Disfunção hepática em animais modelo para a doença de
Alzheimer: o papel do stresse oxidativo e da β-amilóide
Dissertação de Mestrado em Biologia Clínica Laboratorial
JOANA FILIPA ALMEIDA FARIA
Orientadores:
Professora Doutora Maria Manuel Oliveira
Doutor Romeu António Videira
Composição do Júri:
_______________________________________________________
_______________________________________________________
_______________________________________________________
ii
Instituição Universidade Trás-os-Montes e Alto Douro Curso Mestrado em Biologia Clínica Laboratorial
Título “Disfunção hepática em animais modelo para a doença de
Alzheimer: o papel do stresse oxidativo e da β-amilóide”
Autor Joana Filipa Almeida Faria
Orientador Professora Doutora Maria Manuel Oliveira Co - Orientador Doutor Romeu António Videira
iv
“Este trabalho foi expressamente elaborado como dissertação original para o efeito da obtenção do grau de Mestre em Biologia Clínica Laboratorial.”
v
Findada mais uma etapa do meu percurso académico, gostaria de expressar alguns agradecimentos a pessoas especiais, sem as quais não conseguiria chegar até aqui.
À Professora Doutora Maria Manuel Oliveira, pela disponibilidade e amizade demonstrada desde o início do meu percurso universitário e, mais recentemente, pela orientação deste trabalho.
Ao Doutor Romeu António Videira, pela orientação deste trabalho, pelo apoio demonstrado mesmo quando o meu desempenho não correspondia e pela constante aprendizagem.
Ao Professor Doutor Francisco Peixoto, pela disponibilidade e amizade demonstrados durante este percurso académico.
À Fundação para a Ciência e Tecnologia, FEDER e COMPETE por financiar o projeto PTDC/SAL-NMC/115865/2009, do qual surgiu esta dissertação.
Aos meus amigos companheiros de laboratório e de “salinha”, particularmente à Vera Cardoso e Marisa Castro, por todos os momentos partilhados e por fornecerem o melhor escape quando o trabalho não corria da melhor forma.
Aos meus amigos de sempre, porque sem vocês não seria a pessoa que hoje conhecem.
Por último, mas não menos importante, a toda a minha família, em particular ao meu pai José, à minha mãe Isaura e aos meus irmãos Ricardo e Lúcia. Tudo o que faço é por vós e para vós.
vi
Disfunção hepática em animais modelo para a doença de Alzheimer: o papel do stresse oxidativo e da β-amilóide.
A doença de Alzheimer é uma das principais doenças neurodegenerativas predominante entre indivíduos idosos. Clinicamente, é caracterizada por perda de memória e mudanças comportamentais, sendo uma das principais causas de demência afetando milhões de pessoas em todo o mundo. A nível celular, os marcadores patológicos da doença de Alzheimer são a acumulação extracelular de placas senis (maioritariamente formadas pela deposição do peptídeo β-amilóide) e a formação intracelular de tranças neurofibrilares constituídas pela proteína tau hiperfosforilada, associadas a uma redução da densidade sináptica e consequente perda neuronal. Considerando o papel do fígado nos processos de desintoxicação, a produção excessiva de β-amilóide associada à progressão da doença de Alzheimer pode promover a disfunção hepática.
Para investigar esta hipótese, avaliou-se o impacto fisiológico da progressão da doença de Alzheimer na atividade do fígado, usando murganhos triplo-transgénicos (3xTg-AD) com 3, 6 e 12 meses de idade, como animais modelo da doença de Alzheimer, e murganhos da estirpe selvagem (WT) com idades equivalentes, como controlo. Nestes modelos, foi investigado o efeito do envelhecimento na bioenergética mitocondrial, considerando as atividades dos complexos I, II, IV e FoF1-ATPase, bem como alguns
parâmetros relacionados com o stresse oxidativo, como as atividades da catalase e Superóxido dismutase (Cu-Zn-SOD), o conteúdo total de tióis nas proteínas e os níveis de peroxidação lipídica. O efeito da β-amilóide na atividade dos complexos mitocondriais e na peroxidação lipídica foi avaliado em ensaios in vitro, para melhor compreender o papel da β-amilóide no desenvolvimento da doença de Alzheimer.
Os estudos revelaram que os animais 3xTg-AD com 3 e 6 meses de idade devem apresentar disfunção hepática, pois a atividade de enzimas chave relacionadas com bioenergética mitocondrial (complexos I, II e IV) é significativamente inferior à dos correspondentes controlos. No entanto, os efeitos tendem a desaparecer quando os animais atingem um ano de idade. Adicionalmente, nestes modelos as defesas antioxidantes das células do fígado estão comprometidas devido à menor atividade da catalase. Os estudos in
vitro mostraram que a β-amilóide é tóxica para as mitocôndrias, reduzindo progressivamente
a atividade do complexo I e IV até concentração de 2,5 µg/mg de proteína mitocondrial, mas concentrações superiores têm menor efeito. Estes dados sugerem que a toxicidade
vii
Assim, para além da prevista degeneração do cérebro, efeitos em órgãos periféricos como o fígado parecem surgir durante a progressão da doença de Alzheimer, sugerindo que o seu estudo é fundamental não só para compreender os múltiplos aspetos relacionados com a doença como para identificar marcadores específicos para os vários estágios que caracterizam a progressão da doença de Alzheimer.
viii
Liver dysfunction in triple-transgenic mice model of Alzheimer’s disease: the role of oxidative stress and β-amyloid
Alzheimer’s disease (AD) is one of the major neurodegenerative diseases prevalent among older individuals. Clinically, it is characterized by memory loss and behavioral changes, being a major cause of dementia affecting millions of people worldwide. At cellular level, the pathological hallmarks of Alzheimer's disease are the extracellular accumulation of senile plaques (mostly formed by deposition of β-amyloid peptide) and the formation of intracellular neurofibrillary tangles composed by hyperphosphorylated tau protein, associated with a reduction in synaptic density and consequent neuronal loss. Considering the role of liver in detoxification processes, excessive production of β-amyloid associated with Alzheimer's disease progression can promote liver dysfunction.
To investigate this hypothesis, the liver physiological impact of Alzheimer's disease progression was evaluated using triple transgenic mice (3xTg-AD) with 3, 6 and 12 months-old, as Alzheimer's disease animal model, and wild type (WT) mice, as age-matched controls.
In these models, the age-dependent effects on liver bioenergetics were evaluated considering the effects on complex I, II, IV and FoF1-ATPase activities, as well as on key
parameters related to oxidative stress, including catalase and superoxide dismutase (CuZn -SOD) activities, total thiols proteins contents and lipid peroxidation. In vitro assays, β-amyloid effects on the activity of complex I and IV of liver mitochondria isolated from health animals were assessed to evaluate its putative influence on liver mitochondria dysfunction. Additionally, the role of β-amyloid on lipid peroxidation process was evaluated in synthetic and native membranes.
Our studies suggest that 3xTg-AD animals with 3 and 6 months-old may present hepatic dysfunction, since key mitochondrial enzymes related with liver bioenergetics (complexes I, II and IV) exhibit significantly lower activity than in the age-matched controls. However, these effects are vanished when the animals reach 12 months-old. On the other hand, the liver antioxidant defenses in 3xTg-AD animals may be committed due to the low catalase activity. In vitro studies show β-amyloid mitochondrial toxic effects mainly at low concentration, since a progressive reduction of both complexes I and IV activities were detected up to 2.5 µg β-amyloid/mg of mitochondrial protein, while higher concentrations did not have significant additional effects. These data suggests that the mitochondrial β-amyloid toxicity results mainly from its high soluble monomeric structure.
ix
organs may be essential not only to understand specific aspects related with the molecular mechanism underlay disease progression, but also to identify novel biomarkers of each stage of disease.
x
3xTg-AD – triplo-transgénicos para a doença de Alzheimer AD – Doença de Alzheimer
APP – amyloid precursor protein ATP – Adenina trifosfato
BACE – beta-site APP-cleaving enzyme Cu-Zn-SOD – superóxido dismutase DPPC – dipalmitoilfosfatidilcolina DPPS – dipalmitoilfosfatidilserina PS1 – persilina 1
PS2 – persilina 2
ROS – espécies reativas de oxigénio WT – wild type
xi
eletrões I, II, IV e da FoF1-ATPase avaliada em mitocôndrias isoladas de fígado de
murganhos WT e 3xTg-AD com idade de 3 meses, normalizada pela atividade da Citrato sintase.
Figura 2 – Atividade individual dos complexos enzimáticos da cadeia transportadora de eletrões I, II, IV e da FoF1-ATPase avaliada em mitocôndrias isoladas de fígado de
murganhos WT e 3xTg-AD com idade de 6 meses, normalizada pela atividade da Citrato sintase.
24
Figura 3 – Atividade individual dos complexos enzimáticos da cadeia transportadora de
eletrões I, II, IV e da FoF1-ATPase avaliada em mitocôndrias isoladas de fígado de
murganhos WT e 3xTg-AD com idade de 12 meses, normalizada pela atividade da Citrato sintase.
