AGRADECIMENTOS
Ao meu orientador, Doutor Pier Parpot, o meu reconhecido agradecimento pelos conhecimentos transmitidos, apoio na execução experimental e orientação científica prestada, sem os quais não era possível a realização deste trabalho.
Ao Doutor Maurício, o meu reconhecido agradecimento pela sua disponibilidade, apoio e incentivo, tanto ao longo da realização das actividades experimentais, como na elaboração do trabalho final.
À Doutora Ana Paula Bettencourt, expresso o meu agradecimento pela sua disponibilidade e colaboração na realização deste trabalho.
Aos colegas Yolanda, Francisco e Manuela, por toda a colaboração prestada na execução experimental e também pela amizade dispensada.
Aos meus pais, o meu profundo agradecimento por todo o apoio e compreensão com que sempre me recompensaram ao longo deste trabalho.
Ao Rui, Miguel e Pedro, por se encontrarem sempre ao meu lado e pelo carinho e apoio demonstrados ao longo deste tempo.
RESUMO
Os hidratos de carbono podem ser convertidos em produtos químicos, energia, têxteis, materiais de construção, papel e muitos outros produtos industriais. Estes constituem uma matéria prima barata, renovável e facilmente disponível.
No início dos anos noventa, 95% da biomassa produzida eram hidratos de carbono e correspondiam a duzentos biliões de toneladas.
Nos dias de hoje, apenas 3 a 5% desta biomassa são utilizados em termos industriais. Os restantes sofrem degradação e são reciclados segundo processos naturais.
As principais razões para o uso limitado dos hidratos de carbono como matéria prima estão directamente ligadas à sua funcionalização excessiva devido à existência de grupos hidroxilo com reactividade similar, e ainda devido à baixa solubilidade nos vulgares solventes orgânicos. Assim conseguir uma oxidação directa e selectiva dos sacáridos pressupõe uma estratégia preliminar de protecção, sendo esta difícil nos métodos clássicos.
Os métodos electroquímicos surgem como uma alternativa, em que as transformações são realizadas em solução aquosa, associando os conceitos e os métodos da electroquímica interfacial com os da catálise heterogénea.
O presente trabalho tem como objectivo a oxidação regiosselectiva da D-galactose em meio aquoso, utilizando electrocatalisadores que contêm complexos de metais de transição.
A presença de complexos de metais de transição na superfície do eléctrodo pode alterar a selectividade e melhorar o rendimento na oxidação dos monossacáridos.
Este trabalho inicia-se com a síntese e caracterização de complexos estáveis, nomeadamente de ruténio e molibdénio. O ligando utilizado para a síntese destes complexos foi o 1,2-piridilazo-2-naftol (PAN).
A partir destes complexos preparam-se eléctrodos modificados, com os quais se procede a um estudo de voltametria cíclica, com o fim de investigar a oxidação electrocatalítica da D-galactose, tendo sido assim determinados parâmetros cinéticos das reacções electroquímicas.
Por fim foi realizada uma electrolise exaustiva, tendo sido os produtos desta identificados por cromatografia líquida de alta eficiência ( HPLC ).
ABSTRACT
Carbohydrates can be converted in chemical products, energy, textiles, building materials, paper and many other industrial products. Carbohydrates are a very cheap raw material and easily available. .
In the beginning of the nineties, 95% of the biomass produced was carbohydrates and corresponded to two hundred billion tons.
In our days only 3 to 5% of this biomass is industrially used. The remaining decays and recycles along natural pathways.
The main reasons for the limited use of carbohydrates as raw material is related to its overfunctionalization caused by the hydroxyl groups with similar reactivity, and also, to its poor solubility in most of the common used organic solvents. To achieve a direct and selective oxidation of saccharides, a preliminary protection strategy should occur at first time. This is very difficult to get in classical methods.
The electrochemical method appears as an alternative, and the transformations are developed on aqueous solutions, associating the concepts and the electrochemical interfacial methods, with the heterogeneous catalyses.
The goal of this investigation is the regioselective oxidation of D-galactose in aqueous solution, using electrocatalists which have transition metal complexes.
The presence of the transition metal complexes at the surface of the electrode, can change the selectivity and improve the yield in the oxidation of the monosaccharides.
This investigation starts with synthesis and characterization of the stable complexes, like ruthenium and molibdenium complexes. The ligand used in the synthesis is the 1,2-piridilazo-2-naftol (PAN).
Using these complexes, modified electrodes were made and cyclic voltammetric studies were developed, with the special focus on the investigation of electrocatalitic oxidation of D-galactose. Kinetics parameters of the electrochemical reactions were also determined by cyclic voltammetriy.
At last, an exhaustive electrolyse has been effectuated, and the products of reaction obtained were identified and quantified by High Performance Liquid Chromatography ( HPLC ).
ÍNDICE
Agradecimentos ii Resumo iii Abstract iv Índice v Abreviaturas vii Introdução 9Capitulo 1 – Revisão bibliográfica 13
1.1 Hidratos de carbono 14 1.1.1 Oxidação da galactose 16 1.2 Catálise heterogénea 18 1.3 A química de coordenação 19 1.3.1 Complexos de ruténio 20 1.3.2 Complexos de molibdénio 22 1.3.3 Ligandos 22 1.4 Métodos electroquímicos 23 1.4.1 Voltametria cíclica 23 1.4.2 Electrólise 30 1.4.3 Materiais de eléctrodo 35 1.4.3.1 Eléctrodos modificados 36 1.4.4 Electrólito de suporte 38 1.4.5 Remoção do oxigénio 38 1.5 Métodos analíticos 39
1.5.1 Cromatografia líquida de alta eficiência 41
1.5.2 Supressor de efluente para cromatografia iónica 43
Capítulo 2 – Síntese e caracterização de complexos de ruténio e molibdénio 44
2.1 Introdução 45
2.1.1 Síntese dos complexos de ruténio 46
2.2.1 Técnicas de síntese em atmosfera inerte 59
2.2.2 Equipamento utilizado na caracterização 60
2.2.3 Técnica electroquímica 61
2.2.3.1 Tipo de eléctrodos 62
2.2.3.2 Electrólito 63
2.2.3.3 Solventes 63
2.2.3.4 Materiais de partida 63
2.2.4 Descrição experimental da síntese dos complexos de ruténio 64
2.2.5 Descrição experimental da síntese dos complexos de molibdénio 65
Capítulo 3 – Oxidação electroquímica da D-galactose 66
3.1 Dispositivo experimental 67
3.2 Preparação do material e soluções 67
3.2.1 Preparação do material de vidro 67
3.2.2 Preparação das soluções 68
3.2.3 Preparação dos eléctrodos 69
3.2.4 Células de voltametria cíclica 70
3.2.5 Células de electrólise 72
3.2.6 Equipamento utilizado 74
3.3 Discussão dos resultados experimentais 75
3.3.1 Estudo voltamétrico 75
3.3.2 Apresentação dos voltamogramas 75
3.3.3 Análise dos voltamogramas (I) 82
3.3.4 Estudo da velocidade de varrimento de potencial (I) 87
3.3.5 Análise dos voltamogramas (II) 94
3.3.6 Estudo da velocidade de varrimento de potencial (II) 96
3.3.7 Estudo da concentração 101
3.3.8. Análise dos voltamogramas(III) 104
3.4 Preparação para a análise cromatográfica 106
3.5 Electrólise exaustiva 112
Conclusões 118
Bibliografia 120
ABREVIATURAS
A área do eléctrodo (cm2) 0
B
C concentração da espécie B na solução (moldm-3) υ velocidade linear média (ms-1)
C(E) concentração molar do soluto na fase estacionária (moldm-3) C(M) concentração molar do soluto da fase móvel (moldm-3)
CA(0,t) concentração à superfície do eléctrodo da espécie A no instante t (moldm-3) CB(0,t) concentração à superfície do eléctrodo da espécie B no instante t (moldm-3) DB coeficiente de difusão da espécie B (cm2s-1)
e- electrão
E(t) potencial no tempo t (V)
E0 potencial de redução padrão (V) Ef potencial final (V)
Ei potencial inicial (V)
Ep potencial da altura do pico (V) Ep/2 potencial de meia altura do pico (V) Epa potencial do pico anódico (V) Epc potencial do pico catódico (V) F constante de Faraday (96480 Cmol-1) H altura do prato da coluna (m)
HPLC cromatografia líquida de alta eficiência ipa intensidade de corrente do pico anódico (A) ipc intensidade de corrente do pico catódico (A) jp densidade de corrente de pico (Acm-2) i(t) intensidade de corrente no instante t K coeficiente de partição
N número de pratos teóricos da coluna NB número total de moles B
p constante de velocidade da electrólise R constante de gases ideais (8,314 K-1mol-1) T temperatura absoluta (K)
tM tempo de retenção de uma espécie não retida (s) tr tempo de retenção para o soluto (s)
V volume da solução
v velocidade de varrimento (Vs-1) x fracção molar
α coeficiente de transferência anódica ΔE largura do pico do voltamograma
INTRODUÇÃO
(18,19,20)A abundante vida vegetal do nosso planeta é armazenadora da energia solar e de
substâncias químicas, sendo um recurso renovável que chamamos de biomassa.
