palavras-chave Qualidade da água; métodos espectrofotométricos; validação de métodos analíticos.
resumo A legislação Portuguesa indica a espectrofotometria de absorção atómica como metodologia analítica de referência para análise e quantificação de diversos metais em águas de consumo humano (DL 236/98). Este é o caso de metais como cobre, ferro, sódio, cálcio e magnésio, que serão abordados no âmbito deste trabalho.
A escolha destes metais tem por base o facto de a sua quantificação estar sujeita a interferências que dificultam muitas vezes uma quantificação exacta e precisa (ex: ferro), de as suas concentrações na água serem geralmente baixas e, por isso, de difícil quantificação (ex: cobre) ou serem importantes elementos maioritários (ex: sódio, cálcio e magnésio).
Este trabalho pretende comparar uma metodologia relativamente recente - plasma acoplado indutivamente à espectrofotometria de emissão atómica (ICP-AES) - com o método de referência citado na legislação (espectrofotometria de absorção atómica – AAS-F), de modo a averiguar se o ICP-AES traz vantagens. Para atingir este objectivo procedeu-se à validação de ambos os métodos através de avaliação directa e indirecta. Os resultados da validação de ambos os métodos indicam que para os elementos em estudo e nas condições em que o trabalho foi realizado, não há um método que apresente sistematicamente melhor capacidade analítica do que outro e que para muitos dos parâmetros avaliados os dois métodos são semelhantes.
Os dois métodos foram aplicados na análise de 44 amostras de água de consumo humano e verificou-se que os métodos não são significativamente diferentes, conduzindo a resultados semelhantes em amostras reais e que para os metais em estudo, ambos os métodos cumprem com os requisitos que constam na legislação. Assim sendo, o método de plasma acoplado indutivamente à espectrofotometria de emissão atómica (ICP-AES) é um método que pode ser considerado como método analítico alternativo à espectrofotometria de absorção atómica com chama. Para além disto, o ICP-AES permite uma análise multi-elementar, tornando-a bastante mais rápida do que o AAS-F e com maior sensibilidade. Por outro lado, o ICP-AES tem ainda a vantagem de permitir a determinação de elementos vestigiais conjuntamente com a quantificação de elementos maioritários, usando o mesmo equipamento, o que não acontece com o AAS-F.
keywords Water quality; spectrofotometric methods; methods validation.
abstract The Portuguese legislation suggests the atomic absorption
spectrophotometry as the main analytical methodology to be used in the analysis of several metals in water intendend for human consumption (DL 236/98). This is the case of the metals: copper, iron, sodium, calcium and magnesium, which will all be addressed to in the course of this work.
The choice of these specific metals was based on the fact that its quantification is oftenly subject to interferences that hinder an accurate and precise quantification (eg: iron) and on the fact that their concentration in water is, usually, low (and therefore difficult to quantify – eg: copper) or they are considered important major elements – eg: sodium, calcium and magnesium).
This work aims compare a relatively new methodology - the inductively coupled plasma atomic emission spectrophotometry (ICP-AES) - with the one of reference in legislation (atomic absorption spectrophotometry – AAS-F), to try to understand if the use of the ICP-AES has any advantages over the second one. In order to achieve this, both methods were validate through direct and indirect assessment. The results of the validation of both methods indicate that for the studied elements and the conditions of the work, there is no method that sistemetically presents better analytical capacity and that for many parameters the two methods are similar.
The methods were applied in the analysis of 44 drinking water samples which showed they are not significantly different, leading to similar real samples’ results and that for the studied metals both methods fulfill the legislation’s requirements. Thus, the inductively coupled plasma atomic emission spectrophotometry (ICP-AES) may be considered as an alternative analytical method to atomic absorption spectrophotometry – AAS-F. Moreover, ICP-AES allows a multi-elementar analysis, which becomes faster than AAS-F analysis, and whith a better sensitivity. On the other hand, ICP-AES has the advantage of vestigial elements’ determination jointly with majority elements’ quantification, using the same equipment, which does not happen with AAS-F.
- 1 -
Objectivo e estrutura da dissertação
O objectivo deste trabalho é comparar um método relativamente recente - plasma acoplado indutivamente à espectrofotometria de emissão atómica (ICP-AES) - com o método recomendado pela legislação Portuguesa - espectrofotometria de absorção atómica (AAS) - para a análise de metais em águas de consumo humano, de modo a averiguar se o ICP-AES traz vantagens em relação ao método de referência indicado na legislação. Para atingir este objectivo procedeu-se inicialmente à validação de cada um dos métodos através de avaliação directa e indirecta e posteriormente à comparação dos métodos através da análise de ferro, cobre, sódio, magnésio e cálcio em amostras de água de consumo humano.
Este trabalho está dividido em quatro capítulos. No capítulo 1 são apresentadas algumas considerações sobre qualidade da água para consumo humano - critérios de qualidade, legislação e métodos de análise. Os materiais e metodologias utilizados estão descritos no capítulo 2, apresentando-se os resultados e respectiva discussão no capítulo 3. A bibliografia consultada está indicada no capítulo 4. Os Anexos apresentam os resultados da validação para os metais escolhidos.
- 3 -
1. Água para consumo humano – critérios de qualidade, legislação e
métodos de análise
1.1. Importância da análise da qualidade da água
A água é um recurso natural renovável essencial à vida, constituindo um factor indispensável à sobrevivência da biosfera e, portanto, do homem (Peixoto, 1979; Bates, 2000; Virkutyte & Sillanpää, 2006). Constitui um recurso limitado no planeta Terra, não sendo uma realidade estática, uma vez que se integra numa cadeia de subsistemas abertos (entre eles há permuta de massa e energia), que a mantêm numa circulação permanente entre os diversos compartimentos do planeta (Peixoto, 1979; Mendes & Oliveira, 2004), como é demonstrado na Figura 1.
Figura 1: Ciclo hidrológico com indicação dos principais fluxos envolvidos na manutenção do seu equilíbrio (adaptado de Peixoto, 1979).
O corpo humano é constituído, aproximadamente, por 65 a 75 por cento de água, conduzindo uma perda de 15 por cento irremediavelmente à morte (Peixoto, 1979; Cruz & Martins, 1994). Assim, o homem necessita de consumir diariamente 2 a 3 litros de água (Mendes & Oliveira, 2004) e, por isso, a poluição dos recursos hídricos afecta a vida do homem a nível mundial (Suess, 1982).