25
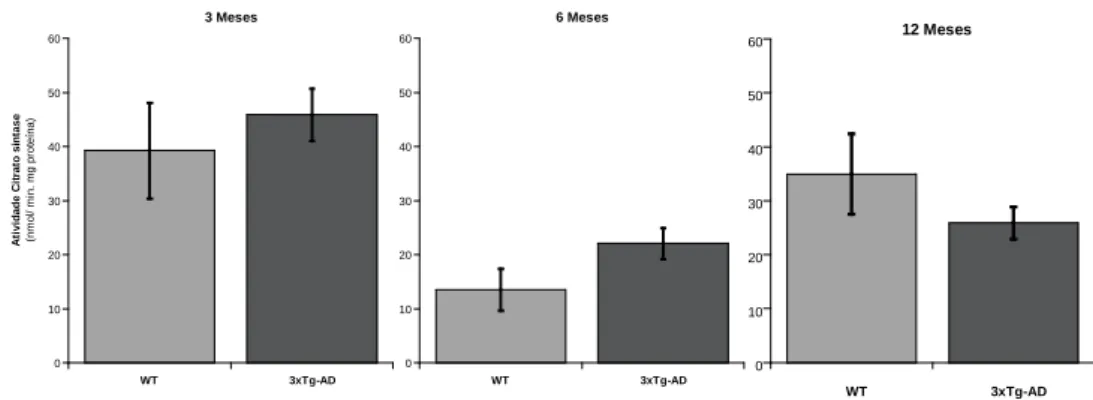
Figura 4 – Atividade da Citrato sintase nas mitocôndrias isoladas de murganhos WT e 3xTg-AD com idades de 3, 6 e 12 meses.
26 Figura 5 – Evolução da atividade dos complexos I, II, IV e FoF1-ATPase de mitocôndrias
isoladas de murganhos WT e 3xTg-AD, ao longo da idade (3, 6 e 12 meses), normalizada pela atividade da Citrato sintase.
27
Figura 6 – Efeito do aumento da concentração de β-amilóide (Abeta) na atividade dos
complexos I e IV.
29 Figura 7 – Atividade da Catalase em nmol de O2 produzidas por minuto por mg de proteína
de no sobrenadante dos homogeneizados de fígado de animais WT e 3xTg-AD com 3, 6 e 12 meses de idade.
31
Figura 8 – Atividade da Superóxido Dismutase (Cu-Zn-SOD), apresentada em unidades
de atividade por minuto por mg de proteína no sobrenadante dos homogeneizados de fígado de animais WT e 3xTg-AD com 3, 6 e 12 meses de idade.
32
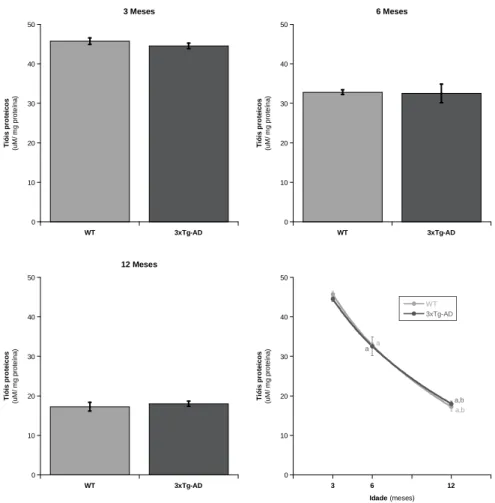
Figura 9 – Quantificação do conteúdo total de tióis proteicos, expressos apresentados em
µM (equivalentes de cisteína) por mg de proteína no sobrenadante dos homogeneizados de fígado de animais WT e 3xTg-AD com 3, 6 e 12 meses de idade.
33
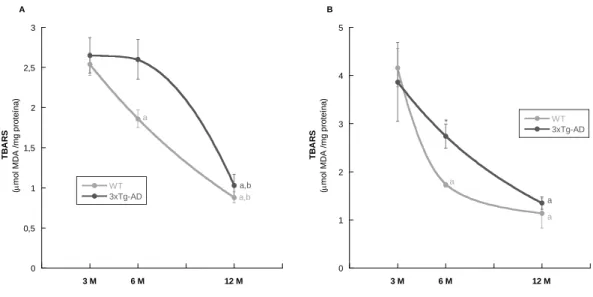
Figura 10 – Efeito da idade na concentração de espécies que reagem com ácido
tiobarbitúrico (TBARS), em equivalente de malonildialdeído (MDA)/ mg de proteína, em dois casos particulares: (A) em sobrenadantes dos homogeneizados de fígado de animais WT e 3xTg-AD; e (B) em mitocôndrias isoladas de fígado de murganhos WT e 3xTg-AD.
34
Figura 11 – Variação da intensidade de fluorescência da sonda cis-parinárico incorporada nas membranas de lipossomas multilamelares de DPPC para concentrações crescentes de β-amilóide, a temperaturas de 35, 41 e 43 o
C.
36
Figura 12 – Variação da intensidade de fluorescência da sonda cis-parinárico encapsulado
em lipossomas multilamelares de DPPC/DPPS (em percentagens 87/13 e 93,5/6,5 simulando constituições membranares assimétricas e simétricas, respetivamente) para concentrações crescentes de β-amilóide e por adição de Ca2+
com concentração final de 1,2 mM. Os ensaios foram realizados a 40 oC.
xii Resumo……….………...vi Abstract……….……….…….viii Lista de Abreviaturas………x Índice de figuras…………..……….…...xi I – Introdução ... 1 1. A doença de Alzheimer ... 2 1.1 Disfunção mitocondrial ... 2 1.2 Stresse oxidativo ... 3 2.Toxicidade da β-amilóide ... 4
2.1 Interação da β-amilóide com as membranas biológicas ... 6
2.2 O papel das membranas biológicas no aparecimento e progressão da doença de Alzheimer ... 7
3. Modelos para o estudo - objetivos ... 8
II – Material e Métodos ...10
1. Reagentes ...11
2. Modelo animal da doença de Alzheimer ...11
2.1 Isolamento de mitocôndrias de fígado ...11
2.2 Preparação dos homogeneizados de fígado ...12
2.3 Quantificação proteína – método de biureto ...12
3. Lipossomas multilamelares – modelos para estudar a interação da β-amilóide com as membranas biológicas ...13
4. Bioenergética Mitocondrial ...14
4.1 NADH:ubiquinona oxidoreductase (Complexo I) ...14
4.2 Succinato:ubiquinona oxidoreductase (Complexo II) ...15
4.3 Citocromo C oxidase (Complexo IV) ...15
4.4 FoF1-ATPsintase (Complexo V) ...16
4.5 Citrato sintase ...17
5. Stresse Oxidativo ...17
5.1 Catalase ...17
5.2 Superóxido Dismutase ...17
5.3 Conteúdo total de tióis nas proteínas ...18
5.4 Avaliação da peroxidação lipídica...18
xiii
IV - Conclusão ...39 V – Referências Bibliográficas ...42
2
1. A doença de Alzheimer
A doença de Alzheimer (AD), inicialmente descrita por Alois Alzheimer em 1907, é a doença neurodegenerativa mais comum entre indivíduos idosos. Apesar de ser, essencialmente, considerada como uma doença esporádica do indivíduo, existem também casos de familiaridade associados a mutações genéticas hereditárias (Moreira et al., 2010). Do ponto de vista clínico, é caracterizada por perda de memória e mudanças comportamentais, sendo uma causa de demência que afeta milhões de pessoas em todo o mundo (Frisardi et al., 2010; Ballard et al., 2011). A nível celular, os marcadores patológicos da AD são a acumulação das placas senis (maioritariamente formadas pela deposição do peptídeo beta-amilóide) e a formação intracelular de tranças neurofibrilares constituídas pela proteína Tau hiperfosforilada (Selkoe, 1994), levando a uma redução da densidade sináptica e consequente perda neuronal (Gotz et al., 2004).
Os mecanismos moleculares subjacentes ao aparecimento e à progressão da doença ainda não se encontram totalmente esclarecidos, contudo, devido aos indivíduos afetados, considera-se o envelhecimento como o principal fator de risco. Também algumas características genéticas são normalmente consideradas como fatores importantes, nomeadamente mutações nos genes que codificam a glicoproteína transmembranar (APP -
amyloid precursor protein), persilina 1 (PS1) e persilina 2 (PS2) (Ballard et al., 2011). No que
respeita a alterações bioquímicas, foram sugeridas perturbações nos metabolismos da proteína APP e β-amilóide, da proteína tau e dos lípidos, bem como uma disfunção ao nível das mitocôndrias das células do sistema nervosos central (Hauptmann et al., 2009, Moreira
et al., 2010, Contestabile, 2011).
