que em Portugal, 1 em cada 6000 novos nados-vivos apresenta a doença. A doença é causada por mutações no gene CFTR (do inglês Cystic
Fibrosis Transmembrane Conductance Regulator) que levam à formação de uma
pro-teína (com o mesmo nome) com função anormal ou reduzida ou até à completa ini-bição da expressão da mesma). A CFTR exerce a sua função de canal de cloreto e de outros aniões na membrana apical de células epiteliais de vários tecidos.
Do ponto de vista clínico, a FQ é caracterizada por um rápido declínio da fun-ção pulmonar devido à obstrufun-ção das vias respiratórias causada por infeções bacte-rianas recorrentes e persistentes. Devido ao ambiente hiper-inflamatório provo-cado por estas infeções, a remodelação do tecido pulmonar ocorre a um ritmo au-mentado, que culmina na formação de fibrose no tecido e na consequente perda de função. Este fenótipo pulmonar é o principal responsável pela morbilidade e morta-lidade dos doentes com FQ. Para além do tecido pulmonar, outros órgãos e tecidos são igualmente afetados, sendo que os pacientes apresentam frequentemente pro-blemas digestivos graves e são geralmente inférteis (todos os homens e uma grande percentagem das mulheres).
Até à data, foram identificadas mais 1900 alterações no gene CFTR sendo a maioria causadora de doença, sendo a deleção do resíduo de fenilalanina na posição 508 da cadeia peptídica a mais comum das detetadas em pacientes e portadores (~90% de todos os casos). Mutações nonsense levam, na maioria dos casos, à degra-dação total ou quase total dos transcritos de CFTR, ao desencadearem o mecanismo de degradação do mRNA denominado NMD (do inglês Nonsense-Mediated mRNA
De-cay).
Estudos recentes, realizados com gene que codifica para a β-globina, demons-traram que a presença de codões stop prematuros em proximidade com o codão de início da tradução não desencadeiam a degradação dos transcritos uma vez que não levam à ativação de NMD. No entanto persiste a dúvida se tal sucederá em genes bastante maiores como é o caso do gene CFTR.
8
O principal objetivo deste trabalho passou, portanto, por uma maior compre-ensão do mecanismo de degradação NMD, no contexto de genes de grandes dimen-sões, usando como modelo o gene CFTR. Mais concretamente, pretendeu-se carac-terizar várias mutações nonsense, previamente detetadas em doentes com FQ e/ou portadores, localizadas em proximidade com o codão AUG (Q2X, S4X e Q39X), usando minigenes de CFTR gerados através de engenharia genética, bem como vali-dar esse modelo para o estudo de NMD, ao induzir este mesmo mecanismo com a mutação G542X, tal como haveria sido reportado em estudos prévios. Pretendeu-se ainda testar capacidade de vários fármacos em induzir o read-through das várias mutações nonsense.
Foram gerados vários plasmídeos codificando minigenes de CFTR, contendo intrões normais e artificialmente construídos, com as várias mutações estudadas e em seguida estabelecido um modelo de expressão estável e isogénica desses mes-mos minigenes em células HEK 293.
Através de RT-PCR foi demonstrado que a presença da mutação G542X na sequência do minigene de CFTR levou à ativação de NMD e consequente degradação dos transcritos de CFTR, enquanto os transcritos dos variantes com mutações pró-ximas do codão de iniciação não foram degradados.
Foi observada, a partir da análise por imunodeteção, a ocorrência da reinici-ação da tradução da CFTR nos variantes resistentes à degradreinici-ação por NMD, e que proteína produzida, apesar de não possuir a região N-terminal era capaz de migrar para a membrana celular. Ensaios de efluxo de iodeto indicaram que a proteína trun-cada apresentava atividade reduzida e retardada.
Não foram no entanto bem-sucedidas as tentativas de promover através de fármacos o read-through dos codões de stop prematuros em nenhum dos variantes.
Palavras-Chave: Fibrose Quística; CFTR; PTC; mutações nonsense próximas
9
III Abstract
Cystic Fibrosis is the most common lethal autosomic recessive disorder in the Caucasian population, affecting 1 in 6000 newborns in Portugal, and is caused by mutations in the CFTR gene which encodes for the CFTR protein.
Since its recognition, more than 1900 CFTR mutations have been identified, being the deletion of a phenylalanine at position 508 the most prevalent of all. Most nonsense mutations lead to complete loss of protein expression due to transcript quick degradation via NMD pathway. However recent studies of the β-globin gene showed some nonsense variants with AUG-proximal PTCs are resistant to this deg-radation mechanism.
The principle aims of this study were to generate and validate a CFTR minigene model that could be used for the study of NMD in the context of CFTR, a far larger gene than β-globin (~190kb vs ~4 kb), to characterize, using said model, naturally occurring AUG-proximal CFTR nonsense mutations, and test the efficacy several pharmacological read-through promoting agents.
CFTR plasmid minigenes containing normal and artificially constructed in-trons as well as several naturally occurring nonsense mutations (Q2X, S4X, Q39X and G542X) were generated and a model for stable and isogenic expression of these minigenes in HEK 293 cells established.
By RT-PCR analysis we showed that the presence of the mutation G542X, was at the minigene sequence was able to activate NMD, while CFTR transcripts with AUG-proximal nonsense mutations were not degraded.
It was shown by western blot essays that AUG-proximal nonsense variants expressed a truncated form of CFTR lacking the N-terminus region which probably resulted from the occurrence of translation re-initiation. Residual levels of this trun-cated form of CFTR were also detected at the cytoplasmic membrane and iodide ef-flux essays indicated that it possessed reduced and delayed channel function.
However, attempts of pharmacologically promote PTC read-through in any nonsense variant were deemed unsuccessful.
10
IV Abbreviations
ABC – ATP-binding cassette
ABCC7 - ATP-binding cassette sub-family C member 7 AFT – arginine-framed-tripeptide
ATP – adenosine triphosphate BSA – bovine serum albumin
CaCC – calcium activated chloride channel cAMP – cyclic adenosine monophosphate cDNA – complementary DNA
CF – cystic fibrosis
CFBE41o-/CFBE – cystic fibrosis bronchial epithelial (cell line) CFF – cystic fibrosis foundation
CFTR – cystic fibrosis transmembrane conductance regulator CHX – cyclohexamide
DAPI – 4',6-diamidino-2-phenylindole dsDNA – double stranded DNA
DMSO – dimethylsulfoxide eIF - eukaryotic initiation factor EJC – exon junction complex
EMEM – Eagle’s minimum essential medium ENaC – epithelial sodium channel
ER – endoplasmic reticulum eRF – eukaryotic release factors
ERQC – endoplasmic reticulum quality control FBS – fetal bovine serum
FLP – flippase
FRT- flippase recombination target GDP – guanosine diphosphate GFP – green fluorescent protein GTP – guanosine triphosphate
11
HBSS - Hank’s balanced salt solution IgG – Immunoglobulin G
IVS – intervening sequence, intron LB – Luria broth
MAPK – mitogen-activated protein kinase MEM – minimal essential medium
mRNA – messenger RNA
MSD – membrane-spanning domain NBD – nucleotide binding domain
NHERF – Na+/H+ exchanger regulatory cofactor NMD – nonsense mediated mRNA decay
ORCC – outwardly rectifying chloride channel ORF – open reading frame
PABPC1 - poly-A binding protein complex 1 PAGE – polyacrylamide gel electrophoresis PBS – phosphate buffered saline
PBS-T – phosphate buffered saline supplemented with 0.1% Tween PCR – polymerase chain reaction
PDZ – post synaptic density protein (PSD95), Drosophila disc large tumor suppressor
(Dlg1), and zonula occludens-1 protein (zo-1) domain PKA – protein kinase A
PM - plasma membrane
PTC – premature termination codon PVDF – polyvinylidene difluoride R-domain – regulatory domain
ROMK – renal outer medullary potassium channel RT – room temperature
RT-PCR – Reverse transcriptase polimerase chain reaction SD – standard deviation
SDS – sodium dodecylsulphate siRNA – small interfering RNA
12
SNAP23 - Synaptosomal-associated protein 23
SNARE – soluble N-ethyl-maleimide sensitive factor Attachment Protein re-ceptors
sq-RT-PCR - semi quantitative reverse transcriptase polymerase chain reac-tion
SYN1A – syntaxin 1A
TM – transmembrane segment uORF - upstream open reading frame UPF - up-frameshift (proteins)
13
V
Notes
The CFTR mutations refered in the text are denominaded using the legacy name1. The CFTR sequence used here was same deposited in the Genbank with the accession number M26886. The exons and introns numbering was done using the legacy numbers (from 1 to 24 including 6a and 6bm 14a and14b and 17a and 17b; and not from 1 to 27).
