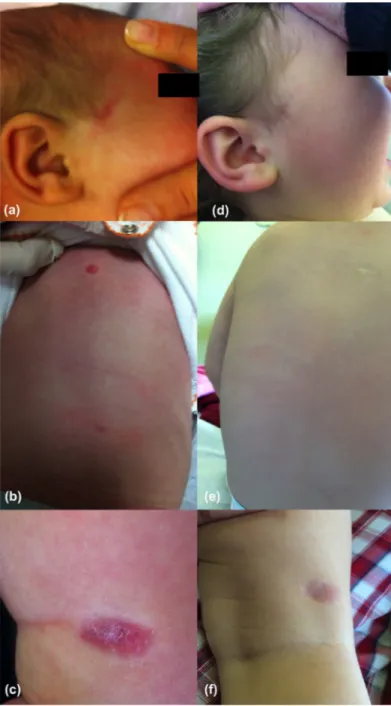
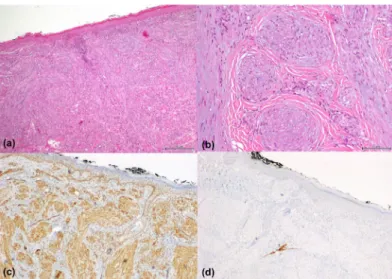
Infantile myofibromatosis - a clinical and pathological diagnostic challenge
5
0
0
Texto
Imagem
Documentos relacionados