As fontes orgânicas que são usadas para produzir energias usando este processo são chamadas de biomassa, pelo que todos os organismos biológicos que podem ser aproveitados como fontes de energia, são chamados de biomassa.(19)
A biomassa é formada pela combinação de dióxido de carbono da atmosfera e água na fotossíntese clorofiliana, produzindo-se os hidratos de carbono. A energia solar é armazenada nas ligações químicas dos componentes estruturais da biomassa.(19) Através da fotossíntese, as plantas capturam energia do sol e transformam essa energia em energia química. Esta pode ser convertida em várias formas de energia tais como electricidade, combustível ou calor. Se a biomassa for queimada de modo eficiente, há produção de dióxido de carbono e água. Portanto, o processo é cíclico e dizemos que a biomassa é um recurso renovável.(19)
A biomassa constitui uma matéria prima renovável para a indústria química.
As principais fontes de energia do século XXI provavelmente serão de origem biológica, produzidas a partir da biotecnologia. Actualmente, responde por 1% da energia eléctrica mundial, mas calcula-se que daqui a vinte anos cerca de 30% do total de energia consumido pela humanidade será proveniente da biomassa.(19)
Além de agrupar várias opções como queima de madeira, carvão vegetal e o processamento industrial de celulose e bagaço de cana-de-açúcar, inclui o uso do álcool como combustível.(19)
Os recursos renováveis representam actualmente cerca de 20% do fornecimento total de energia no mundo, com cerca de 14% proveniente de biomassa.
Podemos considerar várias fontes energéticas de origem natural: - biomassa sólida;
- biocombustíveis gasosos; - biocombustíveis líquidos.(18)
O componente principal da biomassa terrestre é a madeira. Esta pode ser descrita quimicamente como uma mistura de três polímeros interligados, nos quais estão distribuídos os compostos de baixo peso molecular.
Os dois primeiros, celulose e hemicelulose são hidratos de carbono. O terceiro componente, a lenhina é um polímero irregular.
A figura seguinte representa a composição química da biomassa.
Figura 0.1 – Esquema da composição química da biomassa(19).
A biomassa pode ser utilizada em diversas formas e estado para obtenção das mais variadas formas de energia seja por conversão direta ou indireta.
A biomassa é uma fonte natural de hidratos de carbono, cujo nome pode ser glícidos, glucidos, carbo-hidratos ou ainda açucares, pelo que estes são facilmente disponíveis em larga escala.
Neste trabalho a espécie electroactiva utilizada é a D – galactose(+).
A D – galactose (+) é vulgarmente encontrada na lactose ou açúcar do leite. É classificada como um monossacárido, uma aldohexose e é um açúcar redutor(21).
A D – galactose (+) não é doce e difere da glucose somente na posição do grupo hidroxilo, no carbono quatro. A D – galactose (+) é um isómero da glucose(21).
Neste contexto, foi investigada a oxidação electrocatalitica e regiosselectiva de um tipo de açúcar, o monossacárido D-galactose(+).
A funcionalização excessiva, isto é, a existência de grupos hidroxilos com reactividade similar não permite transformações regiosselectivas e constitui a principal limitação ao uso de hidratos de carbono como matéria prima(4).
Os métodos utilizados para a realização deste trabalho, são os métodos electroquímicos. Estes apresentam a vantagem de ser limpos.
Nestes métodos ocorre a substituição de reagentes químicos perigosos, poluentes e/ou caros pelo reagente electrão, não poluente, cujo poder oxidante ou redutor pode variar de uma forma definida e contínua com a gama de valores de potencial escolhida, permitindo uma selectividade que normalmente não é conseguida com outro reagente químico(4).
O presente trabalho encontra-se dividido em duas partes.
Numa fase inicial desta investigação, procedeu-se à síntese de complexos de metais de transição, nomeadamente de ruténio e molibdénio.
Com estes complexos sintetizados, foram preparados eléctrodos modificados que foram utilizados na segunda fase do trabalho experimental, sendo que este trabalho inicial pressupõe uma aplicabilidade destes compostos previamente sintetizados, na oxidação da D-galactose(+), que será assim considerado como uma segunda fase deste trabalho.
O método de preparação dos eléctrodos modificados utilizado nesta investigação, é aquele em que ocorre adsorção do complexo de metal de transição, na superfície de uma placa quadrangular de carbono Toray.
Assim, nesta dissertação, no capítulo 1 faz-se uma revisão bibliográfica sobre hidratos de carbono e da forma como estes podem ser oxidados, técnicas electroquímicas e métodos analíticos, e ainda química de coordenação, complexos de ruténio e molibdénio.
No capítulo 2, são descritas as condições experimentais usadas na síntese, caracterização dos complexos de ruténio e molibdénio, e estudos electroquímicos por voltametria cíclica dos compostos referidos.
caracterizados como descrito no capítulo anterior. São apresentados os resultados obtidos nas técnicas electroquímicas e analíticas utilizadas durante a realização deste trabalho. São realizados estudos electroquímicos por voltametria cíclica para determinação dos parâmetros cinéticos da oxidação da D-galactose utilizando como eléctrodos modificados os compostos atrás referidos. Por último foi realizada uma electrólise exaustiva a potencial controlado. Esta electrólise é realizada, utilizando um eléctrodo de carbono modificado com complexo de molibdénio sintetizado, fracção-2.
Os resultados experimentais obtidos, bem como o seu tratamento e análise, são apresentados no terceiro capítulo.
Os produtos da electrólise foram identificados por cromatografia líquida de alta eficiência, HPLC.
Apresentam-se ainda no final desta dissertação as conclusões obtidas nesta investigação.
CAPITULO 1
1.1 HIDRATOS DE CARBONO(5,8,10)
Hidratos de carbono, glúcidos ou açúcares, são compostos orgânicos constituídos por carbono, hidrogénio e oxigénio. A designação de hidratos de carbono deve-se ao facto de os primeiros glúcidos descobertos obedeceram à fórmula Cx(H2O)y, o que dava a ideia de serem formados por carbono e água(8).
Em 1844, K. Schhmidt propôs o nome de hidratos de carbono ou carbo - hidratos para designar substâncias com as seguintes características(5):
• Serem neutras;
• Apresentarem fórmula geral Cx ( H2O )y na proporção H:O = 2:1, como na água;
• Apresentarem, frequentemente, 6 átomos de carbono, ou múltiplo de 6;
• Reduzirem as soluções de hidróxidos de metais pesados, directamente, ou após aquecimento com ácidos diluídos;
• Apresentarem, frequentemente, sabor doce.
Existiam ainda substâncias que no entanto nem sempre verificavam todas estas propriedades como, por exemplo(5):
1. Compostos com a mesma fórmula geral, como o CH2O, aldeído fórmico, o C2H4O2 , ácido oxálico, e outros, tiveram que ser excluídos, por não apresentarem mais nenhuma propriedade comum, para além da fórmula geral;
2. Substâncias que tinham menor número de átomos de oxigénio como, por exemplo, o amido, tiveram que ser excluídas neste grupo de compostos, porque, embora não tivessem a mesma fórmula geral, possuíam todas as outras propriedades. Com o decorrer dos tempos, a designação de hidratos de carbono foi sendo substituída pelas designações glícidos ou açucares, devido ao seu sabor doce. Na molécula de um glícido existem alguns grupos característicos como o da função álcool, OH e das funções aldeído, HC=O ou cetona , C=O. O sabor doce, tão característico deste grupo de compostos, não apresenta a mesma intensidade para todos eles.
Os hidratos de carbono dividem-se em três grupos: monossacáridos, dissacáridos e polissacáridos(10,15).
- os dissacáridos quando sofrem hidrolise, dão origem a duas moléculas de monossacáridos; - os polissacáridos quando sofrem hidrolise, dão origem a várias moléculas de monossacáridos.