14
1
Introduction
1.1 Cystic Fibrosis – Overview
Cystic Fibrosis (CF, MIM#2109700) is the most common lethal autosomal re-cessive disorder in the Caucasian population2, affecting 1 in 3500 newborns in Eu-rope corresponding to ~30000 patients3. The disease frequency is variable among ethnic groups, being highest in Northeastern Europe and quite rare among oriental populations4. In Portugal it is estimated that 1 in 6000 newborns are affected5.
The first description of CF as a disorder in its own right was made in 1938 by Dr. Dorothy Hansine Andersen, based on autopsy studies of malnourished infants, who also came to find that it had a recessive autosomal pattern of inheritance6. At the time, cystic fibrosis was described as a digestive disorder, since the first detect-able symptoms were intestinal obstruction, and was associated with progressive fi-brosis of the pancreatic tissue7. Since infants affected with the disease died at a very young age, these were the only noticeable symptoms. With the advances in the healthcare, the digestive problems in newborns and infants were gradually over-came, revealing the onset of problems in the respiratory tract with the progression of the disease, which became the major cause for morbidity and mortality for CF patients.
During the 1948 heat wave in New York, Dr. Paul di Sant’Agnese observed that babies presented increased risk for heat prostration, which led to the discovery that CF patient’s presented sweat with an extremely high concentration of salt, which persisted after the heat wave subsided8. In 1959, the sweat test became pri-mary test for CF diagnosis. This excess of salt in the sweat of CF patients, was later identified as a result of defective transport of chloride (Cl-) by the sweat glands9. In other studies, the chloride movement, from epithelia to airway lumen, was found diminished10, while it was possible to observe an increased reabsorption of sodium (Na+) in the epithelium11. This defective chloride transport was also detected in the pancreas and intestinal epithelium, the other most affected tissues4.
In 1989, was identified, using a positional cloning strategy, that a single gene was responsible for onset of the disease. The discovery of the CF gene led to the demonstration that the impaired chloride transport is due to the failure of a
cAMP-15
regulated Cl- channel, which is expressed in a large number of epithelial tissues, and was then cautiously named cystic fibroses transmembrane condunctance regulator (CFTR or ABCC7; MIM# 602421)12,13,14.
All CF patients analyzed, were found to harbor mutations in the CFTR gene, the most common of which being a deletion of three nucleotides encoding for a phe-nylalanine in position 508 (commonly termed as ΔF508 or F508del)13
1.2 Clinical Features of Cystic Fibrosis
As was described in the previous section, CF is caused by mutations in CFTR, an anion selective ion channel, required for the normal function of epithelial lining the airways, intestinal tract, ducts in the pancreas, as well as salivary and sweat glands15, and the absence of its activity results in the failure of ionic and water ho-meostasis at exocrine epithelial surfaces16.
In the respiratory tract, CF is manifested by the obstruction of the airways by thick, dehydrated mucus that prevents proper mucocilliary clearance17. This leads to recurring bacterial infections, especially Pseudomonas aeruginosa and
Staphylococcus aureus species, that generate a hyper inflammation environment in
the lungs of patients from a very early age18, which exacerbates tissue remodeling processes and fibrosis19. This degradation of the lung tissue and loss of its function is the main cause of morbidity and mortality among CF patients20.
In 85% of the CF patients present defects in the gastrointestinal tract, namely pancreatic insufficiency as a result of the obstruction of the pancreatic ducts, intes-tinal obstruction called meconium ileus, and some develop liver disease at some time during the course of the disease. In adults with CF, infertility is almost universal in males, due to congenital bilateral absence of the vas deferens, and is also frequent in females. Patients also have elevated concentrations of sodium chloride in the sweat; in fact, the sweat test, which measures the amount of salt in the sweat of pa-tients, is still one of the fundamental tools for the establishment of a CF diagnosis21.
16
1.3 CFTR structure and function
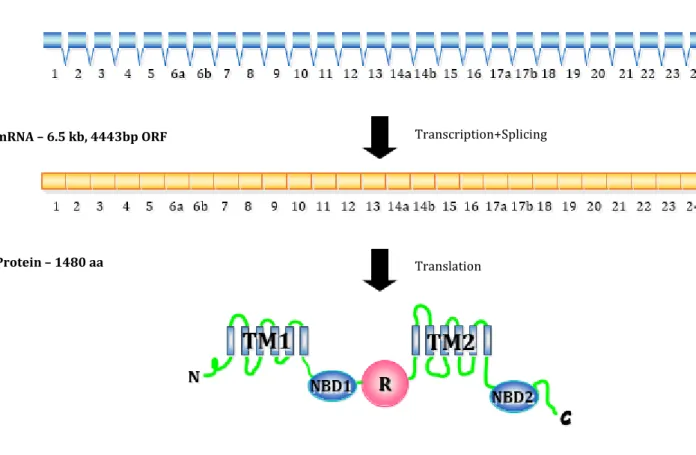
The CFTR gene (or ABCC7) is located on the long arm of chromosome 7, at the region 7q31.212,13,14, and is one of the largest human genes, spanning ~190 kb. After transcription and splicing its mRNA comprises 6129 bp4, of which 4443 bases code to a protein with 1480 amino acid residues, after translation22. The gene con-sists of TATA-less promoter, 27 exons and 26 introns (Figure 1.1)23. The sizes of both exons and introns vary greatly, with the exons ranging from the 38 bp of exon 14b to the 724 bp of exon 13, while the smallest intron (intron 22) comprises 600bp and 28085 bp the largest (intron 10)24.
Figure 1.1 Scheme illustrating the CFTR gene, mRNA and protein. TM - transmembrane segments cluster (TM1
and TM2); NBD – nucleotide-binding domain (NBD1 and NBD2); R- regulatory domain; N – amino terminal; C – Carboxyl terminal; aa – amino acid residue. Adapted from Zielinski and Tsui (1995)25 by MD Amaral
Soon after its discovery, the CFTR protein was identified as a member of the ATP binding cassette (ABC) transporter family due to structure similarity26. Like other ABC transporters, it has two membrane spanning domains (MSDs) with six
mRNA – 6.5 kb, 4443bp ORF
Protein – 1480 aa Gene – 190 kb
Transcription+Splicing
17
transmembrane segments each, portions of which form the pore through which an-ions pass, two nucleotide binding domains (NBD1 and NBD2) and regulatory do-main (RD). Both NBDs bind and hydrolyze ATP, which drives channel opening and closure, respectively27, while the RD, absent in all other ABC transporters, contains consensus sites for phosphorylation by various kinases. Phosphorylation of the RD by PKA in response to cyclic AMP is regarded as the major determinant for opening of the channel28. In the C-terminus, CFTR has a PSD95, Dlg1, ZO-1 (PDZ)-binding motif through which it is involved in complex PDZ-based protein interaction net-works29.
The main function attributed to CFTR is that of an Cl- conducting channel30. However, unlike other ABC transporters, CFTR is unable to drive ion transport con-trary to a gradient, thus functioning as passive channel that allows bidirectional flow of ions when open31.
The gating of the CFTR Cl− channel is tightly regulated by the balance of ki-nase and phosphatase activity in the cell and by cellular ATP levels22.
In addition to chloride, CFTR is also able to transport other anions such as, iodide (I-), bromide (Br-), nitrate (NO3-), bicarbonate (HCO3-) and glu-conate(HOCH2(CHOH)4COO-)32–34, and also glutathione35 and its thiocyanate conju-gates36.