1.1 Disfunção mitocondrial
As mitocôndrias são organelos altamente dinâmicos responsáveis pela produção de energia na forma de adenosina trifosfato (ATP) e, por conseguinte são vitais para a regulação e atividade das células eucariotas, principalmente nos animais. Inúmeros trabalhos suportam a ideia de que a disfunção mitocondrial desempenha um papel importante nas doenças neurodegenerativas. A relação entre a disfunção mitocondrial e a doença de Alzheimer tem sido estudada em vários modelos, considerando principalmente as alterações na atividade das enzimas mitocondriais associadas com o metabolismo bioenergético. Por exemplo, estudos efetuados em tecido cerebral de doentes de Alzheimer, colhido post-mortem, revelaram atividade reduzida da citocromo c oxidase, complexo IV da cadeia respiratória mitocondrial (Mutisya et al., 1994; Kish et al.,1992), bem como uma elevada densidade de mitocôndrias anormais junto das neurites distróficas, empobrecidas
3
em citocromo c oxidase (Perez-Gracia et al., 2008). A menor atividade bioenergética de mitocôndrias de plaquetas recolhidas de doentes diagnosticados com Alzheimer quando comparando com controlos da mesma idade apoiam que a disfunção mitocondrial está associada à patologia estendendo ainda as alterações fenotípicas a tecidos periféricos, isto é, tecidos que não fazem parte do sistema nervoso central (Bosseti et al., 2002). Estudos in
vitro realizados em mitocôndrias isoladas de cérebro de rato mostram que a β-amilóide inibe
a atividade do complexo IV e da FoF1-ATP sintase, bem como das enzimas piruvato
desidrogenase e α-cetoglutarato desidrogenase (Casley et al., 2002). Kim e colaboradores mostraram que a adição de β-amilóide a mitocôndrias isoladas de cérebro de murganhos pode promover a libertação de citocromo c e o intumescimento osmótico mitocondrial, eventos relacionados com a morte celular por apoptose (Kim et al., 2002). Adicionalmente, a dinâmica mitocondrial que inclui fissão e fusão parece afetada nas células dos pacientes com Alzheimer, pelo que a mobilidade intracelular do organelo é afetada conduzindo a uma distribuição intracelular anormal (Wang et al.2009, Santos et al., 2010). Assumindo que a disfunção mitocondrial desempenha um papel importante na doença de Alzheimer, Swerdlow e Khan propuseram a hipótese da cascata mitocondrial, postulando que, no caso da doença esporádica, a disfunção mitocondrial é o evento principal que antecede e promove a deposição da β-amilóide, a degeneração sináptica e a formação de tranças neurofibrilares, eventos que convergem no envelhecimento do cérebro (Swerdlow e Khan, 2009). Por outro lado, Atmna e colaboradores realçam o papel do comprometimento do metabolismo energético mitocondrial (caracterizado por uma reduzida produção de ATP) e o consequente aumento do dano celular por stresse oxidativo (caracterizado por um aumento de espécies reativas de oxigénio) no desenvolvimento e progressão da doença de Alzheimer (Atmna e Frey, 2007).
1.2 Stresse oxidativo
A atividade mitocondrial, em especial o sistema de transporte de eletrões ao longo da cadeia respiratória, é uma das maiores fontes de geração de espécies reativas de oxigénio (ROS), normalmente amplificada pela disfunção mitocondrial (Pamplona e Barja, 2006; Moreira et al., 2007). Adicionalmente, o cérebro é um órgão bastante vulnerável ao ataque de radicais livres, dado que está permanentemente exposto a altas concentrações de oxigénio; as membranares das suas células apresentam altos níveis de ácidos gordos polinsaturados e apresenta uma proteção antioxidante relativamente baixa quando comparada com outros órgãos (e.g. fígado) simultaneamente com um elevado teor de ferro e ascorbato (Moreira et al., 2005). Quando os níveis de radicais livres superam a
4
capacidade de supressão desses radicais pelo sistema de defesa antioxidante celular ocorrem danos celulares, associados à oxidação de DNA, proteínas e lípidos que promovem uma disfunção celular generalizada, incluindo a disfunção mitocondrial e a desregulação da homeostase do cálcio que pode culminar com a morte celular por apoptose ou mesmo por necrose (Reddy e Beal, 2008).
Pelo exposto, o stresse oxidativo, caracterizado por um desequilíbrio entre agentes oxidantes e defesas antioxidantes, pode estar envolvido nos processos neurodegenerativos, incluindo naqueles que caracterizam a doença de Alzheimer. De facto, existem na literatura estudos sugerindo que o stresse oxidativo desempenha um papel central no desenvolvimento da doença, postulando que o dano oxidativo precede qualquer outra alteração fisiológica (Nunomura et al., 2001; Pratico et al., 2001). Apesar da produção de β-amilóide e consequente formação de placas senis ser uma marca patológica da doença de Alzheimer reconhecida por toda comunidade científica, os defensores da hipótese do stresse oxidativo sugerem que a secreção da β-amilóide é uma estratégia compensatória utilizada pelas células para se protegerem do dano oxidativo (Nakamura et al., 2007; Hayashi et al., 2007). Esta hipótese é suportada pelo facto da β-amilóide apresentar atividade antioxidante, tal como demonstrado em estudos efetuados in vitro para avaliar o seu efeito na oxidação de lipoproteínas do plasma (Atwood et al., 1998; 2003). Zhu e colaboradores sugerem que o dano oxidativo precoce e progressivo provoca uma resposta compensatória por parte das células, para que possam sobreviver em ambiente oxidativo, atingindo um “estado oxidativo estacionário” que eventualmente tornará a célula vulnerável a outros ataques, como a deposição de β-amilóide (inicialmente produzida para sequestrar ROS) e a formação de tranças neurofibrilares (Zhu et al., 2001, 2004, 2007). Apoiados nesta hipótese, Bonda e colaboradores propõe um mecanismo de feedback positivo, onde a produção contínua de ROS conduz inicialmente a uma disfunção mitocondrial que posteriormente aumenta a vulnerabilidade da célula à degeneração, cujos efeitos clínicos são o declínio cognitivo e a demência característicos da doença de Alzheimer. Assim, estratégias terapêuticas usando moléculas com atividade antioxidante como a coenzima Q10
e derivados como acetil-L-carnitina estão a ser investigadas (Bonda et al., 2010).
2.Toxicidade da β-amilóide
A β-amilóide é um péptido produzido através da clivagem da glicoproteína transmembranar APP pela β-secretase, enzima proteolítica BACE (beta-site APP-cleaving
enzyme) no terminal N, dando origem a um péptido solúvel sAPPβ que posteriormente sob
5
formação de péptidos de β-amilóide, maioritariamente fragmentos com 40 e 42 aminoácidos, designados respetivamente por Aβ1-40 e Aβ1-42 (Hardy e Higgins, 1992). Estes péptidos são
solúveis e, em condições fisiológicas, encontram-se associados a mecanismos de ativação enzimática (Bogoyevitch et al., 2004; Tabaton et al., 2010) e proteção contra o stresse oxidativo (Zou et al., 2002; Baruch-Suchodolsky et al., 2009). Alguns estudos sugerem que a sua associação com a patologia reside no “misfolding” dos péptidos que favorece a sua agregação e consequente acumulação em placas senis. Aparentemente, a formação destes aglomerados é uma propriedade intrínseca subjacente à estrutura primária da proteína, isto é, à sequência dos aminoácidos (Rochet e Lansbury 2000; Glabe 2006). No entanto, foi já estabelecido que as placas senis, formadas maioritariamente por fibrilas deste péptido, apresentam menor toxicidade do que as formas solúveis constituídas por monómeros e oligómeros do polipeptídeo (Haass e Selkoe 2007). Assim, a formação das placas senis, localizadas no espaço extracelular, é considerada como um produto da doença de Alzheimer, enquanto os oligómeros encontrados também no espaço intracelular apresentam maior relevância toxicológica (Reddy e Beal, 2008). De facto, oligómeros do peptídeo β-amilóide tem sido detetados em regiões afetadas do cérebro, quer em humanos (Gouras et
al., 2000) quer em animais transgénicos usados como modelos para o estudo da doença de
Alzheimer (Oddo et al., 2003; Murphy et al., 2007). LaFerla e colaboradores sugerem que a acumulação de β-amilóide evolui no espaço intracelular, interferindo com a função celular normal (LaFerla et al., 2007).
A toxicidade da β-amilóide tem sido avaliada considerando não só os fragmentos Aβ 1-40 e Aβ1-42, mas também o fragmento Aβ25-35. Este pequeno fragmento com 11 aminoácidos
(Aβ25-35) contém os resíduos de aminoácidos essenciais para o processo de agregação dos
péptidos, apresenta uma cinética de agregação mais rápida e também maior toxicidade (Pike et al., 1993, 1995). Estudos in vitro em mitocôndrias isoladas de cérebro de rato mostraram que fragmentos Aβ1-42 e Aβ25-35 inibem os estados 3 e 4 da respiração
mitocondrial, com efeito direto ao nível da atividade da citocromo c oxidase, piruvato desidrogenase e α-cetoglutarato desidrogenase (Casley et al., 2002). Adicionalmente, a β-amilóide induz a abertura do poro de transição de permeabilidade mitocondrial, podendo desempenhar um papel na morte dos neurónios por apoptose (Moreira et al., 2001, 2002). Clementi e colaboradores detetaram que a adição do fragmento Aβ25-35 a mitocôndrias
isoladas de cérebro de rato induz a libertação de citocromo c e o intumescimento osmótico mitocondrial simultaneamente com uma redução no consumo de oxigénio pela mitocôndria. Estes resultados sugerem que as formas monoméricas e solúveis da β-amilóide são neurotóxicas, promovendo dano mitocondrial e desencadeando os sinais apoptóticos (Clementi et al., 2005).
6
Apesar da toxicidade da β-amilóide, exacerbando as condições patológicas associadas à doença de Alzheimer, estar bem documentada ao nível do sistema nervoso central, também existem estudos realizados em tecidos periféricos que suportam a hipótese da doença de Alzheimer ser uma patologia sistémica com múltiplas consequências ao nível do funcionamento e da atividade de outros tecidos não relacionados com o sistema nervoso (Moreira et al., 2006). Por exemplo, células de tecidos periféricos, como as plaquetas e o músculo esquelético também expressam a proteína percursora da β-amilóide (APP), pelo que a sobreprodução de β-amilóide por estas células pode também contribuir para o fenótipo da doença (Van Nostrand, 1990; Akaaboune, 2000). Adicionalmente, a toxicidade dos oligómeros de β-amilóide está associada a um aumento de cálcio citosólico sugerindo perturbações ao nível da organização e atividade das membranas biológicas, incluindo alterações na permeabilidade das membranas intracelulares como as da mitocôndria (Demuro et al., 2005; Lin et al., 2001).