Os monossacáridos podem ainda subdividir-se. Se contêm um grupo aldeído chamam-se aldoses; se contêm um grupo cetónico, denominam-se cetoses. Consoante o número de átomos de carbono que contêm, o monossacárido denomina-se triose, tetrose, pentose, etc. Uma aldo-hexose, por exemplo, é um monossacárido com seis átomos de carbono que contém um grupo aldeído. Para indicar o sinal do poder rotatório em condições determinadas, é previamente escrito dextro ou levo, ou um dos sinais (+) ou (-). Assim, as modificações racémicas podem ser indicadas pelos prefixos D L ou ±(15). A lactose (+) que é um dissacárido, constitui cerca de 5% do leite humano e do leite de vaca. Obtém-se comercialmente, como subproduto da fabricação do queijo, a partir do soro do leite – a solução aquosa com que se fica depois de se terem coagulado as proteínas do leite. O leite azeda em consequência da transformação da lactose em ácido láctico por acção bacteriana, como por exemplo o Lactobacillus bulgaricus(10).
A lactose (+) tem a fórmula molecular C12H22O11, é um açúcar redutor, forma uma osazona e existe nas formas alfa e beta, as quais apresentam mutarrotação. A hidrólise ácida, ou o tratamento com emulsina, converte a lactose (+) em quantidades iguais de D – glucose (+) e D – galactose (+)(10).
A galactose é um açúcar simples que se encontra na natureza em várias combinações. Combinada com a glucose, constitui a lactose, ou açúcar de leite, e existe nos cerebrósidos em combinação com uma base, a esfingosina, e com ácidos gordos.
Nas cartilagens encontra-se um glucoproteído de que se pode separar o ácido condróitina- sulfúrico. Este, por hidrólise, fornece condrosamina, que é a amina-2-galactose. Nos vegetais existe principalmente em forma de galactanas, de que se liberta por acção dos ácidos diluídos. Os ácidos pépticos têm por núcleo um composto de galactose, o ácido tetragalactusónico(5).
A galactose cristaliza com facilidade, funde a 170-171ºC e com a fenil-hidrazina forma um galacto-sazona cujo ponto de fusão é 201 ºC. Por oxidação converte-se em ácido galactárico, também denominado de ácido múcico(10). É redutora como as outras oses. Sob a
uma enzima existente na mucosa intestinal, a lactase. A galactose absorve-se rapidamente, mais ainda do que a glucose e em compensação a sua transformação em glicogéneo, no fígado, é mais demorada do que a deste último açúcar(5).
1.1.1 OXIDAÇÃO DA GALACTOSE(4,10, 15)
A D – galactose é uma aldo-hexose. Como todos os monossacáridos, aldo-hexose, a D – galactose reduz o reagente de Fehling ou o reagente de Tollens, pelo que é um açúcar redutor. A maioria dos monossacarídeos e dissacarídeos são redutores, excepto a sacarose, sendo este um açúcar não redutor(10,15).
Todos os hidratos de carbono são poli-hidroxi-aldeídos, poli-hidroxi-cetonas ou moléculas que ao sofrerem hidrólise, dão origem a estes compostos. A D-galactose é uma aldo-hexose, pois como já referido contêm um grupo aldeído (10,15).
CHO OH H H HO H HO OH H CH2OH D - galactose
Figura 1.1 – Nome e projecção de Fischer da D-galactose.
A D – galactose como monossacárido, que é tem muitos grupos funcionais reactivos em carbonos adjacentes. Esta apresenta cinco grupos hidroxilo e um grupo aldeído.
A D-galactose pode sofrer várias oxidações, entre as quais:
- Se a oxidação ocorrer no grupo aldeído e no álcool primário, forma-se ácido galactárico:
CHO OH H H HO H HO OH H CH2OH D - galactose OH H H HO H HO OH H COOH COOH Ácido galactárico
O ácido galactárico, também denominado de ácido múcico, forma complexos estáveis com vários catiões, tendo assim uma aplicabilidade como complexante nos detergentes, isto é como sequestrante(10).
- Se a oxidação ocorrer no álcool primário, forma-se ácido galacturónico:
CHO OH H H HO H HO OH H CH2OH D - galactose OH H H HO H HO OH H COOH CHO Ácido galacturónico
- Se a oxidação ocorrer no grupo aldeído, forma-se ácido galactónico:
CHO OH H H HO H HO OH H CH2OH D - galactose OH H H HO H HO OH H CH2OH COOH Ácido galactónico
1.2 CATÁLISE HETEROGÉNEA
As reacções que foram estudadas ao longo deste trabalho foram reacções de catálise heterogénea. Estas, são reacções com interesse industrial que têm de ser rápidas, o que se consegue frequentemente à custa de um catalisador.
Em chinês, a palavra equivalente a catalisador significa casamenteiro, isto é, aquele/aquela que promove o processo mas não entra com protagonista (32).
Quando o catalisador constitui uma fase separada, a catálise é heterogénea. Neste caso a reacção química ocorre na interface entre as fases, e a sua velocidade será em princípio, proporcional à área respectiva. Em catálise heterogénea são possíveis diversas combinações de fases, mas em geral o catalisador é um sólido, enquanto que os reagentes e produtos se distribuem por uma ou mais fases fluidas. A superfície dos catalisadores não é uniforme(29).
As reacções ocorrem em locais específicos da superfície, os centros activos. A reacção catalítica envolve a adsorção transitória de um ou mais reagentes na superfície do catalisador, rearranjo das ligações e desadsorção dos produtos. A adsorção química é a primeira etapa da reacção catalítica, permitindo o enfraquecimento das ligações das moléculas reagentes e facilitando a sua conversão em produtos(29).
A maior parte dos catalisadores selectivos usados industrialmente são óxidos dos metais de transição. Para oxidações que não envolvam a quebra de ligações C-C, os catalisadores contêm geralmente molibdénio(29).
Os factores que determinam a selectividade intrínseca do catalisador são de natureza electrónica e geométrica(29):
Factor electrónico :
A força da ligação da adsorção química, que é governada pelas características electrónicas dos átomos envolvidos – se for muito fraca não há adsorção ; se for muito forte, a desadsorção é lenta.
Factores geométricos :
- os centros activos são átomos isolados da superfície, a reacção exige um número mínimo de centros de coordenação por átomo superficial;
- o centro activo é um conjunto de vários átomos superficiais adjacentes a reacção exige um tamanho mínimo deste conjunto;
- o centro activo poderá ter que satisfazer determinadas condições estereoquímicas para que a reacção seja estereoespecífica.
1.3 A QUÍMICA DE COORDENAÇÃO(6,7)
Originariamente, o termo complexo foi utilizado para designar os compostos formados através da combinação de espécies químicas capazes elas próprias de existência independente.
Estudos sobre o comportamento dos complexos revelaram que iões complexos têm existência como espécies discretas. As ligações nos complexos podem ser covalentes quando envolvem a sobreposição de orbitais do ligante e do ião metálico, mas, algumas vezes implicam a interacção extensamente iónica. O ligando tanto pode ser uma espécie molecular como uma espécie iónica.
Quando um metal forma um ião complexo genericamente representado por MLn, em que M representa o metal, L o ligando, o número máximo, n, de ligandos representa o número de coordenação do ião metálico.
Um ião complexo pode ser definido como um ião que contém um átomo central ao qual se ligam outros átomos, os ligandos, em que na maior parte dos casos esse átomo central é um elemento metálico.
As forças responsáveis por estas ligações são diferentes das encontradas noutro tipo de compostos, mas são devidas à sobreposição de orbitais e emparelhamento de electrões como no caso de compostos mais simples.
Certos complexos têm ligandos que podem associar-se por várias ligações ao ião central. Estes ligandos dizem-se polidentados.
Complexos assim formados chamam-se quelatos, do grego chele (garra, pinça), porque os seus ligandos formam como que uma “garra química”.
Os ligandos polidentados são agentes complexantes poderosos, pois os complexos por eles formados podem ser muito mais estáveis do que aqueles que envolvem ligandos monodentados. A estabilidade do complexo aumenta com o número de ligações entre o ligando polidentado e o metal.
Os metais de transição estabelecem ligações através das orbitais d com diversos ligandos de modo a estabilizarem as suas cargas positivas.
1.3.1 COMPLEXOS DE RUTÉNIO(11,12,13,54)
A química dos metais de transição é a química dos seus múltiplos estados de oxidação e todos os seus fenómenos de redox associados(13).
Se um elemento em particular fosse escolhido para ilustrar este ponto de vista, a escolha seria o ruténio. Este é um elemento que é directamente e indirectamente o centro activo da abundância dos fenómenos redox, rodeado de diferentes estados de oxidação e uma surpreendente diversidade de estruturas e ligações.
Recentes avanços em instrumentos electrónicos e teorias pragmáticas fizeram que se tornassem disponíveis ferramentas electroquímicas poderosas para comprovar as variações redox em células electroquímicas.