While reported, it is still debated whether CFTR is responsible for the transport of HCO3- in vivo37. Permeability of CFTR this anion is quite low when com-pared to that of Cl- (25% of the permeability to chloride), and its main transporter, the Cl-/HCO3- transporter, is also expressed in most secretory epithelia. However, HCO3- transport is defective in patients with CF, which partially accounts for the loss of pancreatic function, which indicates that if not directly transporting bicarbonate, CFTR might act upon the regulation of the process38.
In fact, regulation of other channels present at the cell membrane, such as the epithelial sodium channel (ENaC)39, the outwardly rectifying channels(ORCCs)40, the renal outer medullary potassium channel (ROMK)41 and the calcium activated chloride channels (CaCCs)42, has been attributed to CFTR. However the exact mech-anisms by which CFTR exerts its regulatory function are not yet well established, because it is hard to distinguish between the effects cause by changes in CFTR itself and those that arise from an altered chloride conductance43
18
1.4 Classification of CFTR mutations
As to date, there were described more than 1940 CFTR mutations, a number that comprises disease causing mutations and polymorphisms that do not affect the carriers’ phenotype. From these mutations, 40.2% are missense mutations (782 mu-tations), 15.9% frameshift mutations (309), 11.6% splicing mutations (226), 8.3% nonsense mutations (161), 4.5% are deletions (87) and 0.8% mutations in the pro-moting region (15)1.
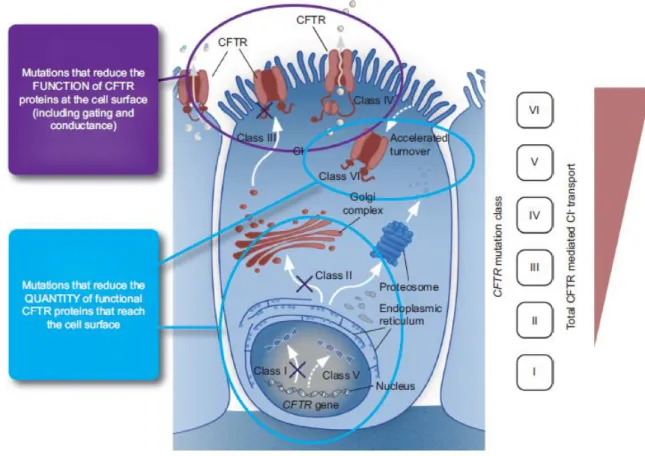
Currently these mutations are also divided in 6 different, classes defined ac-cording to the molecular changes caused by the different CFTR variants (Figure 1.2)44,45.
Figure 2.2 Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations are categorised into
six classes. Mutation classes I, II, V and VI result in an absence or reduced quantity of CFTR protein at the cell membrane, whereas mutation classes III and IV influence the function or activity of CFTR at the cell membrane. Class I mutations are associated with the greatest disruption to CFTR-mediated chloride transport; in general, chloride transport gradually increases through the remaining five classes, with the greatest activity being ob-served in Class IV–VI mutations. Adapted from Derichs (2013)46.
19
1.4.1- Class I – Mutations that lead to no protein production
Class I mutations are those that can give origin to premature terminations codons (PTC), such as nonsense mutations or frameshift mutations, or alter the nor-mal splicing pattern, and completely inhibit CFTR synthesis. These mutations are thus associated with more severe CF phenotypes47.
Modification of the codons that codifies for the glycine in the position 542, arginine in position 553 or for the glutamine in position 637 into a stop codons (G542x, R553X and Q637X, respectively), are examples of class I mutations. G542X and R553X are two of the most common mutations in European countries after the F508del mutation48. These mutations lead to total or near total degradation of the respective transcripts by nonsense mediated mRNA decay (NMD) preventing any protein production. Any residual proteins that might be translated from these tran-scripts correspond to a truncated and/or instable forms that are unable to pass the cellular quality control mechanism, and are quickly degraded. These mutations will be discussed with more depth in section 1.9.
1.4.2- Class II – Mutants that prevent intracellular traffic
CFTR variants with class II mutations are not correctly folded and processed, and are thus incapable to reach apical membrane of epithelial cells. F508del, the most common CF causing mutation, is a representative of this class of CFTR muta-tions. By not being correctly folded, F508del is unable to be fully glycosylated and is sequestered in the endoplasmic reticulum (ER), without ever being transported to the apical membrane.
1.4.3- Class III - Mutations affecting the regulation of the chloride
channel
These mutations cause a defective response of CFTR to the phosphorylation of the RD by PKA after activation by cAMP. These mutations are generally located in the NBDs, affecting their interaction with ATP, and the thus interfering with the cor-rect gating of the chloride channel. Class III mutations effects range from slight loss of function (G551S, glycine to serine), and reduction of the response to cAMP (S1255P, serine to proline), to total loss of function (G551D, glycine to aspartate).
20
1.4.4- Class IV - Mutations that lead to defective chloride transport
Class IV mutations, such as R117H (arginine to histidine), lead to the synthe-sis of proteins able to be transported to the membrane and respond to stimulus, but have reduced chloride transport function. Patients with these mutations an inter-mediate CF phenotype
1.4.5 – Class V – Mutations that lead reduced levels of protein
This class includes missense mutations (A455E, alanine to glutamate) and those that affect the correct splicing of pre-mRNA (3272-26A>G or TGmTn sequence in intron 8), which produce low levels of functional protein and reduced levels of transcripts49,50. These mutations are related to mild CF phenotypes.
1.4.6– Class VI – Reduced stability or altered regulation of separate
ion channels
In this group are also included membrane-rescued F508del, the deletion of the CFTR start site (120del23), N287Y (asparagine to tyrosine), and variants with nonsense or frameshift mutations that originate PTCs in the last exon of the CFTR gene and are translated in truncated proteins near the carboxyl terminus (C-termi-nus).
These proteins while being correctly processed, transported to the mem-brane and presenting normal function are unstable and have an increased turnover at the cell surface51.
1.5 Current and upcoming Cystic Fibrosis treatments
Cystic fibrosis is a life-threatening disease, and CF patients have a short mean life expectancy of ~37 years.Lung disease is the main cause of morbidity and mortality in CF patients, so most of the different therapeutic approaches currently used focus on the ameliora-tion of the respiratory symptoms by antibiotics and anti-inflammatory treatments, that combat chronic infections and consequent chronic lung inflammation, and
21
treatments directed towards restoring the levels of airway surface liquid and reduc-ing mucus thickness52. Lung transplantation is still the ultimate and choice for pa-tients with end-stage lung disease.
However, these treatments are not long-term effective, with pulmonary in-fections recurring even after treatment, and survival rate after 4 years on lung trans-plant is less than 50%53. Thus new therapies must be developed to target not symp-toms, but the basic molecular defect of CF54.
Due to the large spectrum of CF causing mutations, mutation-specific are not a viable option. However, common therapeutic strategies directed towards groups of mutations that lead to similar phenotypes, as mentioned in the previous section, were and are being developed with promising results55.
Aminoglycosides and non-aminoglycosides were described to being able to suppress some class I nonsense mutations, leading to production of full-length pro-tein56,57, a topic that will be further discussed in section 1.8.
For class II mutations, chemical corrector and pharmacological chaperones were shown to being able to stabilize CFTR structure and promote its correct fold-ing, thus correcting the traffic defect58. From these, VX-809 was able correct F508del-CFTR folding and is currently in Phase III clinical trial59.
CFTR activators, such as genistein flavonoids, were tested in class II muta-tions in order to bypass the channel gating defect of these variants, acting thus as potentiators. Ivacaftor, a CFTR potentiatior developed by Vertex Pharmaceuticals, is now approved to be prescribed to patients with the G551D mutation, after success-ful clinical trials showing its ability to restore CFTR function60. The kind of thera-peutic approach might also be implemented in class IV mutations, which also pre-sent reduced CFTR function.