Enquanto composto dotado de características físico-químicas próprias, a água não se encontra no estado incolor, inodoro e insípido na natureza. A sua presença é indissociável de substâncias estranhas, existentes em suspensão e/ou solução, que afectam as suas
- 4 -
características e as suas potenciais aplicações. Daí advém a necessidade de indicação da sua qualidade, a qual é definida tendo em consideração a sua aplicabilidade para determinados fins (Mendes & Oliveira, 2004). Nenhuma água poderá, em princípio, ser usada para todos os fins sendo por isso a sua utilização, função dos objectivos visados pelos seus utilizadores. Exemplo disso é a água destilada – quase pura – que não é adequada para beber. Também para a indústria da pasta de papel branqueada, uma água adequada para consumo humano originaria manchas de ferrugem, devido ao ferro que contém, podendo tornar essa pasta inutilizável (Mendes & Oliveira, 2004).
Considera-se água destinada ao consumo humano toda a água, que no seu estado original ou depois de tratada, é destinada a ser bebida, a cozinhar, à preparação de alimentos, à higiene pessoal ou a outros fins domésticos, independentemente da sua origem e toda a água utilizada na indústria alimentar para fabrico, transformação, conservação ou comercialização de produtos ou substâncias destinados ao consumo humano, assim como a utilizada na limpeza de superfícies, objectos e materiais que podem estar em contacto com os alimentos (DL 306/2007).
Uma das mais importantes razões para estudar a qualidade da água é o aumento da população mundial, o que resultou num enorme consumo das reservas hídricas mundiais (Waite, 1984; Virkutyte & Sillanpää, 2006). A escassez de água pode levar ao declínio da prosperidade e à queda de civilizações. No passado, civilizações foram extintas devido à falta de água potável (Peixoto, 1979; Waite, 1984). Hoje em dia, começa já a sentir-se o perigo do desequilíbrio entre a água necessária e a disponível (Peixoto, 1979). Torna-se então evidente que devemos proteger os recursos hídricos, tratando-os e prevenindo a sua poluição (Cruz & Martins, 1994), uma vez que esta reduz substancialmente a disponibilidade da água: uma pequena porção de água poluída pode tornar inutilizáveis quantidades consideráveis de água potável (Peixoto, 1979; Suess, 1982; Waite, 1984; Mendes & Oliveira, 2004; Virkutyte & Sillanpää, 2006).
A qualidade da água é avaliada numa primeira impressão pelas suas qualidades organolépticas – características que actuam sobre os sentidos do consumidor – e uma água teoricamente boa para que possa ser consumida deverá ser incolor, inodora e insípida (Belitz et al., 2004), mas apresentar estas características pode não ser suficiente para que seja considerada adequada para consumo humano, uma vez que, entre outros, pode estar contaminada com microrganismos patogénicos. Assim, para poder ser consumida sem
- 5 -
restrições, a água terá de respeitar diversas outras exigências que garantam a sua potabilidade (Mendes & Oliveira, 2004). Deverá então ser objecto de cuidados permanentes e adequados, que são traduzidos por critérios de qualidade da água – limites para a alteração não natural da qualidade da água que são considerados compatíveis com o uso dessa e não causam efeitos adversos a humanos pelo seu uso (Howell, 1997; Virkutyte & Sillanpää, 2006). Para isso, é necessário recorrer simultaneamente a técnicas analíticas, físico-químicas, radiológicas e microbiológicas, cujo número e complexidade analítica têm crescido ao longo das últimas décadas (Mendes & Oliveira, 2004). A água deve ser analisada com recurso a métodos experimentais harmonizados para que os resultados obtidos sejam fidedignos, reprodutíveis, compatíveis e, portanto, comparáveis entre laboratórios, por exemplo através de ensaios interlaboratoriais (Suess, 1982; Dijk-Looijaard & Genderen, 2000).
1.2. Critérios de qualidade da água
A água para consumo humano tem de apresentar características de qualidade que garantam a sua potabilidade, e deverá, para isso, ser objecto de cuidados permanentes e adequados que são traduzidos por critérios para a qualidade da água, os quais passaram a ser quantitativos, em vez de qualitativos, apenas nos últimos três séculos (Zuane, 1990). Eles incluem a especificação dos parâmetros analíticos que deverão ser objecto de controlo permanente e eficiente.
Segundo o DL 306/2007 de 27 de Agosto, que entrou em vigor a 1 de Janeiro de 2008, os referidos parâmetros são classificados em três grupos: microbiológicos, químicos e indicadores. A escolha dos parâmetros a quantificar no âmbito deste trabalho foi feita com base no facto de a sua quantificação estar sujeita a interferências que dificultam muitas vezes uma quantificação exacta e precisa (ex: ferro), de as suas concentrações na água serem geralmente baixas e, por isso, de difícil quantificação (ex: cobre) ou por serem elementos maioritários importantes para a saúde humana (ex: cálcio, sódio e magnésio).
De seguida apresentam-se algumas das características dos elementos mencionados e que foram quantificados em águas de consumo humano.
- 6 - 1.2.1 Cobre
O cobre é um metal maleável, dúctil e excelente condutor de electricidade e calor (Zuane, 1990). Está presente em rochas (basalto, xistos), oceanos e plantas (Mendes & Oliveira, 2004), podendo ser encontrado na crusta terrestre na forma de calcopirite, bornite, calcosite (Zuane, 1990; Chang; 1994; Mendes & Oliveira, 2004).
A maioria dos minerais de cobre são relativamente insolúveis e as concentrações normalmente presentes em águas naturais são baixas, devendo-se à erosão das rochas (Suess, 1982; American Water Works Association, 1990; EPA, 2007). Se o cobre estiver presente em concentrações superiores a 1 mg L-1 pode provocar sabor metálico e adstringente, formação de precipitados de hidróxido de cobre, que turvam a água, e escurecimento de alguns alimentos no decurso da cozedura (Suess, 1982; Mendes & Oliveira, 2004). Os valores máximos fixados para o cobre na água de consumo devem-se a razões de carácter organoléptico, não estando directamente relacionados com riscos sanitários (Zuane, 1990; Mendes & Oliveira, 2004).