As reacções de eléctrodo devem ser uniformemente escritas como reduções:
Ox + ne- Red
Correspondentemente, os potenciais de eléctrodo (E0T) são dados como potenciais de redução. O eléctrodo de calomelanos saturado é provavelmente o mais utilizado como eléctrodo de referência.
Em experiências electroquímicas redox, os electrões são usualmente transferidos heterogeneamente, entre a superfície do eléctrodo e a substância electroactiva.
O ruténio é ricamente dotado com orbitais redox e isto consequentemente manifesta-se num largo número de estados de oxidação. Quando o metal está ligado a um ligando o qual é igualmente rico em orbitais redox, tem-se uma situação na qual uma série de passos de transferência de electrões pode ocorrer.
Em experiências voltamétricas as séries redox são expressas na forma de uma sequência de respostas redox.
O ruténio abarca todos os estados de oxidação desde - 2 a + 8, com excepção de -1. É conveniente classificar os dez níveis de oxidação observados em três camadas como alto, médio e baixo, com base no seu grau de oxidação. Em termos de quantidade de conhecidos compostos disponíveis quimicamente, uma segunda classificação pode ser feita tal como, não comum, comum e muito comum. Os dois estados de oxidação muito comuns pertencem ao nível médio, enquanto que o alto e o baixo, a estados de oxidação não comum.
Classificação dos estados de oxidação do metal A. Grau de oxidação - alto +5, +6, +7, +8 - médio +2, +3, +4 - baixo -2, 0, +1 B. Ocorrência - não comum -2, +5, +6, +7, +8 - comum 0, +1, +4 - muito comum +2, +3
Electroquimicamente, a transferência acessível de electrões pode ocorrer num sítio de um ligando coordenado quando o sítio possui uma ou mais orbitais escondidas não ocupadas e/ou um ou mais orbitais ocupadas não muito estáveis.
A química do ruténio com ligandos com anéis aromáticos azotados foi inicialmente desenvolvida com bases de piridina. Numa pesquisa sistemática de novos complexos de ruténio, descobriu-se alguns grupos de ligandos baseados em azopiridinas, que têm o cromóforo N=CN=N(54).
O interesse da química do ruténio reside na sua capacidade para um alargado intervalo de estados de oxidação, de -2 a +8, e as suas várias geometrias coordenadas(11).
Devido a estas características os complexos de ruténio exibem propriedades versáteis de electrotransferência e uma alargada gama de reactividades.
Os complexos de ruténio, especialmente os compostos azo, permitem o metal manter um estado baixo de potencial redox Ru (IV), o qual é adequado para a electrooxidação da moléculas orgânicas(12).
Como resultado de tal, uma larga variedade de complexos de ruténio, têm sido utilizados em estudos de fotossínteses artificiais, projectos fotomolecular, elucidação de estruturas e propriedades de electrotransferências de proteínas e ADN, oxidação catalítica da água e substratos orgânicos e estequiometricos, assim como reacções catalíticas de sínteses orgânicas.
1.3.2 COMPLEXOS DE MOLIBDÉNIO(14, 16)
Para o molibdénio, os estados mais altos de oxidação são os mais comuns e mais estáveis contra fenómenos de redução. Este metal tem uma grande gama de estados estereoquímicos assim como uma variedade de estados de oxidação. Na sua química encontra-se entre o maior complexo dos metais de transição.
O molibdénio ocorre principalmente na forma de MoS2, que é oxidado a MoO3. Mesmo em pequenas quantidades, a presença deste metal em ligas de aço causa um forte incremento na dureza e resistência destas.
O molibdénio é usado em óxidos e outros sistemas como catalisador para uma variedade de reacções, sendo um exemplo a utilização na síntese do acrilonitrilo.
Este metal de transição está presente em algumas enzimas como por exemplo as redutoras do azoto.
Há muitos complexos, onde ocorrem todos os tipos de estado de oxidação desde o estado II a VI. O estado +2 não é muito conhecido excepto nos compostos em que ocorra Mo24+. Molibdénio VI forma vulgarmente espécies dioxo nas quais as duas ligações Mo=O são cis.
Estudos relatam electroredução do acetileno a etileno e etano, utilizando vários catalisadores redox como Mo III em solução aquosa.
1.3.3 LIGANDOS(12,17)
Antes da adsorção dos iões do metal pesado na fase sólida para préconcentração, geralmente os iões metálicos são convertidos numa forma adequada incluindo os metais quelíferos ou os complexos de metais inorgânicos.
O ligando PAN, isto é o 1,2 – piridilazo – 2 – naftol, foi utilizado na síntese dos complexos de ruténio e molibdénio. Este é um ligando bidentado, podendo coordenar através de um par de electrões não ligantes do átomo de azoto.
1.4 MÉTODOS ELECTROQUÍMICOS(1,3,4)
Uma grande parte do trabalho experimental, é relativo ao estudo electroquímico dos novos compostos sintetizados. Assim, serão apresentados os fundamentos das técnicas electroquímicas utilizadas, como a voltametria cíclica.
Neste ponto do presente trabalho é referido o desempenho de células, materiais de eléctrodo, construção e limpeza de eléctrodos, composição da solução e o controlo da instrumentação.
1.4.1 VOLTAMETRIA CICLICA
A voltametria é uma técnica usada como alternativa na análise qualitativa e quantitativa de qualquer composto químico que seja electroactivo, isto é, que possa ser oxidado e/ou reduzido. Com esta técnica podem ser efectuados estudos fundamentais de processos de oxidação e redução em vários electrólitos, processos de adsorção em diferentes materiais, e mecanismos de transferência electrónica em superfícies de eléctrodos modificados quimicamente. Nesta técnica, a intensidade de corrente que flui através de um eléctrodo é medida em função do potencial aplicado a esse eléctrodo(53).
A voltametria cíclica é uma técnica simples e que permite realizar estudos cinéticos e mecanísticos de espécies electroactivas. Esta é uma técnica que dever ser escolhida, quando se inicia o estudo de um sistema desconhecido, isto é, para se estudar electroquimicamente um composto pela primeira vez. A partir do voltamograma traçado, facilmente se recolhem informações quanto aos potenciais em que ocorrem os processos de transferência de electrões, a identificação de fenómenos de adsorção, determinação de constantes de velocidade, estudo da reversibilidade da reacção redox e o número de electrões transferidos numa reacção.
Este método electroquímico consiste na aplicação de um potencial, ao eléctrodo de trabalho ou eléctrodo estacionário, através de um potenciostato que controla o potencial deste eléctrodo em relação a um eléctrodo de referência, variando-o continuamente com o tempo, levando assim à ocorrência de reacções de oxidação ou redução, isto é reacções faradaicas, das espécies electroactivas em solução.
Podendo ser variado anodicamente ou catodicamente, o potencial aplicado ao eléctrodo de trabalho, varia linearmente com o tempo podendo ser representado pela seguinte expressão:
E(t) = Ei ± vt (1.1)
em que
Ei - potencial inicial E(t) - potencial no tempo t v - velocidade de varrimento
± - varrimento do potencial catódico ( - ) ou anódico ( + )
A técnica de varrimento de potencial é normalmente usada em eléctrodos estacionários, mas também pode ser usada em eléctrodos hidrodinâmicos. A sua principal utilização tem sido para diagnosticar mecanismos de reacções electroquímicas, para a identificação de espécies presentes em solução e para análise semiquantitativa de velocidades de reacção.
A partir das curvas voltamétricas podem ser determinados parâmetros cinéticos com exactidão, tais como constantes de velocidade.
Em voltametria cíclica as velocidades de varrimento podem variar entre valores com ordens de grandeza entre mV.s-1 e centenas de V.s-1. O varrimento de potencial é feito apenas numa direcção, parando num valor escolhido Ef, para t = t1. A direcção do varrimento pode ser positiva ou negativa e a velocidade de varrimento pode ter qualquer valor.
De acordo com a figura 1.2, em voltametria cíclica, ao atingir t = t1, a direcção de varrimento é invertida e variada até E min, e depois invertida e variada para Emax.
Os parâmetros a considerar são : - potencial inicial, Ei;
- a direcção de varrimento, v; - potencial máximo, Emax;
- potencial mínimo, Emin; - potencial final Ef.
Apesar de não ser comum, por vezes é conveniente variar os valores de Emax e de Emin entre ciclos sucessivos.
De acordo com o gráfico constata-se que durante a variação de potencial observam-se picos anódicos e/ou catódicos, que correspondem à oxidação e à redução respectivamente do produto em estudo, originando uma curva do tipo i = f ( E ). A corrente eléctrica observada é diferente da corrente no estado estacionário =0
∂ ∂ t
c .