A combination of therapies be implemented of the previous therapies might be implemented in class V and VI mutations, from potentiators to compounds pro-mote the bypass of nonsense mutations in order to enhance CFTR activity and/or increase its protein levels. Drugs that correct splicing defects might also be able to increase the levels of normal CFTR transcripts, and thus a viable therapeutic option for patients with these mutations61.
CF was also proposed as good target for gene therapy, but initial studies were not able to provide encouraging results due to the difficulty of gene transfer into the
22
lung. However, recent studies show that lentiviral vectors may be able to evade the immune system and, thereby, increase gene transfer efficacy. In addition, systemic and topical administration of a variety of stem/progenitor cells, as well as first at-tempts as producing a tissue-engineered lung, are starting to appear as viable cell therapy-based approaches for CF62.
1.6 Nonsense-mediated mRNA decay and CF nonsense
mutations
The expression of protein-coding genes in eukaryotes involves the orchestra-tion of transcriporchestra-tional and posttranscriporchestra-tional processes. To ensure the fidelity of these processes, the eukaryotic cell has evolved several quality control mechanisms. One such mechanism is the nonsense-mediated mRNA decay (NMD) pathway. NMD rids the cell of aberrant mRNAs that have acquired premature translation termina-tion codons (PTCs) and may, once translated give origin to truncated proteins with potentially deleterious function63.
Recent data shows that in addition to mRNA containing PTCs, NMD also tar-gets a subset of endogenous transcripts64. These endogenous NMD targets do not contain PTCs, although some have recognizable features such as upstream open reading frames (uORFs) that would make translation termination events appear as premature65.
Erroneous gene expression such as errors in transcription, abnormal mRNA processing, somatic mutations, abnormal or alternative splicing, or nonproductive programmed DNA rearrangements can originate PTC-containing mRNA. PTCs can also be inherited as in the case of genetic mutations in disease genes, like CFTR66,67. However (as mentioned in section 1.7), not all mRNAs with PTCS in their sequence trigger NMD. Those positioned less than 50 bp close to next exon junction complex (EJC), or near to the normal termination and initiation codons fail to activate NMD68,69.
The core NMD machinery comprises three trans-acting factors, called up-frameshift (UPF) proteins, UPF1, UPF2 and UPF3. During translation, stop codons
23
are recognized by eukaryotic release factors eRF1 and eRF3 present in the transla-tion apparatus, which recruit UPF1, which in turn recruits protein kinase SMG-1, that together with eRFs forms the SURF complex and eventually lead to translation termination and ribosomal dissociation. However recognition of PTC leads to the interaction of the SURF complex with UPF2 and 3 present at the downstream EJC leads to UPF1 phosphorylation that triggers the transcript degradation, as observed in figure 1.369.
Figure 3.3 - Early molecular events preparing an mRNA to be degraded by nonsense-mediated mRNA decay
(NMD). Translation of an mRNA during the pioneer round of translation (step 1; also see Figure 2) leads to recognition of the stop codon by the eukaryotic release factors eRF1 and eRF3, which recruit the NMD factor UPF1 (labeled 1; step 2) (22, 124). UPF1, in turn, recruits the protein kinase SMG-1 (S1), which together with the eRFs forms a transient complex called SURF (step 3) (22). In an aberrant mRNA like the one shown, the SURF complex interacts with an EJC downstream (step 4). This interaction may be an obligate requirement for SMG-1 to phosphorylate UPF1 (step 5), which then probably triggers subsequent steps that ultimately degrade the mRNA (see Figure 4) and recycles release factors and the 40S and 60S ribosomal subunits (step 6). Abbrevia-tions: CBC, cap-binding complex; EJC, exon-junction complex; S1, protein kinase SMG-1; SMG-1, suppressor with morphogenetic effect on genitalia-1; SURF complex, the SMG-1, UPF1, eRF complex; UPF1 (labeled 1), UPF2 (la-beled 2), UPF3b (la(la-beled 3b), up-frameshift proteins. Adapted from Chang (2007)69.
24
In CFTR, several naturally occurring mutations give rise to a PTCs, such as G542X and W1282X and Q39X, thus having transcripts prone to degradation medi-ated by the NMD pathway. However, NMD efficiency has been shown to be variable between CF patients with different nonsense mutations, as presented in figure 1.4.
Figure 4.4 Levels of several CFTR transcripts decay normalized for wt-CFTR, obtained from rectal biopsies
col-lected from CF patients with different PTC-containing mutations in one of the alleles. Each bar corresponds to a different patient. (AS Ramalho, unpublished data, with premission)
1.7 PTC-containing transcripts resistant to NMD
As mentioned in previous sections, not all PTC-containing transcripts un-dergo NMD.
To trigger NMD, at least EJC must be present downstream of the PTC, in fact mRNAs from intronless genes or with a PTC in the last exon or that spans two exons are immune to NMD70–72. The PTC must also be located >50-55 nts upstream of the 3’-most EJC in order to reduce the mRNA abundance73. However, several studies conducted with the β-globin gene that PTCs presenting the needed requisites, but located in proximity to the initiation codon were able to bypass NMD68.
Two models were proposed to explain the NMD resistance presented by these AUG-proximal nonsense mutants. The first, studied in shows that during trans-lation initiation, poly-A binding protein complex 1 (PABPC1) interacts with the eu-karyotic initiation factor 4 (eIF4G). This interaction indirectly tethers PABPC1 to the 40S ribosomal subunit via the interaction of eIF4G with eIF3 subunits. The resulting configuration brings PABPC1 into in the vicinity of the AUG initiation codon the 40S
25
during the initial phase of translation elongation brings it into close contact with an AUG-proximal PTC in a transcript where the ORF is quite short. This proximity to the PTC allows PABPC1 to interact with the release factor eRF3 at the termination complex, thus impairing the association of UPF1 to the ribonucleoprotein complex, resulting in efficient translation termination and inhibition of NMD74.
While other postulates that this immunity NMD, is due to the presence of al-ternative initiation sites downstream of the PTC, and that the presence of ribosome initiating the translation elongation process at those sites, might contribute to the transcript stability and prevents NMD triggering75.
1.9 Premature termination codon read-through
As above-mentioned the presence of in-frame PTCs can lead to the produc-tion of truncated nonfuncproduc-tional or deleterious proteins, or trigger NMD resulting no production of protein at all. Therapeutic approaches aimed at promoting transla-tional read-through of the PTCs (figure 1.5), and thus enable the synthesis and ex-pression of full-length functional proteins were developed with relatively positive results76.
Read-through of PTCs can be achieved by suppressor transfer RNAs (tRNAs), factors that decrease translation-termination efficiency, such as small-interfering RNAs (siRNAs) directed against the translation-termination factors and RNA anti-sense that targets the nonanti-sense mutation region77.
Another extensively studied approach that has reached clinical trials is read-through by drugs affecting the ribosome decoding site, such as aminoglycoside an-tibiotics like G418 and Gentamicin. However, aminoglycosides have severe side ef-fects, such as nephrotoxicity and ototoxicity, when used at high concentrations and/or used long-term78. While searching for non-aminoglycosides capable of pro-moting read-through, a high-throughput screening revealed a small molecule, Ata-luren (previously PTC124), which can read-through PTCs without severe side ef-fects79. This molecule is now in phase 3 clinical trial for CF patients with nonsense
pa-26
tients responded to the treatment and the patients that are being treated with amino-glycosides antibiotic due to bacterial infection prior to the clinical trial were the ones to show worse results81.