2.1 Interação da β-amilóide com as membranas biológicas
No contexto da vida, as membranas celulares são estruturas dinâmicas com cerca de 5 nm de espessura, necessárias para compartimentalização, que emergem da auto-organização de centenas de diferentes moléculas lipídicas em bicamadas com uma capacidade inerente para incluir e discriminar proteínas específicas. A natureza fluida, a assimetria estrutural e a heterogeneidade lateral das membranas são características que desempenham um papel importante na regulação da atividade celular (Videira, 2002). Têm como função manter gradientes de concentração atuando como barreira seletiva de permeabilidade para iões e moléculas. Para além deste papel, também acolhe e regula um grande número de atividades metabólicas e biossintéticas, incluindo os processos de fosforilação oxidativa responsáveis pela síntese de mais de 90% do ATP utilizado pelas células eucariotas (Gennis, 1989). O papel dos lípidos nas membranas biológicas tem merecido grande atenção por parte da comunidade científica nos últimos anos, uma vez que são estruturas de grande versatilidade e diretamente ou através dos seus metabolitos estão envolvidos em quase todos os processos de transdução de sinal. Já passaram mais de 80 anos desde que foram sugeridos como constituintes base das biomembranas, mas ainda não são conhecidos os mecanismos subjacentes à sua distribuição assimétrica numa mesma membrana nem como é que as células regulam a composição específica de cada uma das suas membranas (Hilgemann, 2003; Shevchenko e Simons, 2010). Por exemplo, as membranas biológicas são estruturalmente assimétricas tanto no conteúdo proteico como lipídico (Bretscher, 1972) e a célula funcional mantém a assimetria lipídica com custos
7
energéticos permanentes (Rothman e Lenard, 1977). As espécies de fosfatidilserina, carregadas negativamente, são mantidas, contra o gradiente de concentração no folheto interno da membrana plasmática por um processo de transporte dependente de ATP, para criar uma matriz interfacial com alta densidade de carga negativa que ajudam a regular os fluxos intracelulares de Ca2+, e consequentemente, controlando as múltiplas ações deste segundo mensageiro (Lomasney et al.,2012).
Bokvist e colaboradores (2004) referem que a interação de Aβ1-40 com os fosfolípidos
aniónicos das biomembranas favorece a sua acumulação e agregação na região superficial da membrana com formação de estruturas tóxicas. Assim, foi sugerido que a fosfatidilserina medeia os efeitos neurotóxicos da β-amilóide (Simakova e Arispe, 2007). Adicionalmente, o carácter lipofílico do peptídeo β-amilóide solúvel sugere que a interação deste com as membranas celulares pode promover alterações na organização dinâmica da membrana citoplasmática (alterações ao nível da fluidez e do potencial membranar) que destabilizam a função sináptica, induzindo o processo neurodegenerativo que lhe está associado (Hooijman e Kiliaan, 2008).
2.2 O papel das membranas biológicas no aparecimento e progressão da doença de Alzheimer
A organização membranar não é estabelecida por ligações rígidas e resulta essencialmente do efeito hidrofóbico conduzido de modo irreversível por um aumento da entropia das moléculas de água. A ausência de ligações covalentes entre a maioria dos componentes da membrana é essencial para o seu normal funcionamento, no entanto, também as tornam sensíveis a alterações do meio que as rodeia, sejam elas químicas ou físicas, tais como temperatura, pH, força iónica e estado de hidratação (Videira, 2002). Há ainda que ter em conta o papel do colesterol na organização e dinâmica das membranas, sabendo-se que a sua concentração varia significativamente de membrana para membrana e que, a sua presença a concentrações suficientemente elevadas influencia processos de transição de fase (Bloom e Mouritsen, 1988) regulando a própria organização lateral da membrana (Simons e Ehehalt, 2002). Nas membranas biológicas é possível encontrar microdomínios dinâmicos e altamente ordenados, ricos em colesterol, esfingolípidos e fosfolípidos saturados. Estes microdomínios são designados por “rafts” e constituem autênticas “jangadas” lipídicas cuja composição confere não só a sua diferenciação do meio envolvente, mas também a associação com proteínas específicas. Sugere-se então que estas plataformas serão essenciais para uma série de funções biológicas tanto em situações saudáveis como patológicas (Simons e Ehehalt, 2002; Michel e Bakovic, 2007).
8
A interação da β-amilóide com as membranas compromete a integridade das mesmas com consequente rutura da homeostase do cálcio (Demuro et al.,2005). O cálcio modula muitos processos neurais, incluindo a plasticidade sináptica e a morte celular por apoptose, pelo que a desregulação dos processos de sinalização intracelular mediada pelo cálcio tem sido implicada no aparecimento e na progressão da doença de Alzheimer. Apoiada nestes dados surgiu outra hipótese para explicar a doença de Alzheimer, conhecida genericamente como “hipótese do cálcio” (Arispe et al., 1993).
Videira e colaboradores [PTDC/SAU-NMC/115865/2009] formularam a hipótese da doença de Alzheimer se iniciar por um défice da atividade das mitocôndrias sinápticas com redução da quantidade de ATP e a consequente perda da assimetria lipídica das membranas plasmáticas, nomeadamente da fosfatidilserina (lípido aniónico) e fosfatidiletanolamina. A perda da assimetria caracterizada pela externalização das fosfatidilserinas tem duas consequências principais: i) diminuição da densidade de carga negativa na superfície citosólica da membrana que promove desregulação dos fluxos de cálcio caraterizado por um aumento do Ca2+ citosólico livre, e à toxicidade que lhe está associada, incluindo a hiperfosforilação da proteína Tau; ii) a deslocalização das fosfatidilserinas e das fosfatidiletanolaminas para o folheto externo da membrana deve modificar a sua organização lateral, induzindo a agregação dos constituintes intramembranares dependente das fosfatidiletanolaminas. Estas alterações na arquitetura da membrana deverão tornar a APP acessível à hidrólise pela BACE com a consequente formação da β-amilóide, que devido ao seu efeito sobre as membranas e solubilidade nos fluídos permite que a doença se propague para tecidos para além dos do sistema nervoso.
3. Modelos para o estudo - objetivos
Considerando a hipótese anteriormente exposta, a doença de Alzheimer pode ser considerada uma doença sistémica que afeta todo o organismo manifestando-se essencialmente por disfunções ao nível da atividade das membranas celulares. Escolhemos:
(i) murganhos triplo-transgénicos (3xTg-AD) com 3, 6 e 12 meses de idade, como animais modelo da doença e correspondentes controlos da estirpe selvagem (WT), a fim de avaliar o efeito do envelhecimento sobre o fígado considerando aspetos relacionados com bioenergética mitocondrial. A escolha deste órgão é baseada no papel do fígado nos processos de desintoxicação, uma vez que a produção excessiva de β-amilóide associada à progressão da doença de Alzheimer poderá promover a disfunção hepática.
9
(ii) lipossomas, como modelos que mimetizam várias características das membranas biológicas, para estudar o efeito direto da β-amilóide sobre as membranas considerando aspetos relacionados com a peroxidação lipídica.
11
1. Reagentes
A β-amilóide (fragmento 1-42) e os fosfolípidos dipalmitoilfosfatidilcolina (DPPC) e dipalmitoilfosfatidilserina (DPPS) foram obtidos da Sigma Chemical Co. (St. Louis, USA). Os restantes reagentes utilizados são de grau analítico.
2. Modelo animal da doença de Alzheimer
No presente trabalho foram usados murganhos triplo-transgénicos, com alterações ao nível dos genes Tau, APP e PS1, como modelo da doença de Alzheimer humana (3xTg-AD) e os animais da estirpe selvagem (WT) como controlos. Os animais foram obtidos do Centro de Neurociências e Biologia Celular da Universidade de Coimbra (CNC/UC) que mantém uma colónia em contínuo e mantidos no biotério da UTAD em condições padrão (temperatura de 24 ± 2 ºC, humidade relativa de 55 ± 5% e fotoperíodo de 12 h), em gaiolas de policarbonato até atingirem a idade pretendida. Durante o estudo os murganhos foram alimentados ad libitum com água e dieta padronizada. Nos estudos foram utilizados animais do género masculino com 3, 6 e 12 meses de idade.