O cobre é um elemento essencial para o homem enquanto activador de sistemas enzimáticos e tem também um importante papel na activação ou repressão da transcrição dos genes; no entanto, pode ter efeitos adversos, como danos nos rins e fígado e distúrbios gastrointestinais como inflamações gastrointestinais crónicas, vómitos e náuseas (Suess, 1982; American Water Works Association, 1990; Chapman, 1997; Leeuwen, 2000; Mendes & Oliveira, 2004; Virkutyte & Sillanpää, 2006; Sadhra et al., 2007; EPA, 2007). A sua deficiência ou toxicidade são raros em humanos, podendo ser a ingestão de largas quantidades de compostos de cobre fatal (Sadhra et al., 2007). A quantidade diária necessária estimada é de 1 a 1.5 mg (Belitz et al., 2004). Apesar da água de consumo poder apresentar concentrações significativas de cobre, geralmente esta contribui marginalmente para a ingestão total de cobre, sendo as necessidades do homem fundamentalmente garantidas pela alimentação (Suess, 1982; American Water Works Association, 1990; Zuane, 1990; Mendes & Oliveira, 2004).
Este elemento é muito utilizado em ligas, cabos eléctricos, canalizações, moedas e também na indústria têxtil, fotográfica e cerâmica. Na agricultura recorre-se aos sais de cobre enquanto insecticida, fungicida e algicida, o que provoca a sua variação sazonal nas águas superficiais (Zuane, 1990; Chang, 1994; Mendes & Oliveira, 2004).
- 7 - 1.2.2 Ferro
Elemento metálico cinzento, dúctil, maleável e vital para animais e plantas, o ferro é muito reactivo e corrói rapidamente (Chang, 1994; Zuane, 1990). É um metal pesado essencial aos organismos vivos, sendo a dose diária recomendada 1.5 a 2.2 mg (Leeuwen, 2000; Belitz et al., 2004; Mendes & Oliveira, 2004). A deficiência nutricional deste metal conduz a anemia (Zuane, 1990; Chang, 1994; Mendes & Oliveira, 2004). Já o seu excesso é relativamente tóxico, tendo efeitos mutagénicos, nefrotóxicos e carcinogénicos (Tahán et al., 1994). É muito abundante na crusta terrestre (Chang, 1994; Mendes & Oliveira, 2004), sendo utilizado pelo homem na metalurgia e em muitas outras indústrias químicas. É um elemento presente em rochas (ultramagmáticas, basalto, argila, xisto), minérios (hematite, siderite, magnetite, limonite, pirite), solo, oceanos e plantas (Zuane, 1990; Chang, 1994; Mendes & Oliveira, 2004).
A solubilidade deste metal nas águas naturais é dependente do pH (Suess, 1982; Chapman, 1997; Virkutyte & Sillanpää, 2006) e na ausência de agentes complexantes, os sais só são solúveis em água em quantidades significativas se o pH for inferior a 5 (Suess, 1982; Zuane, 1990; Mendes & Oliveira, 2004). Assim, as águas naturais contêm, geralmente, teores reduzidos de ferro, podendo a sua presença resultar de lixiviações do solo ou de poluição ambiental (Suess, 1982; Zuane, 1990; Mendes & Oliveira, 2004).
Muitas vezes a presença de ferro deve-se quase exclusivamente à corrosão das tubagens, libertando para a água o elemento na forma férrica. Como consequência a água fica avermelhada, frequente causa de queixas por parte dos consumidores (Sarin et al., 2004; Zacheus & Martikainen, 1997). Numa água para consumo, a presença de ferro pode ser indicadora de um tratamento mal conduzido, já que na potabilização de águas brutas se utilizam sais de ferro como agentes coagulação/floculação (Mendes & Oliveira, 2004). Águas com resíduos decorrentes deste caso podem apresentar uma coloração amarela-alaranjada, podendo ter também sabor e odor indesejáveis (Suess, 1982; Zuane, 1990; Bates, 2000; Virkutyte & Sillanpää, 2006).
1.2.3 Sódio
Elemento macio, muito reactivo, o sódio é um metal alcalino muito abundante na crusta terrestre. Ocorre em minerais como a halite (NaCl), o nitrato (NaNO3) ou em
- 8 -
Oliveira, 2004). É um elemento essencial da matéria viva mas pode ser tóxico quando em concentrações elevadas, estando presente nos fluidos celulares e sendo essencial para o equilíbrio osmótico, para funções enzimáticas e para a estabilidade de estruturas celulares (Chang, 1994; Chapman, 1997; Belitz et al., 2004; Mendes & Oliveira, 2004). A dose mínima diária recomendada de sódio varia de 1.3 a 1.6 g (Belitz et al., 2004; Mendes & Oliveira, 2004). Doses excessivas provocam vómitos, doenças cardíacas, problemas gatrointestinais e complicações nos doentes renais (Mendes & Oliveira, 2004).
Nas águas de consumo humano, este metal é um constituinte maioritário, sendo considerado nocivo para pessoas com doenças do foro cardíaco ou renal - está comprovado que o sódio aumenta a pressão sanguínea (American Water Works Association, 1990; Zuane, 1990; Tahán et al., 1994). A alimentação é a fonte primordial de sódio (American Water Works Association, 1990).
O sódio foi muito usado, sob a forma de NaCl, na conservação do peixe através da salga. Actualmente é também utilizado no fabrico de detergentes, em fertilizantes e em gasolinas sem chumbo, sendo os seus sais usados em diversas indústrias químicas, têxteis, de papel, de vidro e de petróleo (Zuane, 1990; Mendes & Oliveira, 2004).
1.2.4 Cálcio
É um metal alcalino-terroso, muito reactivo, abundante na crusta terrestre. É essencial à matéria viva, sendo o componente maioritário dos ossos e dos dentes (Zuane, 1990; Chang, 1994; Chapman, 1997; Belitz et al., 2004; Mendes & Oliveira, 2004; Li et al., 2008). Sob a forma de catião activa diversos processos metabólicos. Desempenha ainda funções vitais na actividade cardíaca, na coagulação sanguínea, na contracção muscular e na transmissão nervosa (Chang, 1994; Belitz et al., 2004; Mendes & Oliveira, 2004; Li et al., 2008; Zhao et al., 2008). Não acarreta preocupação de saúde pública, uma vez que não é tóxico e a dose diária recomendada varia com a faixa etária, sendo 1 g nos adultos (Zuane, 1990; Belitz et al., 2004; Mendes & Oliveira, 2004).