Figure 1.5. The effect of read-through strategies on protein translation. Several ways exist for modifying the nor-mal processes that occur during termination by a premature termination codon (PTC). (a) Nornor-mal translation, (b) In the presence of a PTC, there is no tRNA matching the stop codon. Instead, the release factors eRF1 and eRF3 bind and terminate translation by releasing the polypeptide, which is a truncated protein. (c) When ami-noglycosides, PTC124, or negamycin bind to the rRNA there is no premature termination of translation, despite the presence of a PTC. An alteration of rRNA conformation is induced upon binding of the small molecule, reduc-ing the accuracy of the codon–anticodon interaction. This enables incorporation of an aminoacetylated tRNA, moving translation towards the canonical stop codon and originating a full-length protein. These proteins often contain missense mutations at the PTC location because near-cognate tRNAs may recognize the codon sequence by two nucleotides, allowing the insertion of near-cognate amino acids instead of the cognate amino acid. There-fore, the proteins produced may be functional or non-functional depending on whether or not the missense mu-tations affect conformation and binding to other proteins. The interaction of aminoglycosides, negamycin, and PTC124 with the rRNA may be different. In this picture, and for the sake of simplicity, the interaction between the read-through agent and the rRNA is depicted at the same spot in the three strategies. (d) Depletion of release factors eRF1 and/or eRF3 leaves the A site available to the entrance of any tRNA that can interact with the PTC, promoting missense read-through. This may lead to the production of a full-length protein carrying missense mutations. (e) The suppressor-tRNA anticodon is mutated to be complementary to the PTC, so it is able to rec-ognize the PTC and insert the cognate amino acid. A competition occurs between suppressor-tRNA and the re-lease factors eRF1 and eRF3 for the A site of rRNA. When the suppressor-tRNA enters the A site, with successful interaction with the mRNA PTC, the cognate amino acid is bound to the nascent polypeptide, with read-through of the PTC, and a normal full-length protein is produced. Adapted from Bordeira-Carriço (2012) 82
27
2
Objectives
The main objective of the present work was to gain a better understanding of the NMD mechanism in large genes using as model the CFTR gene.
More specifically, we intended to characterize the naturally occurring CFTR AUG-proximal nonsense mutations, Q2X, S4X and Q39X using novel CFTR mini-genes by testing their ability to escape NMD in comparison to other CFTR nonsense mutations which induce NMD, such as G542X. Furthermore, we aimed also to deter-mine the effect of several pharmacological compounds in read-through transcripts containing these mutations.
To accomplish the main objectives, this work was divided into several tasks that were necessary to achieve, as described below:
1) Generate CFTR minigenes containing several CF-causing nonsense mutations;
2) Validate those minigenes as bonafide NMD models of CFTR transcripts in immortalized human embryonic kidney (HEK293) cells;
3) Test the ability of different CFTR nonsense mutations to trigger NMD; 4) Investigate the possible cause for NMD resistance of CFTR transcripts
containing AUG-proximal nonsense mutations;
5) Test the potential of read-through promoting drugs to restore full-length CFTR expression of nonsense variants.
28
3
Materials and Methods
3.1 Production of vectors to study the susceptibility of
CFTR mutant transcripts to NMD
3.1.1 Bacterial strain
The bacterial strain used for cloning and DNA amplification was XL1-Blue (Stratagene), which is tetracycline resistant. XL1-Blue cells are endonuclease (endA) deficient, which greatly improves the quality of miniprep DNA, and are recombina-tion (recA) deficient, improving insert stability. The hsdR mutarecombina-tion prevents the cleavage of cloned DNA by the EcoK endonuclease system.
The lacIqZΔ M15 gene on the F´ episome allows blue-white color screening. XL1-Blue Genotype: recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F´ proAB lacIqZΔM15 Tn10 (Tetr)]. (Genes listed signify mutant alleles. Genes on the F´ epi-some, however, are wild-type unless indicated otherwise).
3.1.2 Plasmid vectors
pcDNA5/FRT/CFTR wt (kindly provided by the Garry Cutting lab, at Johns Hopkins Hospital,Baltimore,USA), comprises the complementary DNA (cDNA) from the CFTR wild-type gene from position 122 to 4725 (Genebank accession number M28668) in the PmeI site of the multi-cloning site of pCDNA5/FRT vector (Invitro-gen). All the others recombinant vectors are produced using this as template. pcDNA5/FRT/CFTR+IVS4art+IVS5, corresponds to the wt-CFTR cDNA with the in-sertion of artificial constructed intron (IVS) 4 (with approximately ~300nts of the 5’ of the normal IVS4 sequence joined with ~300 nts 3’ of the of the normal IVS4 sequence) between exon 4 and 5 and insertion of normal full-length IVS5 sequence between exon 5 and 6a, pcDNA5/FRT/CFTR+IVS13+IVS14a+IVS14b, wt-CFTR cDNA with the insertion of normal intron 13 sequence, artificial constructed IVS14a (produced as IVS4art) and normal sequence of IVS14b introduced in the correct lo-cation of the introns, and pOG44, a vector which encodes for the flipase recom-binase. These vectors produced in the lab prior to this work. The
29
pCDNA5/FRT/CFTR vectores containing introns will be hereafter referred as CFTR minigenes.
Maps and cloning schemes for these vectors are available in Appendices. All these plasmids contain the ampicillin resistance gene, which was used for selection of transformed bacteria.
The empty vector (pcDNA5/FRT) was produced here thought the removal of
fragment containing the CFTR open reading frame of p
cDNA5/FRT/CFTR+IVS14aart+IVS14b+IVS15, as described in section 3.1.6. Non-sense mutants of CFTR (Q2X, S4X, Q39X and G542X) vectors were obtained by site-directed mutagenesis using as template the CFTR minigenes as described in section 3.1.7.
3.1.3 Production of competent bacteria
Bacteria were plated in LB-agar medium supplemented with tetracycline (25 µg/ml) and a single colony was used to inoculate 10mL of LB medium overnight at 37ºC with adequate shaking (220rpm). Tis pre-inoculum was the used to inoculate at dilution 1/100 100mL of LB medium, grown in the previous conditions to a final concentration of 5 × 107 bacteria/mL (0.3 abs at 600nm). Bacteria were transferred to ice and centrifuged at 100g for 15min at 4ºC. The pellet was the resuspended and incubated on ice for 15 min in 33mL of RF1 buffer (100mM RbCl, 50mM MnCl2, 30mM KCH3COO pH 7.5, 10mM CaCL2, 15% (w/v) glycerol, pH 5.8) and pelleted again in the same conditions following a resuspention and incubation of 15min on ice in 8.3mL of RF2 buffer (10mM MOPS, 10mM RbCl, 15mM CaCl2, 15% (w/v) glyc-erol, pH5.8). 200µL aliquots were then rapidly frozen with liquid nitrogen and stored at -80ºC.
3.1.4 Transformation of competent bacteria
Bacteria were transformed by incubating 200 µL of competent cells with DNA (0.1-10ng of plasmid DNA) for 45 min on ice, followed by heat-shock (2min at 42ºC), 90sec incubation on ice, and then allowing antibiotic resistance to be expressed by growth in antibiotic-free LB medium for 1 h at 37ºC at 220rpm. Bacteria were the
30
pelleted (5000g for 2min), the supernatant was discarded and the pellet resus-pended in the remaining supernatant. The suspension was then plated into LB-agar supplemented with 100µg/mL ampicillin and left to grow o/n at 37ºC.
Transformed bacteria colonies were grown o/n in 10mL of LB medium sup-plemented with 100µg/mL ampicillin at 37ºC at 220rpm and used the following day to extract plasmid DNA.
Individual clones were stored at -20ºC in 1.5mL of LB medium supplemented with 15% (w/v).
3.1.5 DNA extraction and quantification
Small scale plasmid DNA was purified with the QIAprep Spin Miniprep Kit (QIAGEN) according to the manufacturer’s instructions.
DNA concentration was determined by measurement of absorbance at 260nm (one absorbance unit corresponding to 50µg/mL of dsDNA using Nanodrop 200 spectrophotometer (Alfagene) and its purity was evaluated by the assessment of the A260/A280 ratio. Only DNA with a ratio above 1.8 was considered pure enough for further use. Only DNA concentrations above 500ng/µL were considered acceptable for cell transfection.