As reacções químicas podem ser classificadas de reversíveis ou irreversíveis. Uma reacção é considerada reversível quando o produto inicial de oxidação ou de redução formado durante o varrimento é respectivamente reduzido ou oxidado no varrimento inverso.
Na figura 1.3 apresenta-se um voltamograma cíclico representativo de um processo reversível. Ep,a é o potencial anódico, Ep,c é o potencial catódico, ip,a é a intensidade de corrente do pico anódico e ip,c é a intensidade de corrente do pico catódico.
Figura 1.3 – Voltamograma cíclico para uma reacção reversível.(53).
Além de reacções reversíveis e irreversíveis, podem ainda ocorrer reacções quasi-reversiveis. Estas ocorrem quando a velocidade relativa da transferência electrónica comparada com a correspondente ao transporte de massa é insuficiente para manter o equilíbrio de Nernst à superfície do eléctrodo.
Na figura 1.4 apresentam-se os voltamogramas cíclicos típicos para os três casos existentes.
A forma do voltamograma cíclico depende de vários factores tais como a velocidade de transferência electrónica, de transporte de massa onde ocorrem fenómenos de adsorção e desadsorção e ainda de outras reacções que possam ocorrer na superfície do eléctrodo.
Figura 1.5 – Representação esquemática das transformações possíveis de ocorrer numa experiência de voltametria cíclica(4).
Uma vez que a forma do voltamograma está ligada à velocidade de transferência electrónica, é importante considerar os dois processos baseados em transferência electrónica que são:
- Transferência electrónica reversível (rápida) - Transferência electrónica irreversível (lenta)
A reacção de transferência electrónica reversível poder ser representada como a que se segue.
-No início apenas a espécie B está presente em solução. Quando o potencial é varrido no sentido catódico ocorre a reacção redox entre as espécies A e B, dando origem a uma corrente eléctrica catódica com a forma de um pico. Ao fazer o varrimento do potencial no sentido oposto verifica-se o processo inverso, isto é, a espécie reduzida é reoxidada produzindo uma corrente eléctrica anódica.
Assim, num sistema reversível o processo exibe um pico anódico, na oxidação, e o correspondente pico catódico, no sentido contrário de varrimento de potencial, referente à redução.
Num voltamograma cíclico podem ser medidos parâmetros importantes como os potenciais do pico catódico - Epc - e anódico - Epa - as intensidades de corrente do pico catódico - ipc - e do pico anódico ipa .
O pico do voltamograma depende de velocidades de transporte de massa, e da diminuição do reagente na camada de difusão.
Num sistema reversível existe uma diferença nos potenciais dos picos anódico e catódico que se pode representar :
ΔE = Epa - Epc= 0,058/n (V) (1.2)
Relativamente à largura do pico, esta é obtida pela seguinte expressão :
Ep – Ep/2 = 0,058/n (V) (1.3)
Em que:
Ep – potencial do pico
Ep/2 – potencial do pico a meia altura
n - número de electrões por molécula envolvidos no processo
Quando a largura do pico é menor, isto é quando os picos anódicos e catódicos estão pouco afastados ao longo do eixo do potencial, então a transferência electrónica é rápida.
A razão pela qual é comum determinar o valor de potencial do pico a meia altura, tem a ver com uma determinação mais precisa deste, devido ao arredondamento do pico, o que lhe confere alguma imprecisão.
A transferência electrónica ocorre lentamente quando os picos anódicos e catódicos estão mais separados ao longo do eixo do potencial, sendo que quanto mais baixa for a
velocidade de transferência, maior é a separação. A reacção é irreversível quando a velocidade do processo de transferência de carga é baixa quando comparada com a velocidade de transporte de massa.
As concentrações das espécies electroactivas, na vizinhança do eléctrodo, dependem do tempo, por consequência das suas renovações limitadas por transporte de massa – difusão, ou por reacções que ocorrem à superfície do eléctrodo-adsorção.
Quando a velocidade de transferência de carga é elevada, a transferência de massa da espécie electroactiva A é o único processo que determina o fluxo à superfície do eléctrodo. Assim sendo, as concentrações das espécies electroactivas A e B junto ao eléctrodo, são dadas pela lei de Nernst :
E = E0 + nF RT ln ) , 0 ( ) , 0 ( t c t c B A (1.4) sendo:
E0 – potencial de redução padrão;
cA(0,t) – concentração à superfície do eléctrodo da espécie A no instante t; cB(0,t) – concentração à superfície do eléctrodo da espécie B no instante t;
No caso das transferências electrónicas serem controladas ora por difusão, ora por adsorção, as expressões matemáticas correspondentes ao potencial do pico e à densidade de corrente são diferentes.
Transferência Ep jp Reversível E n 0285 , 0 0 + 0 446 , 0 B B C RT nFD nF υ Irreversível ⎥ ⎥ ⎦ ⎤ ⎢ ⎢ ⎣ ⎡ + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + ln 0,780 0 s B K RT nFD nF RT E α υ α 0 496 , 0 B B C RT nFD nF α υ
Nestas expressões matemáticas, temos que Ep corresponde ao potencial de pico (V), E0 representa o potencial formal de redução (V), jp corresponde à densidade de corrente de pico ( A cm-2 ), n representa o número de electrões envolvidos no processo, F é a constante de Faraday ( F=96480 Cmol-1 ), R é a constante dos gases ideais (R=8,314 JK-1mol-1), T corresponde à temperatura absoluta ( K ), υ corresponde à velocidade de varrimento de potencial ( Vs-1 ), 0 corresponde à concentração da espécie B na solução ( moldm
B
C -3 ), DB
corresponde ao coeficiente de difusão da espécie B ( cm s ), K
B
2 -1 s representa a constante de velocidade padrão ( cms ) e α representa o coeficiente de transferência anódica. -1
Transferência Ep jp Reversível 0 E υ 0 2 2 25 , 0 CB RT F n Irreversível ⎟⎟⎠ ⎞ ⎜⎜ ⎝ ⎛ + s K RT nF nF RT E α υ α ln 0 υ α 2 2 0 368 , 0 CB RT F n
Tabela 1.2 – Expressões correspondentes ao potencial de pico e densidade de corrente de pico para uma transferência electrónica controlada por adsorção(4).
1.4.2 ELECTRÓLISE
A electrólise é a conversão de energia eléctrica em energia química de modo a converter substâncias por oxidação ou por redução, em que os produtos são formados como o elemento ou na forma de um composto apropriado(1).
Esta técnica, que é utilizada a nível industrial, permite aos electroquímicos, o controlo de vários parâmetros, tais como o solvente, o electrólito de suporte, a concentração das espécies electroactivas, o movimento da solução, a forma e o material dos eléctrodos, a célula electroquímica, o potencial ou a corrente aplicados e ainda a temperatura(1).
Com o fim de alcançar um bom rendimento é necessário maximizar o contacto entre o eléctrodo e o electrólito, sendo ainda algumas vezes necessário aplicar potenciais bastante elevados para ultrapassar a resistência da solução.
Existem vários tipos de reactores electroquímicos. Os três tipos básicos são (1): - reactor descontínuo;
- reactor tubular;
- reactor contínuo com agitação.
O tipo de reactor utilizado na electrólise realizada, neste trabalho, é o último, ou seja reactor contínuo com agitação, que é denominado ao longo do trabalho de célula de electrólise.
A electrólise pode ser dívida em duas categorias: - electrólise a corrente controlada;
- electrólise a potencial controlado.
Neste trabalho a electrólise foi realizada a potencial controlado. Nesta as reacções químicas ocorrem num eléctrodo onde o potencial se mantém constante.
As técnicas com controlo de potencial são geralmente as mais convenientes pois, neste caso, o potencial de eléctrodo é o parâmetro que controla o processo electrolítico(33).
Com eléctrodos catalíticos, a electrólise a potencial fixo constante não é geralmente exequível, em virtude do fenómeno de envenenamento da superfície que provoca a diminuição da corrente de oxidação e a sua rápida anulação(34). Durante as electrolises prolongadas, e com o objectivo de manter a actividade do eléctrodo suficientemente elevada, é necessário seleccionar programas de potencial de modo a limpar periodicamente a superfície do eléctrodo por oxidação das espécies fortemente adsorvidas, a que se designam de “venenos”(34) .