Na natureza ocorre na rocha calcária, na calcite, no giz e no mármore sob a forma de CaCO3; na dolomite sob a forma de CaCO3.MgCO3; no gesso sob a forma de CaSO4.2H2O
e na fluorite sob a forma de CaF2 (Zuane, 1990; Chang, 1994; Mendes & Oliveira, 2004).
O cálcio dissolve-se a partir das rochas, sendo detectado em todas as águas e contribui para a dureza total destas (Suess, 1982; Chapman, 1997; Mendes & Oliveira, 2004).
- 9 -
O cálcio está geralmente presente nas águas de consumo humano, sob a forma de bicarbonatos. São os produtos lácteos que representam a maior fonte deste elemento na alimentação humana, sendo reduzido o contributo da água (Mendes & Oliveira, 2004).
O cálcio metálico tem utilizações limitadas mas a “cal viva” (CaO) é usada em metalurgia e a “cal apagada” (Ca(OH)2) é usada no tratamento de águas, sendo os óxidos
de cálcio utilizados na construção civil e nas indústrias de papel, açúcar, petróleo e curtumes (Chang, 1994; Mendes & Oliveira, 2004).
1.2.5 Magnésio
É um metal alcalino-terroso, maleável, dúctil, bom condutor térmico, pouco resistente à corrosão e abundante na crusta terrestre. É muito reactivo e ocorre em minérios como a peridase (MgO), a brucite (Mg(OH)2), a dolomite (CaCO3.MgCO3) e a epsomite
(MgSO4.7H2O). É um componente comum das águas naturais contribuindo para a dureza
total das águas (Zuane, 1990; Suess, 1982; Chang, 1994; Chapman, 1997; Mendes & Oliveira, 2004; Zhang et al., 2008).
É essencial para a vida de animais e plantas, desempenhando várias funções biológicas importantes (Zuane, 1990; Chang, 1994; Chapman, 1997; Mendes & Oliveira, 2004; Wan et al., 2008). Está presente nos fluidos celulares, sendo essencial para o bom funcionamento de várias enzimas (Chang, 1994; Belitz et al., 2004; Mendes & Oliveira, 2004). A dose diária necessária varia de 300 a 400 mg (Belitz et al., 2004).
Nas águas de consumo humano o magnésio pode provocar irritações gastrointestinais e efeitos laxantes, quando em elevadas concentrações. Pode também conferir à água um sabor amargo, não representando um problema sério, excepto em pessoas que sejam insuficientes renais (Mendes & Oliveira, 2004).
As utilizações mais importantes do magnésio são ligas para protecção catódica, síntese orgânica, baterias, flashs fotográficos, pirotecnia e bombas incendiárias (Zuane, 1990; Chang, 1994).
1.3. Legislação sobre a qualidade da água para consumo humano
Já no século XVI, Filipe Aurélio Teofrasto Bombastus von Hohenheim, conhecido como Paracelso pensava que “tudo é remédio, tudo é veneno”, ou seja, que “é a dose que
- 10 -
faz o veneno”. Este pensamento justifica toda a problemática da definição dos quantitativos acima dos quais determinadas substâncias são tóxicas ou capazes de induzir consequências negativas. Todos os dias surgem novos compostos nos contextos científico e industrial, o que aumenta a necessidade de desenvolver cada vez mais e melhores técnicas para efectuar o seu controlo (Suess, 1982). Legislar sobre este domínio é, portanto, difícil e para fixar determinados limites em relação a um composto específico, um legislador tem de se basear em estudos relativos à sua toxicidade, bem como às interacções que esse composto apresenta com outras substâncias de ocorrência previsível, para que o seu impacto ambiental possa ser previsto (Waite, 1984; Mendes & Oliveira, 2004).
Em Portugal, a legislação sobre a qualidade da água tem vindo a ser actualizada, já tendo sofrido diversas alterações (Mendes & Oliveira, 2004). Em 1998, foi publicado o DL 236/98, uma primeiratentativa de regularização da situação nacional, no que diz respeito à legislação da água para consumo humano (Mendes & Oliveira, 2004). Este DL transpôs para o direito interno, a Directiva Comunitária 80/778/CEE, relativa à qualidade da água e à protecção das águas superficiais e subterrâneas contra a poluição provocada por certas substâncias perigosas (DL 236/98; Guerreiro & Pereira, 2002). Este diploma refere a espectrofotometria de absorção atómica como método analítico de referência para a análise dos metais aqui abordados, em águas para consumo humano. No entanto, como previsto no DL 306/2007, os laboratórios de ensaios podem recorrer a métodos analíticos alternativos desde que comprovem, junto da autoridade competente, que os resultados obtidos são, no mínimo, tão fiáveis como os que seriam obtidos pelos métodos especificados. O DL 243/2001 transpôs para o direito interno a Directiva 98/83/CE, de 3 de Novembro, e tem por objectivo proteger a saúde humana dos efeitos nocivos resultantes de qualquer contaminação da água destinada ao consumo humano, assegurando a sua salubridade e limpeza.
O DL 306/2007 (Lei Quadro da Água) entrou em vigor no dia 1 de Janeiro de 2008 e procedeu à revisão do DL 243/2001, registando algumas alterações como a introdução de novos parâmetros no controlo da qualidade da água e tendo por objectivo proteger a saúde humana dos efeitos nocivos resultantes da eventual contaminação dessa água e assegurar a disponibilização tendencialmente universal de água salubre, limpa e desejavelmente equilibrada na sua composição. Nele foram também estabelecidos novos limites para os
- 11 -
parâmetros de controlo. No caso dos metais estudados neste trabalho estes valores estão resumidos na Tabela 1.
Tabela 1: Valores paramétricos dos parâmetros em estudo para água de consumo humano (adaptado do DL 306/2007).
Parâmetro Valor paramétrico Unidade
Cálcio 100 mg L-1
Ferro 200 µg L-1
Cobre 2.0 mg L-1
Sódio 200 mg L-1
Magnésio 50 mg L-1
Segundo este DL, o valor paramétrico é o valor máximo ou mínimo para cada um dos parâmetros a controlar. No caso do cálcio, ferro, sódio e magnésio o valor estabelecido deve ser considerado como valor guia, uma vez que estes elementos estão classificados como parâmetros indicadores (DL 306/2007).