3.1.6 Cloning-out
In order to produce the empty vector pcDNA5/FRT, the fragment containing
the CFTR gene was cloned-out from the plasmid
pcDNA5/FRT/CFTR+IVS14aart+IVS14b+IVS15. The plasmid was hydrolized with
PmeI endonuclease (Fermentas) and the resulting fragments were separated by
aga-rose gel electrophoresis. The band containing the fragment corresponding to the empty vector were excised from the gel and frozen for 10 min at -80ºC following a centrifugation of 5 min at 10000 g. The agarose gel was then discarded and the liq-uid resulting from the previous centrifugation was incubated with T4-DNA ligase and ligase buffer (Roche) for 5 min at room temperature. Bacteria were then trans-formed with the ligation product, plated into LB-agar supplemented with 100 µg/mL ampicillin and left to grow o/n at 37ºC.
31
Single colonies were then grown o/n in 10 mL of LB medium supplemented with 100 µg/mL ampicillin for plasmid extraction and sequencing.
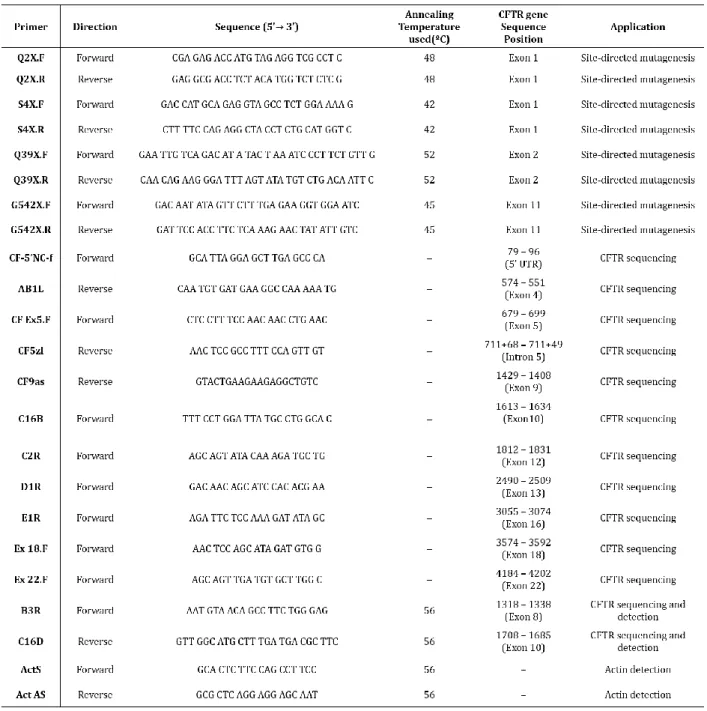
3.1.7 Site-directed Mutagenesis
Nonsense mutations were introduced into pcDNA5/FRT/CFTR+IVS4art+ IVS5 and pcDNA5/FRT/CFTR+IVS14aart+IVS14b+IVS15 using the KOD Hot Start Kit (Toyobo) with complementary pairs of mutagenic primers described in ta-ble 3.2. The PCR programs used are displayed in tata-ble 3.1. The amplification of plas-mids that underwent site-directed mutagenesis was confirmed by agarose gel elec-trophoresis, and the PCR products incubated with DpnI, a restriction enzyme that specifically hydrolyzes methylated and hemi-methylated DNA, in order to com-pletely degrade the template DNA, that is heavy methylated due to its bacterial origin, giving origin to pure sample of the mutagenized plasmid that was synthetized
in vitro.
After hydrolysis, bacteria were transformed with PCR products. Following transformation, plasmid DNA was extracted and the presence of each mutation was confirmed by automatic DNA sequencing.
32
3.1.8 DNA sequencing
Plasmid DNA were purified as described in section 3.1.5. The sequencing reac-tion were outsourced to StabVida, using BigDye Terminator 3 (Applied Biosystems) and analyzed in an ABI 3730XL sequencer (Applied Biosystems). The sequencing pri-mers used are in table 3.1.
For sequence analysis, the sequences obtained were analyzed through com-parison with the reference human CFTR sequence (Genebank accession number M28668) using the software Geneious® or alternatively Bioedit®.
33
3.2 Production of transient and stable cell lines
3.2.1 Characterization, culture and maintenance of cell lines
Experiments involving transient transfections were performed in HEK293 cells (Human Embryonic Kidney 293 cells, further referred simply as HEK) while ex-periments involving stable transfections were performed in Flp-In T-REx 293 cells (Life technologies) (further referred as HEK Flp) a cell line derived from HEK293 cells and containing both Flp-In and T-REx systems. The HEK293 cell line is the most com-monly used cell model used in molecular biology studies due to its easy maintenance, manipulation and high transfection rate, section 3.2.2, while HEK Flp allows the es-tablishment of stable cell lines overexpressing the gene of interest in an isogenic way which enhances the experiments reproducibility, see section 3.2.3.
Cells were cultured in Minimal Essential Medium supplemented with L-alanyl-L-glutamine (MEM+Glutamax)(Gibco) or alternatively with Eagle Minimal Essential Medium supplemented with L-glutamine(EMEM, Lonza) both supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco). For selection of parental HEK Flp cells, the culture medium was also supplemented with 200µg of zeocin (Invitrogen).
Continuous growth was made possible by pre-confluence enzymatic dissocia-tion with trypsin (Gibco). After dissociadissocia-tion, cells were resuspended and redistrib-uted into new flasks or plates. Cell lines were stored in cryogenic vials in aliquots in 40% (v/v) MEM or EMEM, 50% (v/v) FBS and 10% (v/v) DMSO (Sigma-Aldrich) in liquid nitrogen for long storage periods and at -80ºC for shorter storage periods. In order to minimize cell damage, freezing was performed in such a way that the cooling speed was 1ºC/min. Thawing was done by transferring the frozen content of the vials was transferred to EMEM at RT following a centrifugation of 3min at 1600rpm, su-pernatant was discarded and the pellet was resuspended a seeded with the adequate medium.
34
3.2.2 Transient transfections
HEK293 cells were submitted to liposomal transfection.
The liposomal transfection, commonly known as lipofection, is based on the ability of cationic lipids to form unilamelar liposomes, which adsorb nucleic acids molecules to their surface and are capable to be internalized by the cells, resulting in protein expression, when using plasmids, or down-regulation of target genes, when using siRNAs. To achieve transient transfections of HEK293 cells, were used either Lipofectamine 2000 (Invitrogen) or Fugene HD (Promega). Lipofectamine 2000 (8µL) and 2µg of DNA (pcDNA5 plasmids) were separately incubated for 5 min in 100µL of OPTIMEM (Invitrogen), then mixed together and allowed to incubate for 15 min at room temperature (RT). The mixture was then added to 70-80% confluent cells which were plated 24h prior to transfection in either MEM+Glutamax or EMEM without FBS. Alternatively, Fugene HD (6µL) and 2µg of DNA (pcDNA5 plasmids) were added and mixed together in 200µL of either MEM+Glutamax or EMEM and then added to 70-80% confluent cells which were plated 24h prior to transfection in either MEM+Glutamax or EMEM without FBS. Medium was changed after 6 h (Lipofectamine 2000) or 24h (Fugene HD) to either MEM+Glutamax or EMEM, both supplemented with 10%(v/v) FBS, and the experiments were performed 48h post-transfection.
3.2.3 Flp-In system for the establishment of stable cell lines
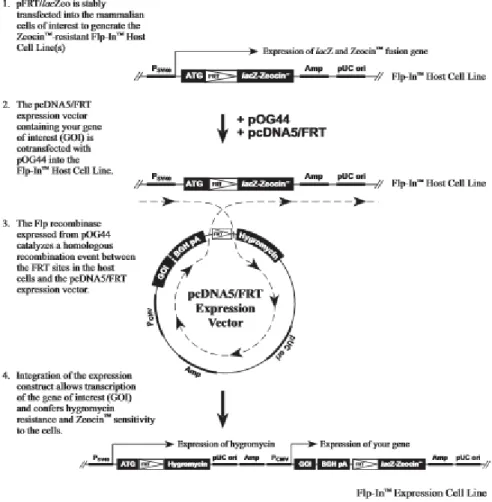
The establishment of isogenic stable cell lines was accomplished by lipofec-tion of HEK Flp cells with pOG44 and each one of the desired constructs, simultane-ously in a proportion of 9:1 for a total 2µg of DNA, as described in section 3.2.1, and cell selection started 24h after transfection by changing the medium to a medium supplemented with 200µg of Hygromycin B (Life Technologies). The creation of each of the isogenic stable cells is made possible by the Flp-In system (Life Technol-ogies). In this study the cells used were the HEK Flp from Invitrogen that already con-tain the flippase recombination target (FRT) site linked to zeocin resistance gene in their genome. By co-transfecting these cells with both the flippase (Flp) recombinase plasmid, pOG44, and an expression plasmid containing the gene of interest flanked by FRT site in both end, and the hygromycin resistance gene.