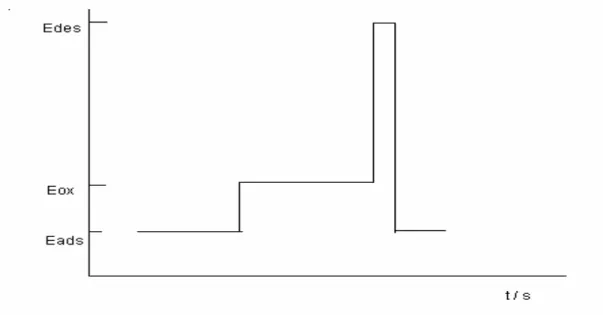
O programa de potencial é constituído por três diferentes patamares, conforme a figura 1.6.
Figura 1.6 – Programa de potencial - tempo utilizado na electrolise exaustiva a potencial controlado.
O primeiro patamar, correspondente ao potencial de adsorção da espécie electroactiva. O segundo patamar corresponde à oxidação da espécie electroactiva, tendo uma duração de tempo variável. O terceiro patamar é o potencial de desadsorção e tem um tempo de aplicação muito curto.
Na célula da electrólise existe uma membrana que separa os dois compartimentos que a constituem. Esta é uma membrana selectiva que deixa passar somente certos iões.
As membranas iónicas permeáveis são polímeros orgânicos que contêm grupos que fazem a permuta iónica(23).
Estas têm como requisitos(23) :
- alta selectividade a um determinado ião; - baixo transporte de solvente;
-estabilidade química aos reagentes, produtos da electrólise, electrólito e temperatura;
- estabilidade mecânica; - alta condutividade iónica;
- permanecerem inalteradas perante alta densidade de corrente.
Durante a electrólise, os iões passam através da membrana, devido ao potencial aplicado. Através deste movimento migratório, uma carga deve passar pela membrana por
cada electrão transferido na superfície do eléctrodo. Com membranas catiónicas os catiões passam do ânodo para o cátodo. Com membranas aniónicas os aniões passam do cátodo para o ânodo(23).
O grau de conversão num processo electrolítico envolvendo uma reacção de transferência electrónica reversível, pode ser calculado através do potencial aplicado(33).
Consideremos a seguinte reacção química :
B A + ne- ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ + = B A C C nF RT E E 0 ln (1.5)
As espécies B e A são ambas solúveis. E0 corresponde ao potencial padrão do par redox. Considerando que no inicio não existe espécie A, CB é a concentração de B no inicio. A fracção de B oxidado a A é designado de x, ao potencial E e Vs corresponde ao volume da solução. Assim, o número de moles de B no equilíbrio é igual a VsCB(1-x)
.
A uma temperatura de 25ºC, temos:
⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ + = x x nF RT E E 1 ln 0 (1.6) ( ) 1 059 , 0 0 10 1 − − − ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ + = E E n x (1.7)
A equação seguinte dá a expressão da intensidade de corrente, ao considerar a electrólise da espécie B, com concentração inicial 0.
B C ) (1.8) ( ) ( 0 t C nFAm t i = B B
Em que A é a área do eléctrodo e mB o coeficiente de transporte de massa.
Para 100 % de eficiência, a corrente pode ser calculada a partir da velocidade de consumo da espécie B, conforme a seguinte expressão:
⎥ ⎦ ⎤ ⎢ ⎣ ⎡ − = dt t dN nF t i( ) B( ) (1.9)
em que NB é o número total de moles de B, no sistema.
Considerando a solução homogénea, vem:
V t N t
CB0( )= B( ) (1.10) em que V é o volume total da solução.
⎥ ⎦ ⎤ ⎢ ⎣ ⎡ − = dt t dC nFV i B( ) 0 (1.11) ) ( ) ( ) ( 0 0 0 t pC t C V A m dt t dC B B B B ⎟ =− ⎠ ⎞ ⎜ ⎝ ⎛ − = (1.12)
em casos simples a intensidade de corrente eléctrica diminui durante a electrólise de acordo com a seguinte relação:
) (1.13) exp(
0 pt
i
i= −
onde p é dado por,
V A m
p= B (1.14)
i é a corrente electrolítica no tempo t, i0 é a corrente electrolítica no instante t=0, A é a área do eléctrodo de trabalho e V é o volume da solução.
A carga total consumida Q(t), em Coulomb, durante a electrólise é dada por:
(1.15) idt t Q t 0 ) ( =∫
1.4.3 MATERIAIS DE ELÉCTRODO (1)
Quando se opta por um determinado material de eléctrodo, a opção é feita em função da zona de potenciais úteis deste no solvente a utilizar.
Esta zona de potenciais úteis está limitada por factores, como, a dissolução do eléctrodo, formação de uma camada na sua superfície de uma substância isoladora, decomposição do electrólito de suporte e decomposição do solvente.
Os materiais mais utilizados como eléctrodos em voltametria são os metais, o carbono, óxidos metálicos e sais orgânicos condutores, tendo sido no passado o mercúrio, o material de eléctrodo mais usado.
Os metais que são mais utilizados são a platina, o ouro e a prata. Estes metais apresentam vantagens como a elevada condutividade, reprodutibilidade, e simplicidade de construção do suporte do eléctrodo, assim como a facilidade de polimento. Como desvantagem teremos o elevado custo.
Relativamente ao carbono, as reacções electroquímicas são normalmente mais lentas em eléctrodos deste elemento do que em eléctrodos metálicos. A transferência de electrões durante a reacção depende da estrutura e preparação da superfície do respectivo eléctrodo. O carbono tem uma actividade de superfície elevada, o que explica a sua susceptibilidade para o envenenamento por compostos orgânicos.
Existem vários tipos de carbono que são usados como eléctrodos. Como exemplo temos o carbono negro, fibras de carbono, carbono vítreo, grafite e pasta de carbono. A pasta de carbono consiste em partículas de grafite incorporadas numa matriz inerte. O mais usado é o carbono vítreo que é isotrópico. O fabrico de eléctrodos é difícil devido à fragilidade e dureza do carbono(1).
1.4.3.1 ELÉCTRODOS MODIFICADOS
Conforme, já referido, as reacções químicas de catálise heterogénea, ocorrem na presença de um catalisador. Os eléctrodos utilizados neste trabalho são eléctrodos de carbono modificados com complexos de metais de transição.
Um processo electroquímico envolve vários passos físicos e químicos, como a transferência electrónica, adsorção / desadsorção e transporte de massa na interface do eléctrodo que consiste numa estrutura estratificada consistindo em níveis de difusão e de transferência eléctrica ao longo da superfície do eléctrodo e no seio da solução. Tal ocorre mesmo no modelo mais simplificado(22).
O controle da reacção deve ser o mais importante no processo electroquímico, pelo que este tem sido convencionalmente realizado com a intenção de seleccionar as condições electrolíticas, tais como o potencial do eléctrodo, o material do eléctrodo, o electrólito de suporte, o solvente, a densidade de corrente, etc. Recentemente, a utilização de eléctrodos com as superfícies modificadas com substâncias químicas funcionais, têm sido desenvolvidas para síntese electroorgânica, utilizando uma variedade de sistemas redox. Assim, desenvolvimento de novos conceitos e metodologias para a modificação da interface do eléctrodo são exigidos para estes novos desafios da electroquímica (22).
O eléctrodo cuja superfície é modificada com substâncias químicas funcionais, foi alvo de interesse desde 1960 e tem acumulado resultados valiosos desde os seus aspectos práticos e fundamentais, tais como a electrocatálise, electroreacção enzimática, electroreacção assimétrica, etc. A modificação química da superfície do eléctrodo, não é só por si, suficiente para uma maior eficiência e precisão no controle da reacção. Além da modificação química da superfície do eléctrodo, também modificações físicas e energéticas devem ter como objectivo o controle da reacção nos processos electroquímicos(22).
Novos métodos para modificações da superfície dos eléctrodos utilizando complexos metálicos foram desenvolvidos por Aramata(23). Um complexo de ósmio quiral é ligado com sucesso na monocamada, a nível molecular num eléctrodo de platina em soluções não aquosas. A ligação dos iões revela um fenómeno de interacção repulsiva entre cada um na platina, na formação do campo quiral e também nas reacções redox Os (II/III), enquanto os complexos racémicos interagem menos repulsivamente entre si(22).
O estudo aqui apresentado é realizado, com a utilização de eléctrodos modificados, como eléctrodos de trabalho.
Eléctrodos modificados são aqueles, cuja superfície é intencionalmente alterada. Esta alteração de superfície pode ser realizada de várias formas, tais como: adsorção, recobrimento físico, ou ligação de espécies especificas(1).