1.4. Métodos espectrofotométricos de absorção e emissão atómica
Em 1900, Max Planck propôs a teoria quântica para explicar as propriedades da radiação emitida por corpos aquecidos, que foi depois estendida a outros tipos de processos de emissão ou absorção (Skoog et al., 1998). A teoria incluía dois importantes postulados: i) átomos, iões e moléculas apenas podem existir em estados (electrónicos) discretos, caracterizados por definidas quantidades de energia. Quando uma espécie muda de estado, absorve ou emite uma quantidade de energia exactamente igual à diferença de energia entre os dois estados; ii) Quando os átomos, iões ou moléculas absorvem ou emitem radiação na transição de um estado de energia para outro, a frequência ou o comprimento de onda da radiação relaciona-se com a diferença de energias entre os estados (Christian, 1994; Skoog et al., 1998).
À temperatura ambiente os átomos de uma amostra estão predominantemente no estado fundamental (Dean, 1997). Se é fornecida a um átomo energia suficiente (térmica ou eléctrica), certas frequências podem ser absorvidas selectivamente. Este processo de absorção pelo qual a energia electromagnética é transferida para átomos, iões ou moléculas constituintes de uma amostra é responsável pela passagem dessas partículas a estados de
- 12 -
maior energia – estados excitados (Dean, 1997; Skoog et al., 1998; Vogel, 2002). Isto pode acontecer no calor de uma chama ou plasma (Dean, 1997; Vogel, 2002). Pela teoria quântica, as partículas têm um número limitado de níveis energéticos discretos e para a absorção ocorrer a energia do fotão incidente deve coincidir exactamente com a diferença de energia entre um estado excitado e o estado fundamental – estado electrónico de menor energia (Christian, 1994; Skoog et al., 1998). Um estado excitado é instável, sendo o seu tempo-de-vida breve (tipicamente 10-8 s). Assim, o electrão regressa a um estado de menor energia, sendo esse regresso acompanhado por emissão de radiação (Dean, 1997). Essa emissão de radiação electromagnética corresponde à libertação da energia em excesso sob a forma de fotões – partículas discretas constituintes da radiação electromagnética (Skoog et al., 1998).
As técnicas espectrofotométricas baseiam-se na emissão ou absorção de radiação electromagnética (Dean, 1997). Neste trabalho será descrito o método de espectrofotometria de absorção atómica com chama (AAS-F) e o método de plasma acoplado indutivamente à espectrofotometria de emissão atómica (ICP-AES), pois apesar da espectrofotometria de absorção atómica ser a metodologia de referência prevista no DL 236/98 para a determinação dos elementos seleccionados neste trabalho, é permitida a utilização de métodos analíticos alternativos. Os dois métodos mencionados são dos melhores métodos para quantificar vários elementos que constam da legislação sobre águas e considerou-se pertinente proceder à sua comparação com a finalidade de avaliar se o ICP-AES poderá ser utilizado de forma vantajosa como método analítico alternativo.
1.4.1 Espectrofotometria de absorção atómica com chama
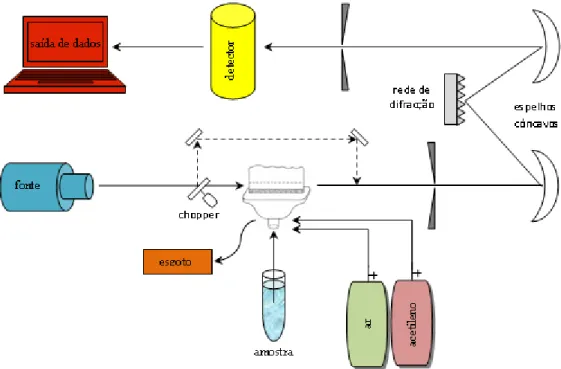
Instrumentos típicos de espectrofotometria contêm cinco componentes: uma fonte de radiação estável; um suporte para a amostra/célula de atomização; um sistema que isola uma restrita região do espectro; um detector de radiação que converte energia radiante num sinal, normalmente eléctrico; um processador e leitor de sinal que mostra/expõem o sinal traduzido (Loon, 1980; Christian, 1994; Dean, 1997; Skoog et al., 1998). No caso da espectrofotometria de absorção atómica com chama, os componentes são colocados como mostra a Figura 2.
- 13 -
Figura 2: Instrumentação de um espectrofotómetro óptico com atomização por chama.
As fontes mais utilizadas em espectrofotometria de absorção atómica com chama, são as lâmpadas de cátodo oco (Christian, 1994; Skoog et al., 1998; Vogel, 2002). Estas consistem num ânodo de tungsténio e num cátodo cilíndrico selados num tubo de vidro, com uma janela de quartzo e cheio de néon ou árgon à pressão de 1 a 5 torr, como mostra a Figura 3 (Christian, 1994; Dean, 1997; Skoog et al., 1998; Vogel, 2002). O cátodo é construído do metal que está a ser medido ou serve para suportar uma camada desse metal (Skoog et al., 1998; Vogel, 2002). Estas lâmpadas são idealizadas para o elemento de interesse, isto é, emitem radiação característica do analito, sendo depois o detector sintonizado para esta frequência (Loon, 1980; Suess, 1982).
Figura 3: Lâmpada de cátodo oco (adaptado de Dean, 1997).
Para iniciar a actividade da lâmpada, uma diferença de potencial (2 a 30 mA) é aplicada entre os eléctrodos. A passagem de corrente ioniza o gás nobre de enchimento,
Pinos conectores Cátodo oco Envelope de vidro Janela de quartzo Árgon Ânodo
- 14 -
Esgoto
sendo os catiões formados acelerados para o cátodo. A energia do impacto é suficiente para excitar os átomos do cátodo, que emitem radiação (Dean, 1997; Vogel, 2002).
Geralmente são usados monocromadores para seleccionar o comprimento de onda, tendo estes como principal função estreitar a gama de comprimentos de onda emitidos pela fonte que chegam ao detector, isolando a linha de ressonância do elemento, resultante da transição directa do estado fundamental para um estado excitado (Loon, 1980; Suess, 1982; Vogel, 2002). O monocromador serve também para minimizar a energia emitida pelo enchimento inerte de gás da fonte, uma vez que esta energia pode modificar a relação quantitativa entre a concentração de analito e a absorvância medida (Suess, 1982). É aconselhável manter as fendas do monocromador tão estreitas quanto possível para rejeitar linhas indesejadas das lâmpadas de cátodo oco e da chama (Loon, 1980).