2.1 Isolamento de mitocôndrias de fígado
As mitocôndrias foram isoladas a partir do fígado de animais 3xTg-AD e WT por centrifugação diferencial de acordo com o método descrito por Gazotti e colaboradores (Gazotti et al, 1979), com algumas modificações (Peixoto, 2002). O animal foi morto por deslocamento cervical e decapitado para sangrar rapidamente. O fígado foi retirado e colocado num copo com tampão de homogeneização (Sacarose 250 mM, HEPES 10 mM, EGTA 0,2 mM, BSA 0,1%, pH=7,4) a cerca de 4 ºC. O fígado foi cortado em pequenos pedaços e lavado com meio de homogeneização para remover o excesso de sangue. De seguida, o tecido cortado foi colocado em cerca de 10 volumes de meio de homogeneização num homogeneizador de vidro tipo “Potter Elvehjem” e homogeneizado rodando o pistão de teflon a cerca de 500 rotações por minuto, utilizando um agitador IKA EUROSTAR power basic. O homogeneizado foi transferido para tubos de centrífuga e centrifugado a 1000 x g, durante 10 minutos, numa centrífuga refrigerada Sigma 2 – 16 K, à temperatura de 4 ºC. Este processo permite a sedimentação de núcleos, células intactas e fragmentadas e outros componentes mais densos. A gordura acumulada na superfície dos tubos foi aspirada e o sobrenadante foi cuidadosamente decantado para novos tubos de centrífuga, arrefecidos a 4 ºC, e submetido a nova centrifugação utilizando as mesmas condições (1000 x g, durante 10 minutos) para maximizar a remoção das partículas densas.
12
O sobrenadante obtido foi decantado para novos tubos e de seguida centrifugado a 12000 x g, durante 15 minutos, para sedimentar as mitocôndrias e descartar o sobrenadante. O sedimento (fração mitocondrial) foi ressuspenso em meio de lavagem (Sacarose 250 mM, HEPES 10 mM, pH=7,4) e centrifugado a 12000 x g, durante 15 minutos, para lavagem da fração mitocondrial. O sedimento obtido foi então ressuspenso num volume mínimo de meio de lavagem e transferido para um pequeno tubo cónico (eppendorf), o qual foi mantido a uma temperatura de 4 oC até à quantificação da proteína mitocondrial. A fração mitocondrial restante foi congelada em azoto líquido e guardada à temperatura de -20 ºC para posterior utilização nos estudos da atividade das enzimas da cadeia respiratória.
2.2 Preparação dos homogeneizados de fígado
Para estudar a possível relação entre a doença de Alzheimer e a disfunção hepática relacionada com o stresse oxidativo preparam-se homogeneizados de fígado. Após a determinação da massa de cada fígado numa balança analítica, o órgão foi transferido para um copo e o volume de tampão de homogeneização (160 mM Sacarose, 10 mM Tris-HCl, pH=7,4), previamente arrefecido a 4 ºC, necessário para se atingir a proporção massa de órgão: volume de 1:10 foi adicionado, isto é, 1 mL de tampão por cada 0,1 g de fígado. O fígado foi cortado em pequenos pedaços e homogeneizado num homogeneizador de vidro tipo “Potter Elvehjem” rodando o pistão de teflon a cerca de 500 rotações por minuto, utilizando um agitador IKA EUROSTAR power basic. De seguida, o homogeneizado foi submetido a ultrassons, quatro ciclos de 5 segundos com intervalos de 10 segundos entre ciclos, para aumentar a eficiência de lise das células e novamente homogeneizado no homogeneizador de vidro do tipo “Potter Elvjhem”. O homogeneizado assim obtido foi centrifugado a 16000 x g durante 10 minutos, numa centrífuga refrigerada Sigma 2 – 16 K, à temperatura de 4 ºC, para sedimentar eventuais células intactas, núcleos e vesículas grandes e recolher o sobrenadante com uma pipeta.
2.3 Quantificação proteína – método de biureto
A quantidade de proteína das preparações mitocondriais e dos homogeneizados de fígado foi determinada usando o método de biureto (Gornall et al, 1949). Este método baseia-se na reação do reagente de biureto, o qual é constituído por uma mistura de cobre e hidróxido de sódio na presença de agente complexante que estabiliza o cobre em solução. O cobre, em meio alcalino reage com as ligações peptídicas das proteínas, formando um
13
complexo de geometria quadrangular plana, de cor violeta com um pico de absorção a 540 nm.
As amostras (50 µL) foram solubilizadas pela adição de 50 µL de dodecilsulfato de sódio (SDS) a 10%. A esta mistura adicionou-se água desmineralizada até perfazer um volume de 0,5 mL e, em seguida, 2 mL de reagente de biureto (CuSO4.5H2O 0,15%,
NaKC4H4O6.4H2O 0,6%, NaOH 3%, KI 0,1%). Simultaneamente, foram preparados padrões
de BSA (0, 0,8, 1,6 e 2,4 mg de proteína) nas mesmas condições das amostras. Ao fim de 15 minutos, determinou-se a absorvância das amostras e dos padrões a 540 nm, num espectrofotómetro Varian cary 50 bio.
3. Lipossomas multilamelares
– modelos para estudar a interação da
β-amilóide com as membranas biológicas
No presente trabalho foram usados lipossomas multilamelares como modelos de biomembranas para estudar o efeito da β-amilóide1-42 na oxidação dos lípidos membranares.
Os lipossomas foram preparados com um fosfolípido neutro, dipalmitoilfosfatidilcolina (DPPC) na sua forma pura, ou misturando-o com um fosfolípido aniónico, dipalmitoilfosfatidilserina (DPPS), em proporções específicas, de modo a simular a influência da carga superficial das membranas com uma organização assimétrica (funcional) e com uma organização simétrica (não funcional) na interação com a β-amilóide.
Os lipossomas foram preparados pelo método de De Gier e colaboradores (De Gier, 1968) com algumas alterações (Videira, 2002). Quantidades apropriadas de lípido, de acordo com a constituição pretendida, foram dissolvidas em 5 mL de clorofórmio num balão de fundo redondo. O solvente foi evaporado sob vácuo, num evaporador rotatório (Heidolph 51111), de modo a obter-se uma película fina e homogénea de lípido na parede do balão. Os lipossomas foram formados por hidratação dos lípidos com 10 mL de uma solução tampão (NaCl 100 mM, HEPES 5 mM, pH=7,2) previamente aquecida a uma temperatura superior à temperatura de transição fase. O balão, mergulhado num banho de água termostatizado a uma temperatura superior à da transição de fase do lípido, foi agitado manualmente, com movimentos amplos e sincopados, até que todo o lípido seja removido da parede do balão e a mistura adquira um aspeto leitoso. Após este processo, foi adicionado o volume de solução tampão necessário para obter a concentração de lípido pretendida. A suspensão de lipossomas foi ainda sujeita a sonicação moderada para dispersar microagregados e reduzir a opacidade, a qual foi controlada espectrofotometricamente (Varian cary 50 bio) e mantida em valores de absorvância entre 0,18 e 0,2 a 600 nm.
14
4. Bioenergética Mitocondrial
Os ensaios de bioenergética mitocondrial foram efetuados com as frações mitocondriais isoladas dos fígados de murganhos controlo e 3xTg-AD com 3, 6 e 12 meses de idade. Antes da realização dos ensaios, cada fração mitocondrial foi submetida a 3 ciclos de congelamento/descongelamento para aumentar o acesso dos substratos ao local ativo das respetivas enzimas. Para cada enzima, os ensaios foram efetuados em três preparações mitocondriais independentes obtidas de animais com a mesma idade.
4.1 NADH:ubiquinona oxidoreductase (Complexo I)
A NADH:ubiquinona oxidorreductase (E.c. 1.6.5.3.), também conhecida simplesmente por complexo I, é um complexo proteico transmembranar localizado na membrana interna da mitocôndria que catalisa a transferência de eletrões do NADH para a coenzima Q (CoQ). A atividade deste complexo enzimático foi avaliada por espectrofluorimetria, monitorizando a diminuição na fluorescência a 450 nm, causada pela oxidação de NADH, fixando a excitação a 366 nm, num espectrofluorímetro Varian Cary Eclipse, como previamente descrito (Melo et
al., 2011). Numa cuvete de quartzo com quatro faces polidas foram colocados 2,5 mL de
meio (KH2PO4 25 mM, MgCl2 10 mM e pH=7,4), 0,3 mg de proteína mitocondrial e 20 µL de
KCN 50 mM. A mistura foi incubada, durante de 4 minutos, a 30 ºC sob agitação constante e o ensaio iniciou-se com a adição de 10 µL de NADH 10 mM. Após estabilização da intensidade de fluorescência resultante da adição do NADH, adicionaram-se 13 µL de dodecilubiquinona 25 mM, com a consequente queda na intensidade de fluorescência, graficamente expressa por uma reta cujo declive possibilita a determinação da atividade do complexo I. Dois minutos após a adição da dodecilubiquinona, adicionaram-se 3 µL de rotenona 1,5 mM (um inibidor específico do complexo I) para a garantir que as taxas de decaimento de fluorescência resultam da atividade do complexo I. Assim, a atividade da complexo I, em unidades arbitrárias, foi determinada pela diferença entre os declives das retas antes e após a adição de rotenona, usando o software associado ao espectrofluorímetro. A atividade do complexo I de cada extrato mitocondrial, expressa em unidades arbitrárias/min. mg proteína, foi normalizada pela atividade da citrato sintase determinada nesse mesmo extrato para evitar que as diferenças de atividade resultem de diferentes graus de pureza das frações mitocondriais isoladas.
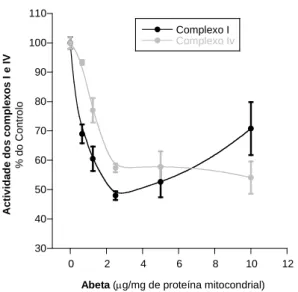
O efeito do aumento da concentração de β-amilóide (0 a 10 µg de β-amilóide/mg proteína mitocondrial) na atividade do complexo I de mitocôndrias isoladas dos animais controlo foi também avaliado. Para o efeito, as mitocôndrias foram incubadas com a β-amilóide à temperatura de 30 ºC, durante 5 minutos, antes de se utilizar o procedimento
15
descrito acima. A β-amilóide foi adicionada a partir de soluções concentradas em DMSO (1 mg/mL).