O resultado desta modificação na superfície do eléctrodo, consiste em bloquear o acesso directo ao eléctrodo. A modificação pode, conferir maior selectividade, sendo normalmente a camada de modificador electroactiva. As correntes obtidas com eléctrodos modificados, são geralmente mais elevadas do que na ausência do modificador. As aplicações de eléctrodos modificados são variadas, destacando-se a catálise do ruténio IV imobilizado dentro de PVP em reacções orgânicas como do propano-2-ol para a acetona. A caracterização de eléctrodos modificados pode ser efectuada por métodos electroquímicos como a voltametria cíclica, espectroscópicos e microscópicos.
De seguida apresentam-se alguns dos métodos usados na preparação de eléctrodos modificados(1).
1 – Modificação química : a espécie electroactiva é imobilizada na superfície do eléctrodo por reacção química.
2 – Adsorção : geralmente é utilizada na preparação de eléctrodos modificados por polímeros. A solução de polímero é pintada na superfície do eléctrodo, sendo o solvente evaporado.
3 – Electroadsorção : é realizada uma adsorção com um potencial aplicado ao eléctrodo.
4 – Plasma : é utilizado para limpar a superfície do eléctrodo. São deixados na superfície átomos não ligados, o que leva à existência de uma superfície activada.
O método de preparação do eléctrodo modificado utilizado nesta investigação, pode ser uma forma adaptada do segundo método atrás referido. Neste, a solução preparada com o complexo de metal de transição a estudar, recobriu a superfície de uma placa quadrangular de carbono Toray, previamente colada a um fio de platina. A substância utilizada na colagem do eléctrodo, consiste numa pasta de carbono electricamente condutora, que foi também utilizada para revestir a platina.
1.4.4 ELECTRÓLITO DE SUPORTE(1)
O electrólito de suporte, tem como função minimizar o fenómeno da migração dos iões electroactivos, causada pelo campo eléctrico e atribuir a diferença de potencial interfacial à distância de maior aproximação de iões solvatados ao eléctrodo. Assim é adicionada à solução o electrólito de suporte, que consiste na adição de uma concentração elevada de um electrólito inerte.
Relativamente à concentração do electrólito de suporte, esta é pelo menos cem vezes mais concentrada do que as espécies electroactivas, uma vez que este electrólito é a fonte de espécies iónicas electricamente condutoras.
Vulgarmente a concentração de electrólito de suporte varia entre 0,01M e 1,0M, sendo a concentração das espécies electroactivas de 5mM ou menos.
Para escolher um electrólito de suporte devem ser considerados alguns factores, como as propriedades do solvente empregue. Este electrólito pode ser um sal inorgânico ou orgânico, um ácido ou uma base, ou ainda uma solução tampão como acetato, citrato ou fosfato(1).
No presente trabalho o electrólito de suporte predominantemente utilizado foi o NaHO 0,1 mol.dm-3, e a espécie electroactiva, a D-galactose tinha uma concentração de 10mM.
1.4.5 REMOÇÃO DO OXIGÉNIO(1)
O oxigénio deve ser removido da solução, pois caso contrário este pode reagir com os reagentes e/ou produtos da reacção de eléctrodo que está a ser estudada, falseando assim resultados. As reacções possíveis de ocorrer encontram-se descritas de acordo com o pH do meio.
Em solução ácida : O2 + 2H+ + 2 e- H2O2
H2O2+ 2H+ + 2 e- 2H2O
Em solução alcalina : O2 + 2H2O + 2 e- 2OH-+ H2O2
1.5 MÉTODOS ANALÍTICOS
Aos produtos obtidos na electrólise, fizeram-se análises de identificação e quantificação.
A técnica utilizada para tal foi a cromatografia. Esta técnica consiste num método analítico que é largamente usado para a separação, identificação e determinação quantitativa de componentes químicos em misturas complexas, muitas das quais não podiam ser resolvidas de outro modo(25).
Em todas as separações cromatográficas a amostra é dissolvida numa fase móvel, a qual pode ser um gás ou um líquido. Esta fase móvel é então transportada através de uma fase estacionária imiscível, a qual está imobilizada numa coluna ou numa superfície sólida(25).
As duas fases são escolhidas de modo que os componentes da amostra se distribuem entre a fase móvel e a fase estacionária de modos diferentes(25) :
- componentes fortemente retidos pela fase estacionária; - componentes fracamente retidos pela fase estacionária.
Assim, as separações baseiam-se nas diferentes velocidades de migração dos componentes da amostra.
Os métodos cromatográficos podem ser classificados em dois grupos(25):
- Classificação baseada no processo físico pelo qual as fases móvel e estacionária são postas em contacto:
- cromatografia em coluna; - cromatografia planar.
- Classificação baseada na natureza das fases móvel e estacionária: - cromatografia em fase líquida – a fase móvel é um líquido; - cromatografia em fase gasosa – a fase móvel é um gás.
A eficiência de uma coluna cromatográfica para separar dois solutos depende das velocidades relativas, às quais as duas espécies são eluidas e estas velocidades são por sua vez determinadas pela razões de partição dos solutos entre as duas fases(25).
O coeficiente de partição ou razão de partição é dado por:
) (
Em que :
C(E) – concentração molar do soluto na fase estacionária; C(M) – concentração molar da fase móvel.
Outro parâmetro de identificação dos componentes de uma mistura é o tempo de retenção.
Temos que:
tr – tempo de retenção para o soluto;
tm – tempo de retenção de uma espécie não retida pela coluna. A velocidade linear média do soluto é dada por :
r
t L
v= (1.17)
Sendo L – comprimento da coluna cromatográfica.
A eficiência de colunas cromatográficas pode ser quantificada através de dois parâmetros:
H – altura do prato ;
N – número de pratos teóricos.
Os dois parâmetros estão relacionados pela seguinte expressão:
H L
N = (1.18)
A eficiência de uma coluna cromatográfica aumenta quando aumenta o número de pratos desta e quando a altura do prato diminui(25).
Se um detector que corresponde à concentração do soluto é colocado no final da coluna e este sinal é traçado em função do tempo, obtém-se uma série de picos simétricos – os cromatogramas(25).
A análise quantitativa que se pode obter na cromatografia, baseia-se na comparação de alturas ou áreas dos picos do cromatograma da amostra com os correspondentes valores padrões. As alturas e as áreas são proporcionais à concentração do componente.
A calibração com padrões pode ser realizada por dois métodos diferentes: o método directo e o método interno.
O método directo consiste na preparação de uma série de soluções padrão cuja composição se aproxima da composição da amostra; obtenção dos cromatogramas dos padrões e da amostra; traçado do gráfico das alturas ou áreas dos picos como uma função da concentração; análise da amostra baseado no gráfico anterior. Este foi o método utilizado neste trabalho de investigação.
O método do padrão interno consiste em uma quantidade de substância padrão interna medida cuidadosamente ser introduzida em cada padrão e amostra e a razão da área ou altura do pico da amostra e a área ou altura do pico do padrão interno é o parâmetro analítico(25).
1.5.1 CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA
No presente trabalho, foi utilizada a cromatografia líquida de alta eficiência HPLC. Nesta a fase móvel é um líquido. As principais razões para o crescimento da HPLC são a sensibilidade do método, adaptação efectiva a determinações quantitativas precisas, capacidade para separar espécies não voláteis ou termicamente estáveis, e ainda uma larga aplicação a substâncias de primordial interesse industrial(25).
A HPLC pode ser classificada em (25):
- cromatografia de partição ou líquido - líquido; - cromatografia de adsorção ou líquido – sólido; - cromatografia de troca iónica;
- cromatografia de exclusão de tamanho.
Os processos de troca iónica baseiam-se em equilíbrios de troca iónica em solução e iões do mesmo sinal na superfície de um sólido essencialmente insolúvel e de massa molecular elevada. Há competição de iões. Iões de carga semelhante são separados por eluição através da coluna. Os iões da amostra são introduzidos no topo de uma coluna empacotada com uma resina de troca iónica apropriada. A eluição é então realizada com uma solução que contém um ião que compete com os iões da amostra para grupos iónicos na superfície da resina(25).
A capacidade de troca iónica de uma resina é medida pela sua capacidade de trocar iões. A capacidade total é definida como a quantidade de carga e grupos potencialmente carregados por grama de resina seca(26).
Estas colunas têm as resinas de troca iónica, que são materiais poliméricos de massa molecular elevada contendo muitos grupos funcionais iónicos por molécula. As resinas de troca iónica podem ser de troca aniónica ou de troca catiónica(25).
As resinas de troca aniónica são do tipo base e consistem em grupos de aminas quaternárias ( –N(CH3)3+OH-) ou grupos aminas primárias (-NH3+OH-).
As resinas de troca catiónica são do tipo ácido e consistem em grupos de ácido sulfónico (-SO3-H+ ) ou de grupos de ácido carboxílico (-COOH)(25).