A célula de atomização é o local onde a amostra é introduzida e dissociada dos constituintes da amostra de forma a obter átomos livres no estado fundamental (Loon, 1980; Dean, 1997). Este processo de conversão da amostra em vapor atómico denomina-se atomização (Loon, 1980; Skoog et al., 1998). O método mais comum é a chama. As soluções são introduzidas num atomizador por nebulização, onde a amostra é convertida num aerossol por uma corrente de gás, que leva depois a amostra a uma região onde se dá a atomização (Skoog et al., 1998). Na atomização por chama o principal tipo de queimador encontrado é o de fluxo laminar, onde o gás oxidante e o combustível são misturados anteriormente à entrada no queimador (Dean, 1997; Vogel, 2002) e cujo esquema é representado na Figura 4.
- 15 -
A luz absorvida pelos átomos no estado fundamental é então medida (Suess, 1982). Este método de atomização é mais adequado quando o analito está presente em altas concentrações e há suficiente volume de amostra disponível. O tempo que um átomo está presente na chama é limitado, levando a limites de detecção elevados (Christian, 1994; Dean, 1997). No entanto, este método apresenta boa reprodutibilidade (Christian, 1994).
A absorção atómica obedece à Lei de Beer-Lambert (Christian, 1994; Skoog et al., 1998), em que a absorvância é directamente proporcional à concentração, para radiação monocromática:
A=εbc
onde, ε é a absortividade, b é a espessura do percurso óptico e c é a concentração dos átomos na célula (Loon, 1980; Dean, 1997).
No entanto, existem desvios à linearidade resultantes de interferências. Nos métodos de absorção atómica são encontradas interferências espectrais, interferências químicas e interferências físicas.
1.4.2 Plasma acoplado indutivamente à espectrofotometria de emissão atómica
A utilização de plasma como fonte de excitação para emissão atómica tornou-se importante nos anos recentes, sendo actualmente um dos modos mais importantes de excitação de átomos (Christian, 1994; Vogel, 2002).
O plasma acoplado indutivamente à espectrofotometria de emissão atómica (ICP-AES) é um método utilizado para a determinação quantitativa de metais numa ampla variedade de amostras (Carvalho, 2007). O ICP-AES é constituído por vários componentes (Figura 5): gerador de radiofrequências, indutor para formar o plasma, tocha, sistema de introdução da amostra (nebulizador, câmara de nebulização e injector), colimador (lentes e espelhos), sistema de dispersão e um detector (Carvalho, 2007).
- 16 - Figura 5: Instrumentação essencial do ICP-AES.
Uma fonte de ICP é constituída por uma tocha (Figura 6) que consiste em tubos de quartzo concêntricos e através dos quais passa uma corrente de gás árgon (Christian, 1994; Dean, 1997; Skoog et al., 1998; Vogel, 2002; Carvalho, 2007; Coimbra, 2007). A ionização do fluxo de árgon é iniciada por uma faísca, interagindo os iões resultantes com o campo magnético produzido por uma bobina indutora. Esta interacção origina um fluxo de electrões e iões dentro da bobina (Christian, 1994; Dean, 1997; Skoog et al., 1998; Vogel, 2002; Carvalho, 2007), sendo o plasma sustentado por um campo de radiofrequências (Coimbra, 2007). Um plasma é portanto uma nuvem quente de gás (6000 K a 10000 K), parcialmente ionizada, condutora de electricidade e contendo uma concentração significativa de catiões e electrões (Skoog et al., 1998; Coimbra, 2007).
Figura 6: Plasma acoplado indutivamente (adaptado de Vogel, 2002).
Bobina de radiofrequência Tubos concêntricos de quartzo Árgon auxiliar Aerossol da amostra em árgon
- 17 -
Geralmente o plasma é de gás árgon, que é um gás do grupo 18 da Tabela Periódica, quimicamente inerte e com vantagens como: emissão de espectro relativamente simples; capacidade de atomizar, ionizar e excitar grande parte dos elementos da Tabela Periódica e baixo custo relativamente a outros gases raros (Carvalho, 2007).
A amostra tem de chegar ao plasma na forma de aerossol. Uma vez que as amostras usadas são líquidas é necessário um nebulizador (Coimbra, 2007), cuja função essencial é converter uma amostra aquosa num aerossol com a ajuda de um gás de arraste (Dean, 1997). As gotículas são arrastadas para o plasma através do tubo mais interior da tocha com auxílio do gás nebulizador (Dean, 1997; Skoog et al., 1998; Vogel, 2002; Coimbra, 2007). O sistema de nebulização utilizado está representado na Figura 7.
Figura 7: Nebulizador utilizado no ICP-AES.
Este tipo de atomização é vantajoso porque as amostras podem ser introduzidas em forma de solução através de um nebulizador, o que torna a análise quantitativa e o manuseamento da amostra relativamente fáceis. A alta temperatura do plasma elimina a maioria das interferências químicas presentes na chama e a maior parte dos elementos são rapidamente excitados (Christian, 1994; Vogel, 2002). São também vantagens o facto de a atomização ocorrer num ambiente quimicamente inerte, o que aumenta o tempo de vida do analito porque previne a formação de óxidos, e a temperatura ser relativamente uniforme, o que origina curvas de calibração lineares para diversas gamas de concentração (Skoog et al., 1998).
A luz emitida pela tocha é dirigida para redes de difracção, que dispersam o feixe policromático através de sulcos. Cada sulco dispersa a luz incidente num ângulo, obtendo-se os comprimentos de onda individuais constituintes do feixe, tal como obtendo-se obobtendo-serva com o arco-íris (Skoog et al., 1998; Dean, 1997; Vogel, 2002; Carvalho, 2007). O espectrómetro vai separar a radiação emissora em linhas espectrais, que correspondem às transições electrónicas dos elementos presentes na amostra. A intensidade luminosa das linhas é
- 18 -
medida e fornece informação quantitativa da concentração de cada elemento na amostra uma vez que são proporcionais – o número de fotões emitidos é proporcional ao número de átomos do elemento (Vogel, 2002; Carvalho, 2007).