4.2 Succinato:ubiquinona oxidoreductase (Complexo II)
A succinato:ubiquinona oxidorredutase (E.c. 1.3.5.1.), vulgarmente designada por complexo II, é um complexo enzimático do ciclo dos ácidos tricarboxílicos, associada à membrana interna da mitocôndria, que catalisa a oxidação do sucinato a fumarato com a redução de ubiquinona a ubiquinol. Não obstante, esta enzima não se considera como parte integrante da cadeia respiratória, dado que funciona apenas como subsidiária da cadeia. O método utilizado para determinar a sua atividade baseia-se na deteção espectrofotométrica da redução do DCIP (2,6-diclorofenolindofenol), acompanhada pela perda de coloração azul, associada à atividade da enzima, como previamente descrito (King, 1967).
Assim, os ensaios foram realizados a 30 oC com 0,1 mg de proteína mitocondrial em 2,2 mL de tampão fosfato 25 mM pH=7,4, enriquecido com 10 µL KCN 50 mM, 6 µL rotenona 2,5 mM, 10 µL antimicina A 1,5 mM, 12,5 µL DCIP 10 mM e 10 µL dodecilubiquinona. A reação foi iniciada com 50 µL de succinato 1 M e acompanhada durante 3 minutos, pela variação da absorvância a 600 nm num Varian cary 50 bio. A reação foi inibida pela adição de 120 µL de oxaloacetato 100 mM. Os declives obtidos antes e depois da adição do oxaloacetato foram usados para determinar a atividade do complexo II expressa em nmol/ min.mg proteína. Tal como descrito para o complexo I, a atividade do complexo II de cada extrato mitocondrial foi normalizada pela atividade da citrato sintase determinada nesse mesmo extrato.
4.3 Citocromo C oxidase (Complexo IV)
A citocromo c oxidase (E.c. 1.9.3.1.), também conhecida como complexo IV, é o último complexo da cadeia respiratória. Este complexo catalisa o passo terminal da respiração aeróbia, transferindo os eletrões provenientes do citocromo c para o oxigénio molecular, com a consequente formação de água. A sua atividade foi avaliada polarograficamente pelo consumo de O2, utilizando um eléctrodo de oxigénio tipo Clark (Hansatech) acoplado a um
computador, como previamente descrito (Brautigan et al, 1978; Melo et al., 2011). Os ensaios foram realizados a 30 oC em 1 mL de meio (Tris-HCl 10 mM, KCl 120 mM, pH=7) enriquecido com 3 µL rotenona 1,5 mM, 3 µL antimicina A 1,5 mM e 0,25 mg proteína mitocondrial. A reação iniciou-se pela adição de 10 µL ascorbato 0,5 M, 10 µL de tetrametilfenilenodiamina (TMPD) 25 mM e 15 µL de citocromo C 1 mM e o consumo de oxigénio acompanhado durante dois minutos. A reação terminou com a adição de 3 µL de
16
KCN 50 mM. A diferença entre o consumo de O2 antes e após a adição de KCN foi usada
para determinar a atividade enzimática do complexo IV, expressa e nmol O2 / min.mg
proteína mitocondrial. Para cada extrato, a atividade do complexo IV foi normalizada pela atividade da citrato sintase determinada nesse mesmo extrato.
O efeito do aumento da concentração de β-amilóide (0 a 10 µg de β-amilóide/mg proteína mitocondrial) na atividade do complexo IV de mitocôndrias isoladas dos animais controlo foi também avaliado. Para o efeito, as mitocôndrias foram incubadas com a β-amilóide à temperatura de 30 ºC, durante 5 minutos, antes de se utilizar o procedimento descrito acima.
4.4 FoF1-ATPsintase (Complexo V)
A FoF1-ATPsintase (EC 3.6.3.14), também conhecida como complexo V, é composta
por duas subunidades: i) a subunidade Fo, constituída por 4 tipos de cadeias polipeptídicas,
é um segmento hidrofóbico que atravessa a membrana interna da mitocôndria formando um canal para protões; ii) a subunidade F1, constituída por 5 tipos de cadeias polipeptídicas, é a
componente catalítica que sintetiza ATP a partir de fosfato inorgânico e ADP, utilizando o gradiente eletroquímico transmembranar de protões gerado pela cadeia respiratória. Esta enzima também é capaz de clivar ATP para gerar um gradiente eletroquímico de protões através da membrana e, neste caso, é designada por FoF1-ATPase. A clivagem de ATP
liberta protões. Então, a atividade da FoF1-ATPase foi avaliada por um método
potenciométrico (eléctrodo de pH) como descrito por Madeira e colaboradores (Madeira et
al, 1974), registando continuamente as variações de pH no meio de ensaio. Os ensaios
decorreram a 30 ºC, numa câmara termostatizada, com agitação magnética constante, em 1,5 mL de meio (Sacarose 130 mM, KCl 50 mM, MgCl2 5 mM, HEPES 0,5 mM e pH=7),
suplementado com 0,3 mg de proteína mitocondrial e 4 µL de rotenona (1,5 mM). As reações iniciaram-se, após 2 minutos de incubação, pela adição de 1,5 µL de ATP-Mg (100 mM). Três minutos após o início do registo, foram adicionados 3 µL de oligomicina (um inibidor da FoF1-ATPase), que aboliu por completo a produção de protões. No final de cada
ensaio, a calibração interna do sistema foi efetuada por adição de 5 µL de uma solução HCl padrão com a concentração 0,016 M. A atividade FoF1-ATPase de cada extrato, expressa
em nmol de H+/min. mg proteína, foi normalizada pela atividade da citrato sintase desse extrato.
17
4.5 Citrato sintase
A citrato sintase (E.c. 4.1.3.7) é uma das enzimas que participa no Ciclo de Krebs, sendo responsável pela conversão de oxaloacetato e acetil-CoA em citrato. Encontra-se localizada na matriz mitocondrial, pelo que a sua atividade é normalmente utilizada como indicador quantitativo do conteúdo de mitocôndrias intactas numa amostra biológica. A atividade da citrato sintase foi avaliada por espectrofotometria, registando as variações de absorvância a 412 nm num Varian cary 50 bio, como descrito por Sere (1969). O ensaio foi realizado a 30 oC numa cuvete contendo em 2,2 mL de meio (Tris-HCl 200 mM, pH=8) suplementado com 0,2 mg de proteína mitocondrial, 13 µL triton 10%, 12 µL oxaloacetato 100 mM e 12 µL de DTNB 1,01 mM. A reação foi iniciada pela adição de 6 µL de Acetil-CoA 10 mM. A atividade foi expressa em nmol/min. mg proteína.
5. Stresse Oxidativo
Os ensaios de stresse oxidativo foram realizados em homogeneizados de fígado obtidos de animais 3xTg-AD e WT com 3, 6 e 12 meses de idade. No entanto, a peroxidação lipídica foi também avaliada nas frações mitocondriais isoladas dos fígados dos animais em estudo, usando o método de TBARS. Adicionalmente, o efeito da β-amilóide na oxidação lipídica foi avaliada, usando o cis-parinárico, em lipossomas multilamelares. Todos os ensaios foram realizados em triplicado.
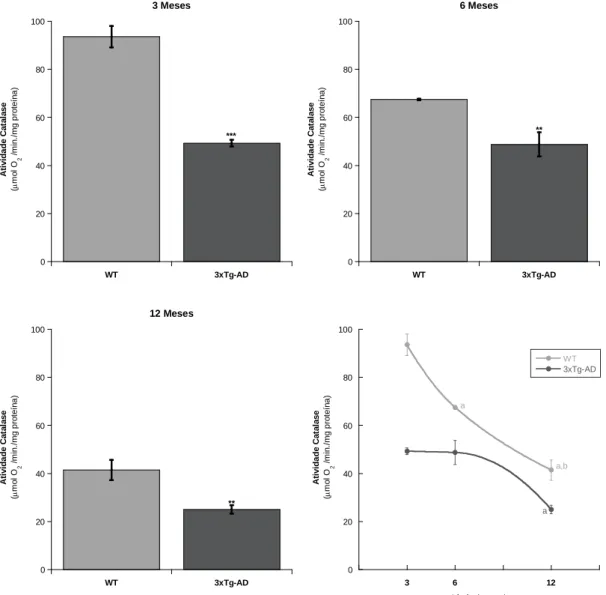
5.1 Catalase
A atividade da catalase foi avaliada polarograficamente, com um eléctrodo de oxigénio do tipo Clark (Hansatech, sistema Oxygraph). Este método é baseado na determinação da velocidade inicial da produção de oxigénio resultante da decomposição do peróxido de hidrogénio (H2O2) pela catalase (Del Río et al, 1977).
A reação decorreu numa câmara de reação aberta termostatizada sob agitação magnética em de 1 mL de tampão fosfato 50 mM, pH=7 suplementado com 0,75 µg de proteína. Após 5 minutos de incubação a 25 oC, a reação iniciou-se pela adição de 17 µL de H2O2 10 mM. A cinética da reação foi acompanhada durante cerca de 1,5 minutos e a
atividade da catalase foi expressa em µmol O2/ min. mg proteína.