35
The presence of the Flp recombinase induces the integration of the fragment in the plasmid flanked by the FRT site in recombinase target site present in the cells genome leading to the stable expression of the gene of interest, loss of the zeocin resistance and acquisition of resistance to Hygromycin B (Figure 3.1). By this system is only integrated a single copy of the fragment of the plasmid of interest per cell, originating in a cell population with isogenic expression pattern of the gene of inter-est, after selection with Hygromycin B.
Figure 3.1 – Schematic representation of the general mechanism for generating cell lines with constitutive
ex-pression of a gene of interest via Flp-In system. Adapted from the product manual provided by the manufacturer (Invitrogen).
36
This avoids the need for single clone selection and the establishment of iso-genic stable cell lines was accomplished by lipofection of HEK Flp cells with pOG44 and each one of the desired constructs, simultaneously in a proportion of 9:1 for a total 2µg of DNA, as described in section 3.2.1, and cell selection started 24h after transfection by changing the medium to a medium supplemented with 200µg of Hy-gromycin B.
3.3 RT-PCR analysis of CFTR PTC-containing transcripts
3.3.1 RNA extraction
RNA from HEK293 and HEK Flp was extracted with the nucleospin RNA II kit (Macherey-Nagel) following the protocol provided by the manufacturer. Every step was executed in RNAse free environment to avoid RNA degradation. Following ex-traction were quantitated using the Nanodrop 200 spectrophotometer, and purity was assessed by the A260/A280 ratio. Only RNA samples with a ratio above 2.0 were deemed acceptable. The extracted samples stored at -80ºC.
3.3.2 cDNA synthesis
For the synthesis of cDNA, 500ng of the previously extracted mRNA were in-cubated with 1µL of oligo-dTs (short sequence of deoxy-thymine nucleotides) (Invi-trogen) for 10 min at 60ºC. This first step promotes the denaturation of the second-ary structure of the RNA facilitating the binding of the oligo-dTs to the poly-A (poly-adenosine) strand of mRNAs and enhancing reverse transcription yield. Afterwards the pre-mix containing the RNA and the oligo-dTs was put on ice and was added 6.9µL of an RT (reverse transcription) mix containing, 1x 1st strand Buffer (4µL of a 5x stock) (Invitrogen), 0.1M DTT (1µL of 1M stock)(Invitrogen) , 40U/µL RNase In-hibitor (0.5µL) (Invitrogen), 25mM dNTP mix (0,4µL)(Invitrogen), and ddH2O (bi-distillated water)(Invitrogen) for a final volume of 19µL. This mixture was then in-cubated at 42ºC for 2min, followed by the addition of 1µL of Superscript II RT en-zyme (200U/µL, Invitrogen), and further incubation at 42ºC for 1h. The reaction was
37
stopped by inactivating the enzyme with an incubation of the mixture at 70ºC for 15min. Samples were stored at -20ºC.
3.3.3 Polymerase chain reaction
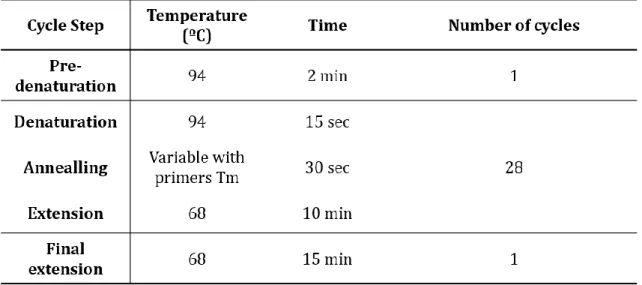
After RNA extraction and cDNA synthesis, see sections 3.3.1 and 3.3.2, detec-tion and semi-quantitative analysis of CFTR transcripts were done by regular poly-merase chain reaction (PCR) and a semi-quantitative variant of this method, respec-tively.
The PCR is a widely spread technique used in most research labs focused on medical and biological research due to large spectrum of applications that goes from gene expression detection to genetic engineering. It consists on cycles of repeated heating and cooling of the reaction for DNA melting, primers annealing and enzy-matic replication of DNA by a DNA polymerase.
For the regular PCR method, a pre-reaction mix was prepared, containing 1µL of each of the reverse primer and forward primer, (10pmol/µL)(primers pairs in table3.2, which are complementary with the interest sequences, 0.2µL of Taq Poly-merase (NZYtech), 5 µL of polyPoly-merase Buffer solution (10x concentrated, NZYtech), 3µL MgCl2 (50 mM), 0.2 µL dNTPs mix (25 pmol/µL of each dNTP) and water for a final volume of 49µL, per reaction, being possible to scale up or down according to the experiments necessities. All these steps were executed in DNA free environment to avoid contamination of the reagents and on ice to avoid unspecific amplification. Afterwards, 1µL of cDNA was added to each reaction. The PCR reactions were exe-cuted in a T-Professional thermo-cycler (Biometra) following the heating-cooling cycles stated on table 3.3. After the thermo-cycling, the contents of each reaction were analyzed by agarose gel electrophoresis. A 2% (m/v) agarose (Lonza) gel in TAE buffer (…) with 1U of RedSafe(Chembio was runned for at least 40 min at 120 Volts. After electrophoresis the PCR products were visualized by exposing the gel to UV light in a dark container coupled to a Kodak photographic machine (Kodak EDAS 290 Electrophoresis Documentation System). The images capture and analysis was done using the Kodak EDAS 290 Electrophoresis Documentation System software.
38
3.3.3.1
Semi-quantitative analysis
For semi-quantitation of expression of the CFTR gene by each of the nonsense mutants, there were introduced variations to the PCR method. Instead of letting the reaction conclude all the heating-cooling cycles as in a regular PCR, 5µL of each re-action, were collected after 20, 22, 24, 26, 28 and 30 cycles. Afterwards, the ampli-fied DNA was separated and detected, as described previously (section 3.3.3), and signal intensity of the bands corresponding to CFTR and β-actin were quantified with the IMAGE J software. This allowed for a comparative analysis of the quantity of CFTR transcripts between each of the cell lines expressing the different CFTR var-iants at the exponential phase of replication which occurred between cycles 22 and 28, when CFTR signal was detected.
Table 3.3 – PCR program used for the detection of CFTR and β-actin transcripts by RT-PCR
3.4 Biochemical and functional characterization of CFTR
nonsense variants
3.4.1 Preparation of total protein extracts
For Western blot (WB), protein extracts were prepared by cell lysis with a lysis buffer with following composition; 1.5% (w/v) SDS (Sodium dodecyl sul-fate)(Gibco) ; 5% (v/v) glycerol; 0.01% (w/v) bromophenol blue; 0.05 mM dithio-treithol; 0.095M Tris pH 6.8). In order to reduce sample viscosity, DNA was sheared
39
by enzymatic action of 5U/mL benzonase (Sigma-Aldrich) in the presence of 2.5mM MgCl2.
Samples total protein concentration was assessed by Lowry protein assay83.