As interferências que ocorrem na espectrofotometria de emissão atómica podem também ser classificadas em espectrais, químicas e físicas (Christian, 1994).
- 19 -
2. Materiais e metodologias
2.1. Lavagem do materialTodo o material usado neste trabalho foi descontaminado. Primeiramente, foi lavado com detergente Decon a 3% durante 24 horas, seguindo-se uma passagem por água da torneira. Depois, colocou-se o material numa solução de ácido nítrico a 50% (V/V), onde permaneceu 3 dias. Finalmente os frascos foram lavados com água ultra-pura 5 a 6 vezes. A secagem foi feita deixando os frascos secar de boca para baixo.
2.2. Preparação de soluções-padrão
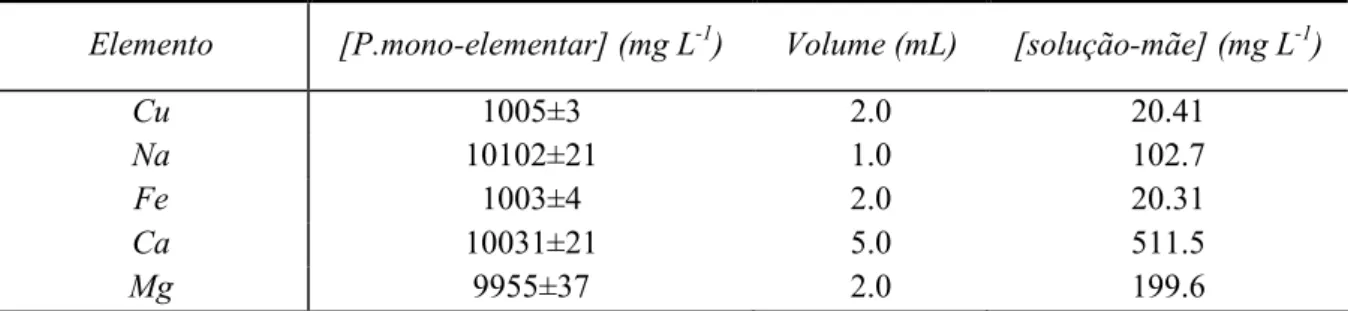
Preparou-se uma solução-mãe multi-elementar, na balança analítica, a partir de padrões mono-elementares dos elementos de interesse para um frasco de plástico de 100 mL e mediram-se os volumes apresentados na Tabela 2. Adicionou-se 1 mL de ácido nítrico, para a solução ficar acidificada a 1%, e perfez-se o volume a 100 mL.
Tabela 2: Concentração dos elementos de interesse na solução padrão-mãe.
Elemento Padrão mono-elementar (mg L-1) Volume (mL) [solução-mãe] (mg L-1)
Cu 1005±3 1.0 9.471
Na 1003±1 20 184.7
Fe 1003±4 1.2 11.40
Ca 10031±21 2.0 189.1
Mg 9955±37 1.0 93.89
A partir da solução-mãe prepararam-se, também na balança analítica, 6 padrões de calibração em frascos de plástico de 100 mL. Os volumes medidos são apresentados na Tabela 3. Adicionou-se também 1 mL de ácido nítrico, para as soluções ficarem acidificadas e perfez-se o volume a 100 mL.
- 20 - Tabela 3: Concentração dos padrões de calibração.
Elemento Padrão Cu (µg L -1 ) Na (mg L-1) Fe (µg L-1) Ca (mg L-1) Mg (mg L-1) P1 93.82 1.855 113.6 1.833 0.8973 P2 185.8 3.673 224.9 3.630 1.777 P3 381.2 7.537 461.4 7.449 3.646 P4 474.2 9.374 573.9 9.266 4.535 P5 759.0 15.01 918.7 14.83 7.260 P6 952.3 18.83 1153 18.61 9.108
Para a espectrofotometria de absorção atómica com chama, foi ainda preparado na balança analítica um outro padrão multi-elementar para verificar a sensibilidade do equipamento (este equipamento tem uma absorvância de 0.200 para uma concentração específica de cada elemento) (Tabelas 4 e 5).
Tabela 4: Soluções padrão-mãe para avaliação da sensibilidade para AAS-F.
Elemento [P.mono-elementar] (mg L-1) Volume (mL) [solução-mãe] (mg L-1)
Cu 1005±3 4.0 40.84
Na 1003±1 0.50 5.022
Fe 1003±4 6.0 60.97
Ca 10031±21 0.40 40.02
Mg 9955±37 0.030 2.978
Tabela 5: Soluções padrão para avaliação da sensibilidade para AAS-F.
Elemento Cu (mg L-1) Na (mg L-1) Fe (mg L-1) Ca (mg L-1) Mg (mg L-1)
Psensibilidade 4.170 0.5201 6.263 4.000 0.2934
Depois de proceder à análise dos padrões de calibração preparados, verificou-se que estes não apresentavam homogeneidade de variâncias e, portanto, as gamas escolhidas não estavam adequadamente ajustadas. Escolheram-se novas gamas - mais estreitas e adaptadas a cada método e a cada elemento - preparam-se novas soluções-mãe e os padrões correspondentes (Tabelas 6 a 9).
- 21 -
Tabela 6: Concentração dos elementos de interesse na solução padrão-mãe para ICP-AES.
Elemento [P.mono-elementar] (mg L-1) Volume (mL) [solução-mãe] (mg L-1)
Cu 1005±3 2.0 20.64
Na 10102±21 3.7 383.6
Fe 1003±4 2.0 20.51
Ca 10031±21 20 2042
Mg 9955±37 3.5 353.3
Tabela 7: Concentração dos padrões de calibração para ICP-AES.
Elemento Padrão Cu (µg L -1 ) Na (mg L-1) Fe (µg L-1) Ca (mg L-1) Mg (mg L-1) P1 62.65 1.145 62.64 6.067 1.035 P2 82.82 1.514 82.80 8.020 1.368 P3 108.7 1.987 108.7 10.52 1.795 P4 135.6 2.480 135.6 13.13 2.241 P5 164.0 2.997 163.9 15.88 2.709 P6 207.4 3.791 207.3 --- 3.426 P7 249.9 4.569 249.9 --- 4.129
Tabela 8: Concentração dos elementos de interesse na solução padrão-mãe para AAS-F.