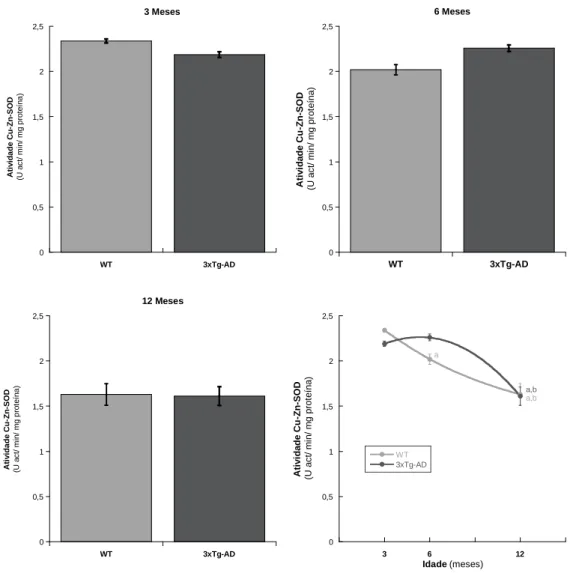
5.2 Superóxido Dismutase
A atividade da superóxido dismutase (Cu-Zn-SOD) foi avaliada espectrofotometricamente, utilizando o método descrito por Payá e colaboradores (Payá et
18
al, 1992), com algumas alterações, recorrendo ao sistema xantina - xantina oxidase na
presença do cromóforo cloreto de azul de nitrotetrazólio (NBT).
Os ensaios foram realizados a 25 °C, em meio de reação (KH2PO4 100 mM, EDTA 1
mM, pH=7.4) enriquecido com 10 µL hipoxantina (10 mM), 10 µL de NBT (10 mM) e 0,596 mg de proteína, perfazendo um volume total de 1,995 mL. Após uma incubação de 2 minutos, a reação foi iniciada pela adição de 5 µL xantina oxidase (5 U/mL) e acompanhada durante 3 min num espectrofotómetro Varian cary 50 bio, a 560 nm. A atividade é expressa em U/min.mg proteína; uma unidade de atividade corresponde a uma redução de 50% da atividade de um ensaio realizado sem amostra (branco).
5.3 Conteúdo total de tióis nas proteínas
O conteúdo total de tióis foi determinado usando o método colorimétrico de Ellman (Riddles et al, 1979). De acordo com este método, os grupos sulfidrilo (SH) das proteínas reagem com o 5,5’-ditiobis-(2-ácido nitrobenzóico) (DTNB) originando um derivado de coloração amarela com um máximo de absorção a 412 nm, o qual pode ser quantificado por espectrofotometria.
Em tubos de rosca contendo 1,85 mL de tampão fosfato 100 mM (pH=8) adicionaram-se 100 µL de DTNB 10 mM e 50 µL de cada um dos sobrenadantes dos homogeneizados de fígado. As misturas foram homogeneizadas em agitação em vórtice e incubadas durante 15 minutos, no escuro, à temperatura ambiente. Em simultâneo, preparou-se uma série de padrões de cisteína (0 a 100 µM) utilizando as mesmas condições das amostras. Após a incubação, as absorvâncias das amostras e dos padrões foram avaliadas, a 412 nm, num espectrofotómetro Varian cary 50 bio. O conteúdo total de tióis de cada amostra foi determinado a partir da curva padrão de cisteína e expresso em equivalentes de cisteína µM / mg proteína.
5.4 Avaliação da peroxidação lipídica 5.4.1 TBARS
A peroxidação lipídica foi estimada pelos TBARS utilizando o método inicialmente descrito por Buege em 1978. É um método baseado na determinação da concentração de malonaldeido, produzido na peroxidação dos sistemas biológicos, pela reação com o ácido tiobarbiturico, a qual, promove a formação de um complexo de cor rosa salmão que absorve luz com comprimento de onda de 535 nm (Buege et al, 1978).
19
Assim, um volume total de 500 µL contendo 1 mg de proteína mitocondrial (ou sobrenadante dos homogeneizados de fígado) em tampão Tris-HCl 10 mM, pH=7,2, foi incubado durante 5 minutos a 30 oC, com agitação no vórtice. De seguida, adicionou- se a cada tubo, contendo amostras ou brancos, 1 mL de reagente de TBARS [ácido tiobarbitúrico (TBA) 0,38% (p/v), ácido tricloroacético (TCA) 37,5% (p/v) e (BHT) 0,015% (p/v)]. Posteriormente, tapados com esferas de vidro, os tubos foram incubados num banho de água a 100 ºC, durante quinze minutos, para promover a reação com o TBA. De seguida, os tubos foram arrefecidos em gelo, sendo o seu conteúdo transferido para eppendorfs de 1,9 mL e centrifugados a 3000 rotações por minuto, 10 minutos, numa centrífuga da Sigma 2-16 K. O sobrenadante foi recolhido e foi avaliada a sua absorvância a 530 nm num espectrofotómetro Varian cary 50 bio.
5.4.2 Cis-parinárico
O ácido 9,11,13,15-octadecatetraenóico (cis-parinárico) devido às suas quatro ligações duplas conjugadas é uma molécula fluorescente quando inserido em ambientes apolares como os das membranas biológicas. Tal como os ácidos gordos insaturados dos fosfolípidos membranares, o cis-parinárico também é sensível à oxidação, sendo a sua degradação considerada um sinal das etapas iniciais da peroxidação lipídica do sistema membranar ao qual foi incorporado. Portanto, a diminuição da intensidade fluorescente da sonda associada à degradação oxidativa das suas ligações duplas pode ser acompanhada por espectrofluorimetria, como previamente descrito (Dinis et al, 1994).
No presente trabalho seguiram-se duas estratégias para avaliar os efeitos da β-amilóide na indução e/ou proteção da oxidação dos lípidos membranares, utilizando lipossomas com composições definidas como modelos de membranas biológicas, nomeadamente:
i) Preparou-se uma série de tubos contendo 2,5 mL de suspensões de lipossomas com a composição indicada nas figuras. A cada tubo adicionou-se, sob agitação em vórtice, 2,5 µL da solução etanólica concentrada de sonda (1,5 mM). A concentração da sonda e a razão sonda/lípido foram respetivamente 1,5 µM e 1/300. À serie de tubos contendo os lipossomas e a sonda foram adicionadas quantidades crescentes de β-amilóide para obter uma série de concentrações entre 0 a 6,64 µM. Os tubos foram rolhados e incubados no escuro, à temperatura de 25 ºC, durante cerca de 14 horas (overnight) antes de se avaliar a intensidade de fluorescência num espectrofluorímetro
Varian Cary Eclipse. A excitação da sonda foi fixa a 324 nm e a emissão foi detectada a
20
ii) 2,5 µL da solução etanólica concentrada de sonda (1,5 mM) foram adicionados, sob agitação a uma célula de quartzo contendo 2,5 mL de lipossomas. A mistura foi incubada durante dois minutos, à temperatura de 25 ºC, para que a sonda incorporasse nas membranas dos lipossomas. A degradação oxidativa foi induzida pela adição de 25 µL de uma solução de ADP/Fe2+ (25 mM/10 mM). A formação de radicais hidroxilo pelo indutor promoveu a oxidação da sonda e, consequentemente, o decréscimo da intensidade de fluorescência, acompanhado num espectrofluorímetro Varian Cary Eclipse. A excitação da sonda foi fixa a 324 nm e a emissão foi detectada a 413 nm. Ensaios para avaliar o efeito do aumento da concentração de β-amilóide (0 - 6,64 µM) foram efetuados utilizando o mesmo procedimento, tendo-se incubado os lipossomas contendo sonda com a β-amilóide durante dois minutos antes da indução do processo oxidativo com o par ADP/Fe2+. Para comparação dos resultados, normalizou-se a intensidade de fluorescência, considerando para todos os ensaios unitária a fluorescência no momento da indução do processo oxidativo.
22
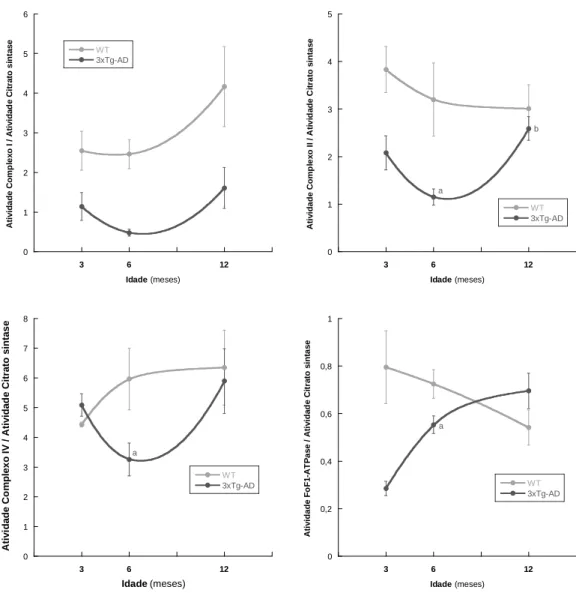
1. Efeitos ao nível da bioenergética mitocondrial
A doença de Alzheimer é uma disfunção neuronal com múltiplas implicações fisiológicas que se estendem para além do sistema nervoso central. Por exemplo, a produção excessiva de β-amilóide pelo sistema nervoso central (Growdon, 1999) e por células de tecidos periféricos como as plaquetas (Di Luca et al., 1996) pode promover a sua libertação para a corrente sanguínea e, consequentemente, a acumulação e metabolização no fígado. A função fisiológica do fígado, incluindo a metabolização de compostos tóxicos, está altamente dependente da atividade das suas mitocôndrias. Assim, o efeito da idade na atividade das enzimas relacionadas com a bioenergética mitocondrial foi avaliada em murganhos 3xTg-AD (animais que desenvolvem de um modo progressivo o processo neurodegenerativo tal como os pacientes humanos atingidos com doença de Alzheimer) e nos correspondentes controlos (WT) para estudar a possível influência da progressão da doença na atividade fisiológica do fígado e selecionar, eventuais, marcadores biológicos capazes de refletir os diferentes estágios da doença.