3.4.2 Western blot
Protein extracts were separated by SDS-PAGE (polyacrylamide gel electro-phoresis) on 7% polyacrylamide mini-gels for 2-3hours at 120mV. After separation, the proteins were transferred from the gel onto Immobillon polivinylidene difluo-ride (PVDF) membranes (Milipore). Membranes were then blocked with 5% skimmed milk (Nestlé) in phosphate-buffered saline with Tween detergent (Bio Rad) (PBS-T, NaCl 137mM; KCl 2.7mM; KH2PO4 6.5mM, pH 7.4, 1% (v/v) Tween) for 1h at RT. After blocking the membranes were incubated, o/n at 4ºC, with primary antibodies, 596 (CFF, mouse, 1:3000), α-calnexin (Milipore, mouse, 1:3000), MM13-4 (Milipore, mouse, 1:1000), in 5% (w/v) skimmed milk in PBS-T, followed by 3x15min washes with PBS-T, and then incubated for 1h at RT with the horseradish peroxidase-conjugated secondary anti-mouse IgG antibody (Bio Rad, Donkey, 1:10000), diluted in 5% (w/v) skimmed milk in PBS-T. Membranes were again washed 3x15min in PBS-T. Blots were developed with the ImmunStar Western C Chemiluminescence kit (BioRad) using the Chemidoc XRS+ analyser (BioRad) for sig-nal detection and caption.
Signal quantification was performed with the ImageLab software (BioRad).
3.4.3 Immunofluorescence
Cells were grown in 10x10mm coverslips placed in P24 well plates coated with 0.001% (w/v) poly-L-lysine, until 40-60% confluence was reached. The cells were washed 3x10min with HBSS (Hank’s balanced salt solution) (Gibco) with 140rpm agitation, and in order to avoid unspecific binding of the antibodies, a block-ing step with 1% (w/v) BSA in HBSS was performed for 30min. Then the cells were incubated with TexasRed-conjugated wheat germ agglutinin (WGA)(Sigma Aldrich) diluted 1:200 (v/v) in HBSS with 0.5% (w/v) BSA, and washed 5x5min with HBSS
40
with 140rpm agitation. All the steps until fixation were executed on ice as to avoid WGA internalization by the cells.
Cells were fixed for 45min with 2% (v/v) formaldehyde, washed 3x10min with PBS supplemented with CaCl2 and MgCl2 (PBS+/+), and permeabilized by incu-bating with 0.2% (w/v) Triton X-100 (BioRad) in PBS+/+ for 15min at RT. After washing 3x10min with PBS+/+ at 140rpm at RT, cells were incubated, o/n at 4ºC in a humid chamber, with anti-CFTR antibody 570 (CFF) diluted 1:500 (v/v) in PBS+/+ with 0,5% (w/v) BSA, and washed 3x10min with PBS+/+ at 140 rpm. This was fol-lowed by an incubation with an anti-mouse IgG antibody conjugated with alexa 488 (Bio Rad), diluted 1:1000 (v/v) in PBS+/+, for 1h at RT. Then the cells were washed 3x10min at 140rpm with PBS+/+. Finally the coverslips were mounted in Vec-tashield (Vector labs) with 1.5µg/mL 49,6-diamidino-2-phenyl-indole (DAPI) for nuclear detection on glass slides and visualized in wide-field fluorescence micros-copy with the microscope (Zeiss, Jena) and images were captured, edited and ana-lyzed with ZEN 9 microscopy software (Zeiss).
3.4.4 Iodide Efflux
Cells were grown in P60 plates coated with 0.001% (w/v) poly-L-lysine. After achieving total or near total (90-95%) confluence, cells were gently washed two times with 2.5 ml of an I- loading buffer (136 mM sodium iodide, 3 mM potassium nitrate, 2 mM calcium nitrate, 11 mM glucose and 20mM HEPES, pH 7.4) previously warmed to 37ºC. Afterwards the cells were incubated, for 1h at 37ºC, in 2.5mL of loading buffer
Following the incubation cells were slowly and gently washed ten times for 1 min with 2.5 ml of an I- free efflux buffer (136 mM sodium nitrate, 3 mM potassium nitrate, 2mM calcium nitrate, 11mM glucose and 20mM HEPES, pH 7.4) also warmed to 37ºC, equilibrating for 5 min in I- free efflux buffer after the last wash.
After the equilibration period, the buffer was removed and replaced with fresh efflux buffer which was collected to P35 plate protect from incident light, and substituted with fresh efflux buffer after a 1 min incubation. This step was repeated from 0-4 min time-points.
41
At time point 0 min the fresh efflux buffer added was supplemented with 10µM of forskolin and 50µM of genistein, two CFTR agonists that stimulate the chan-nel opening. The collection and renewal step with the efflux buffer containing the CFTR agonists was repeated until the 4 min time-point.
After minute 4 the efflux buffer was substituted for efflux buffer without ag-onists, and the cycle repeated until time point 10.
Afterwards iodide concentrations from each collected samples were meas-ured with an I- selective electrode (Orion).
Simultaneously, were prepared, p35 plates, solutions with crescent concen-trations of NaI; 1µM; 10 µM; 50 µM and 100 µM. These solutions were used to deter-mine the standard curve of voltage values, measured by the I- selective electrode, versus iodide concentration, which will allow calculating iodide concentration of each collected sample.
3.5 Pharmacological treatments
3.5.1 Pharmacological indirect inhibition of Nonsense-mediated
mRNA decay
In order to induce indirect inhibition of NMD, HEK Flp cells, stably expressing different CFTR variants, were grown in P24 wells plates coated with 0.001% of poly-L-lysine, and incubated for 5h at 37ºC, with serum free EMEM or MEM+GlutaMAX supplemented with 100 µg/ml of cyclohexamide (CHX) (Sigma-Aldrich). After the incubation period cells are lysed. RNA extracted and analyzed by RT-PCR, as de-scribed in section 3.3.
3.5.2 Pharmacological induction of PTC read-through
In order to study the efficacy of some of these drugs in the induction of PTC read-through on CFTR transcripts containing in their sequence on different posi-tions, HEK Flp cells, stably expressing different CFTR variants were in treated, for 24h at 37ºC, with 50µg/ml of G-418, 50µg/ml of gentamicin, 1mM of Ataluren (PTC124) or 0.2% (v/v) of DMSO. After treatment cells were submitted to western blot and mRNA analysis, as described in previous sections.
42
4
Results
4.1 Production of plasmid vectors
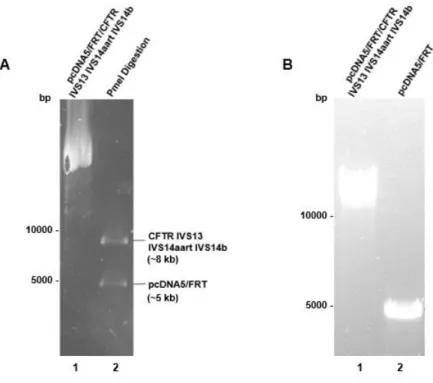
In order to study several naturally occurring AUG-proximal nonsense muta-tions in the CFTR gene, five new plasmid vectors were produced. Three CFTR minig-enes vectors were used as templates to produce these new vectors as follows: i) pcDNA5/FRT/CFTR IVS4art+IVS5 (a pCDNA5/FRT vector containing the full-length CFTR coding sequence as well as the natural occurring CFTR intron 5 (IVS5); ii) and an artificially constructed intron 4 (IVS4art, with just ~200bp from the 5’ end and 200 bp from the 3’end of the natural occurring intron); and iii) pcDNA5/FRT/CFTR IVS13+IVS14aart+IVS14b produced in the lab prior to this work.
The vectors produced in this work were:
1) CFTR mutants minigenes containing the naturally occurring nonsense mutations: Q2X (glutamine to stop codon), S4X (serine to stop codon) and Q39X (glutamine to stop codon);
2) A G542X (glycine to stop codon) mutant with CFTR introns 13, 14a (artificially constructed similarly to intron 4 art) and 14b, which is re-ported to naturally induce NMD84.
3) An empty vector (pcDNA5/FRT), in order to disregard vector interfer-ence;
The template vectors have a multicloning site that includes two Pme I re-striction sites bisecting the CFTR ORF (opreading frame), and an gene that en-codes for ampicillin resistance in bacteria and another for hygromycin B resistance in mammalian cells, as well as the short flippase (Flp) recognition target (FRT) sites flanking the CFTR sequence.