As resinas de troca iónica dos ácidos fracos (-COOH) têm restrição no intervalo do valor do pH, devendo este encontrar-se entre 5 e 15(26).
Os detectores mais comuns para HPLC são de Absorção UV, Absorção IR, Índice de Refracção, Espectroscopia de Massa, e Condutividade.
Os detectores utilizados neste trabalho foram o Índice de Refracção para a D - Galactose e o de condutividade para os ácidos orgânicos provenientes da oxidação da D – Galactose ao longo da electrólise.
O detector de Índice de Refracção (RI) é um dos mais próximos do detector ideal e universal. O índice de refracção de fase móvel deverá ser alterado pela presença de algum soluto que tenha um valor de índice de refracção diferente do da fase móvel. Assim comparando o valor do RI da fase móvel pura com o eluente da coluna, indica a presença de um soluto eluido(27).
Com o detector RI, qualquer soluto pode ser detectado de acordo com a maior diferença do valor do RI entre o soluto e a fase móvel(28).
Os detectores de condutividade são vulgarmente utilizados, quando os solutos eluidos são iónicos como por exemplo ácidos e bases. A maior aplicação deste tipo de detectores é para aniões e catiões inorgânicos, depois da sua separação numa coluna cromatográfica de troca iónica(27).
Estes detectores medem a condutância, isto é, o inverso da resistência preferencialmente à condutividade, isto é, o inverso da resistividade, do eluente apesar de serem vulgarmente conhecidos por detectores de condutividade(28).
Um dos maiores problemas destes detectores é o facto de a fase móvel utilizada conter iões e pode ter uma condutância que será alta comparada com a condutância devida aos iões do soluto(28).
1.5.2 SUPRESSOR DE ELUENTE PARA CROMATOGRAFIA IONICA(30)
Neste trabalho, a coluna cromatográfica que foi utilizada, foi uma coluna de troca iónica, à qual se encontrava associada um supressor.
Uma cromatografia típica de troca iónica, é constituída por vários componentes. O eluente que é condutor, é distribuído utilizando-se para tal uma bomba de alta pressão. A amostra é introduzida, seguindo para a coluna analítica de troca iónica, onde ocorre a separação pela troca iónica. O supressor reduz a condutividade residual do eluente sendo só a condutividade dos componentes a analisar detectada.
No caso do presente trabalho, o ião sódio é removido do eluente pelo supressor, e substituído pelo ião hidrónio.
Figura 1.7 – Diagrama da supressão do anião do eluente pela regeneração química(30).
Este ião hidrónio combina-se com o ião hidroxilo ou carbonato que são provenientes do eluente para formar água ou ácido carbónico, os quais têm um valor de condutividade mais baixo quando comparados com os do hidróxido ou carbonato do eluente(30).
No caso concreto do presente trabalho, o que se formou foi água durante o processo da troca iónica.
CAPITULO 2
SÍNTESE E CARACTERIZAÇÃO DE COMPLEXOS DE
RUTÉNIO E MOLIBDÉNIO
2.1 INTRODUÇÃO
Compostos de coordenação contendo derivados de piridina têm suscitado elevado interesse devido à sua actividade catalítica, estando vários trabalhos referenciados na literatura( 35-37 ).
O interesse na química do ruténio e do molibdénio deve-se à elevada reactividade dos metais por adquirirem um número elevado de estruturas estáveis com os metais em diferentes estados de oxidação. Esta versatilidade confere-lhes propriedades electrónicas que lhes dão diversas aplicações no domínio da catálise, da optoelectrónica e telecomunicações e em processos biológicos( 38-41).
Neste trabalho é explorada a química da coordenação do ruténio e do molibdénio, com o ligando 1,2–piridilazo– 2–naftol (PAN), esquema1:
N=N OH
N
Esquema 1 – Estrutura do ligando 1,2 – piridilazo – 2 – naftol .
Os complexos são caracterizados por espectroscopia de IV, UV–visível, RMN de 1H e análise elementar.
De seguida, é descrito o comportamento electroquímico dos complexos estudados por voltametria cíclica.
Este estudo é interessante devido a que o objectivo do trabalho é utilizar eléctrodos modificados como materiais catalíticos para a oxidação electroquímica dos açucares.
2.1.1 SÍNTESE DOS COMPLEXOS DE RUTÉNIO
O complexo dicloro-bis(1–(2–piridilazo)–2–naftol)–ruténio(II), [RuCl2(PAN)2], foi sintetizado por adaptação do método descrito na literatura para a síntese de complexos análogos(42,43), a partir do complexo triclororuténio(III), esquema 2: + Ru N N N N HO N N OH Cl Cl N2 N=N OH N RuCl3 MeOH r.t. refluxo Estrutura I [Ru Cl2 H2 (C15H11N3O)2] Hidreto de ruténio Esquema 2 – Síntese do complexo de ruténio.
Após oito horas de refluxo obteve-se uma mistura a qual foi levada à secura. O sólido foi solubilizado em dimetilformamida (DMF), e por cromatografia em coluna obtiveram-se três fracções 1, 2 e 3.
A caracterização espectroscópica das fracções 1 e 3 indica tratar-se do mesmo composto, estrutura I, esquema 2.
A caracterização espectroscópica da fracção 2 indica a presença do hidreto de ruténio, esquema 2.





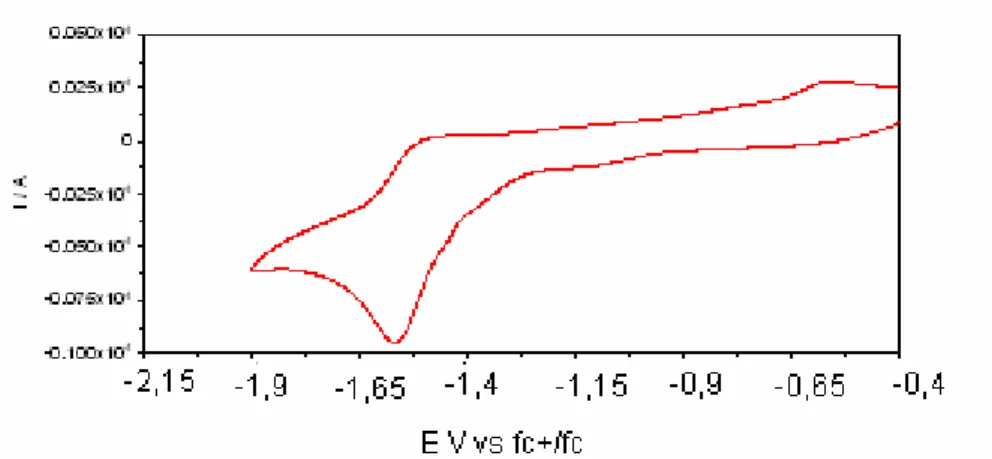
![Figura 2.4 – Voltamograma cíclico para uma solução do complexo [Ru Cl 2 (C 15 H 11 N 3 O) 2 ] – fracção 1 (10 -3 mol.dm -3 ) em dimetilformamida (DMF), num eléctrodo de carbono vítreo, v=0,1V.s -1 , entre -1,9 e -0,4 V vs fc + /fc](https://thumb-eu.123doks.com/thumbv2/123dok_br/17962642.854514/53.892.124.639.267.497/figura-voltamograma-cíclico-solução-complexo-fracção-dimetilformamida-eléctrodo.webp)
![Figura 2.6 – Voltamograma cíclico para uma solução do complexo [Ru Cl 2 H 2 (C 15 H 11 N 3 O) 2 ] – fracção 2 (10 -3 mol.dm -3 ) em dimetilformamida (DMF), num eléctrodo de carbono vítreo, v=0,1V.s -1 , entre -1,8 e -0,4 V vs fc + /fc](https://thumb-eu.123doks.com/thumbv2/123dok_br/17962642.854514/54.892.156.649.572.795/figura-voltamograma-cíclico-solução-complexo-fracção-dimetilformamida-eléctrodo.webp)
![Figura 2.7 – Voltamograma cíclico para uma solução do complexo [Mo(C 5 H 5 ) 2 (C 15 H 11 N 3 O)]Cl 2 (10 -3 mol.dm -3 ) em dimetilformamida (DMF), num eléctrodo de carbono vítreo,v=0,1V.s -1 entre -0,4 e 0,6 V vs fc + /fc](https://thumb-eu.123doks.com/thumbv2/123dok_br/17962642.854514/55.892.126.626.337.551/figura-voltamograma-cíclico-solução-complexo-dimetilformamida-eléctrodo-carbono.webp)