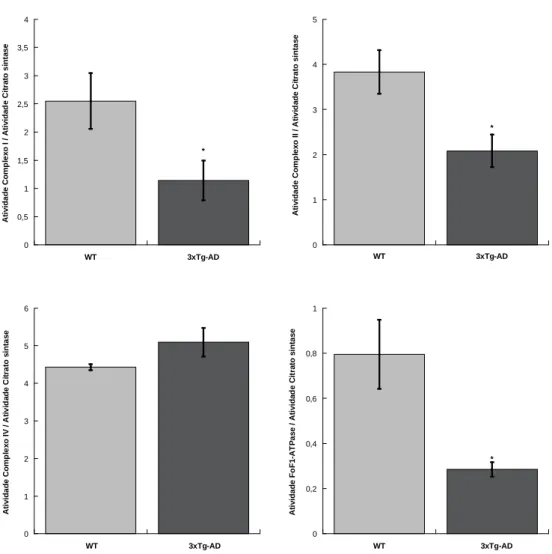
A figura 1 mostra a atividade de enzimas chave da cadeia respiratória (complexos I, II e IV) e da FoF1-ATPase (complexo V), avaliada em mitocôndrias isoladas do fígado de
animais controlo (WT) (barras a cinzento) e triplo-transgénicos (3xTg-AD) (barras a preto) com três meses de idade. As atividades destes complexos enzimáticos, localizados na membrana interna da mitocôndria, foram normalizadas pela atividade da citrato sintase (enzima da matriz mitocondrial) determinada no correspondente extrato mitocondrial, para evitar que possíveis diferenças detetadas entre os dois grupos possam resultar de diferentes graus de pureza das preparações mitocondriais. Os resultados da figura mostram que nas mitocôndrias do fígado dos murganhos 3xTg-AD os complexos I, II e da FoF1-ATPase
apresentam um atividade significativamente inferior do que nas mitocôndrias dos animais controlo. Assim, a atividade do complexo I decresce cerca de 55%, a do complexo II cerca de 45% e a atividade da FoF1-ATPase decresce cerca de 65%. Em relação ao complexo IV,
não foram detetadas diferenças significativas entre os dois grupos (Fig. 1). Estes resultados sugerem que, aos três meses de idade, a atividade bioenergética do fígado dos murganhos 3xTg-AD deverá estar significativamente comprometida com repercussões na atividade global dos hepatócitos. De facto, a mitocôndria suporta, do ponto de vista energético, a regulação de múltiplas funções celulares, nomeadamente, a formação de intermediários metabólicos, o fluxo de iões, lípidos e proteínas e o processo da contração muscular. Adicionalmente desempenha um papel importante tanto nos processos relacionados com a proliferação celular como nos relacionados com morte celular programada (apoptose) pois a mitocôndria é responsável pela síntese de mais de 90% do ATP utilizado pela célula
23
(Wallace, 1999; Videira, 2002). Portanto, a disfunção do fígado deverá ocorrer em simultâneo com os primeiros estágios da doença de Alzheimer caracterizados, neste modelo animal, por disfunção neuronal.
Figura 1 – Atividade individual dos complexos enzimáticos da cadeia transportadora de eletrões I, II, IV e da FoF1-ATPase avaliada em mitocôndrias isoladas de fígado de murganhos WT e 3xTg-AD
com idade de 3 meses, normalizada pela atividade da Citrato sintase. As barras verticais representam o erro padrão e * representa diferenças estatisticamente significativas relativamente ao grupo WT, (p <0,05).
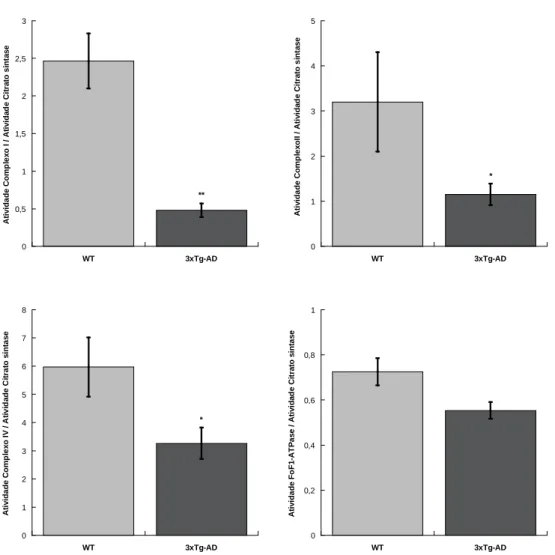
A atividade dos complexos I, II e IV e da FoF1-ATPase nas mitocôndrias isoladas do
fígado de murganhos WT e 3xTg-AD com 6 meses de idade estão representadas na figura 2. Como expectável nas patologias progressivas, a disfunção do fígado é acentuada com o avanço da idade, como mostram as diferenças entre os animais 3xTg-AD e os animais WT , detetadas nas atividades dos complexos I e II. Assim, nas mitocôndrias do fígado dos animais 3xTg-AD a atividade do complexo I decresce cerca de 80% e a do complexo II
0 0,5 1 1,5 2 2,5 3 3,5 4 WT 3xTg-AD A ti v id a d e C o mp le x o I / A ti v id a d e C it ra to s in ta s e * 0 1 2 3 4 5 6 WT 3xTg-AD A ti v id a d e C o mp le x o I V / A ti v id a d e C it ra to s in ta s e 0 1 2 3 4 5 WT 3xTg-AD A ti v id a d e C o mp le x o I I / A ti v id a d e C it ra to s in ta s e * 0 0,2 0,4 0,6 0,8 1 WT 3xTg-AD A ti v id a d e F o F 1 -A T P a s e / A ti v id a d e C it ra to s in ta s e *
24
decresce cerca de 64%, em relação aos controlos. Ao contrário do detetado nos animais com 3 meses idade, a atividade do complexo IV nos animais 3xTg-AD é significativamente inferior (cerca de 45%) à dos controlos. Adicionalmente, aos seis meses de idade, as diferenças na atividade da FoF1-ATPase entre os dois grupos deixam de ser significativas
(Fig.2).
Figura 2 – Atividade individual dos complexos enzimáticos da cadeia transportadora de eletrões I, II, IV e da FoF1-ATPase avaliada em mitocôndrias isoladas de fígado de murganhos WT e 3xTg-AD
com idade de 6 meses, normalizada pela atividade da Citrato sintase. As barras verticais representam o erro padrão e * representa diferenças estatisticamente significativas relativamente ao grupo WT, (p <0,05).
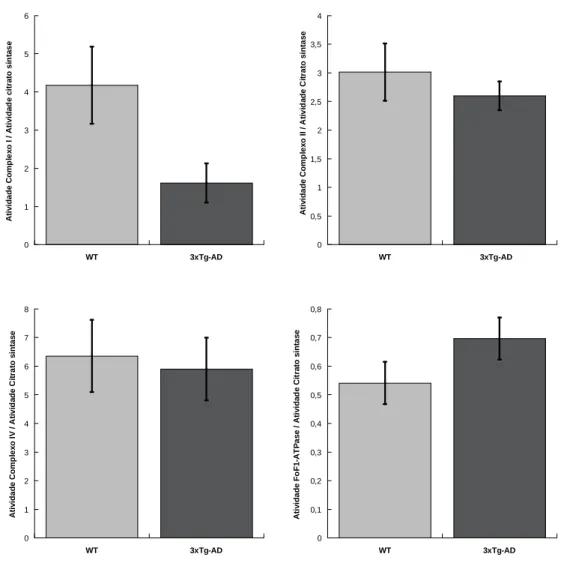
A figura 3 mostra a atividade dos complexos I, II e IV e da FoF1-ATPase nas
mitocôndrias isoladas do fígado de murganhos WT e 3xTg-AD com 12 meses de idade. Os resultados da figura mostram que nos animais 3xTg-AD com 12 meses de idade apenas a atividade do complexo I é significativamente inferior à avaliada nos correspondentes
0 0,5 1 1,5 2 2,5 3 WT 3xTg-AD A ti v id a d e C o mp le x o I / A ti v id a d e C it ra to s in ta s e ** 0 1 2 3 4 5 6 7 8 WT 3xTg-AD A ti v id a d e C o mp le x o I V / A ti v id a d e C it ra to s in ta s e * 0 1 2 3 4 5 WT 3xTg-AD A ti v id a d e C o mp le x o II / A ti v id a d e C it ra to s in ta s e * 0 0,2 0,4 0,6 0,8 1 WT 3xTg-AD A ti v id a d e F o F 1 -A T P a s e / A ti v id a d e C it ra to s in ta s e