Elemento [P.mono-elementar] (mg L-1) Volume (mL) [solução-mãe] (mg L-1)
Cu 1005±3 2.0 20.41
Na 10102±21 1.0 102.7
Fe 1003±4 2.0 20.31
Ca 10031±21 5.0 511.5
Mg 9955±37 2.0 199.6
Tabela 9: Concentração dos padrões de calibração para AAS-F.
Elemento Padrão Cu (µg L -1 ) Na (mg L-1) Fe (µg L-1) Ca (mg L-1) Mg (mg L-1) P1 154.4 0.7641 154.5 3.787 1.457 P2 255.9 1.267 256.1 6.277 2.414 P3 412.1 2.040 412.5 10.11 3.888 P4 565.3 2.798 565.8 13.87 5.333 P5 730.6 3.616 731.2 17.92 6.893 P6 876.4 4.337 877.2 21.50 8.268 P7 1032 5.109 1033 --- 9.739
No caso da AAS-F foi possível obter gamas de calibração com homogeneidade de variâncias com pelo menos cinco padrões, com a excepção do cálcio, que precisou de
- 22 -
padrões intermédios entre P3 e P6. Sendo assim, prepararam-se mais dois padrões a partir da solução padrão-mãe apenas para este elemento. As gamas dos padrões de trabalho escolhidas estão resumidas na Tabela 10.
Tabela 10: Gamas de concentração das soluções padrão de trabalho escolhidas para AAS-F. Elemento Padrões Cu (µg L -1 ) Na (mg L-1) Fe (µg L-1) Ca (mg L-1) Mg (mg L-1) P1 --- 0.7641 154.5 --- --- P2 255.9 1.267 256.1 --- 2.414 P3 412.1 2.040 412.5 9.966 3.888 P4 565.3 2.798 565.8 14.12 5.333 PA --- --- --- 16.11 --- P5 730.6 3.616 731.2 17.63 6.893 PB --- --- --- 19.67 --- P6 876.4 4.337 877.2 21.25 8.268 P7 1032 --- 1033 --- 9.739
Quanto ao ICP-AES, com excepção do cálcio, as gamas escolhidas apenas cumpriam o teste de homogeneidade de variância para quatro padrões, pelo que foi necessário preparar novos padrões a partir da solução padrão-mãe com concentrações intermédias. As gamas de concentração dos padrões de trabalho são apresentadas na Tabela 11.
Tabela 11: Gamas de concentração dos padrões de trabalho escolhidas para ICP-AES. Elemento Padrões Cu (µg L -1 ) Na (mg L-1) Fe (µg L-1) Ca (mg L-1) Mg (mg L-1) P2 82.82 1.514 82.80 8.020 --- Pα 95.81 1.752 95.80 9.279 --- P3 108.7 1.987 108.7 10.52 1.795 Pβ 125.4 2.293 125.4 12.15 2.072 Pγ 131.1 2.396 131.0 12.69 2.165 P4 135.6 2.480 135.6 13.13 2.241 P5 164.0 2.997 163.9 15.88 2.709 Pθ 178.3 3.259 178.2 17.26 2.945 Pδ 188.0 3.437 187.9 18.20 3.106 P6 207.4 3.791 207.3 --- 3.426 PΩ 239.0 4.369 238.9 --- 3.948 P7 --- 4.569 249.9 --- ---
- 23 - 2.3. Preparação de amostras
Foram recolhidas 44 amostras de águas destinadas ao consumo humano em vários locais do país e com várias origens: poços, furos, rede de abastecimento e também águas comercializadas. Todas as amostras foram acidificadas de forma semelhante aos padrões.
2.4. Controlo de qualidade dos resultados
Uma das medidas de controlo e garantia de qualidade dos resultados é a utilização de materiais de referência. A validação dos resultados deste estudo foi obtida pela utilização de materiais de referência da RELACRE - Associação de Laboratórios Acreditados de Portugal.
Para além disto, como recomendado no Guia RELACRE 3, foram feitas réplicas de 10% das amostras, bem como testes de recuperação de modo a poder avaliar os possíveis efeitos de matriz.
2.5. Condições de operação dos equipamentos
2.5.1 ICP-AES
O espectrofotómetro de emissão atómica com plasma acoplado indutivamente usado neste trabalho é da marca Jobin Yvon, modelo ActivaM.
Energia do plasma– 1000 W Velocidade da bomba – 20 rot min-1 Fluxo de gás do plasma – 16 L min-1 Fluxo de sheath– 0.2 L min-1
Tempo de estabilização de sheath – 3 s Fluxo de nebulização – 0.02 L min-1 Pressão do nebulizador – 1.0 bar Tempo de lavagem da agulha – 20 s Tempo de transferência – 15 s Tempo de estabilização – 20 s
- 24 - Tempo de integração – 1 s Slit de entrada – 20 µm
2.5.2 AAS-F
O espectrofotómetro de absorção atómica com chama usado neste trabalho é da marca Perkin-Elmer, modelo AAnayst 100.
Fluxo de aspiração – 0.26 mL seg-1 Energia da lâmpada: Cu – 10 mA
Fe – 30 mA Ca – 15 mA Na – 15 mA Mg – 6 mA
- 25 -
3. Apresentação e discussão de resultados
Tendo em consideração que o objectivo deste trabalho consistia em validar dois métodos analíticos (AAS-F e ICP-AES) para análise de águas de consumo humano e proceder à sua comparação com vista a verificar se o último poderia ser considerado método analítico alternativo ao mencionado no DL 306/2007, iniciou-se o trabalho fazendo a validação dos dois métodos para os elementos seleccionados.
3.1 Parâmetros usados na validação dos métodos analíticos
Exprimir resultados de análises de uma forma consistente tem uma exigência cada vez maior, verificando-se uma crescente necessidade de obter dados analíticos comparáveis, que não apresentem qualquer dúvida razoável no que diz respeito à sua exactidão e que possuam uma precisão adequada para o fim a que se destinam.
Primeiramente deve-se validar o método analítico para que esteja assegurado que o método está sob controlo (Carvalho, 2007; Coimbra, 2007). Os requisitos mínimos para a validação de métodos de ensaio compreendem o estudo da gama de trabalho/linearidade, limiares analíticos (detecção/quantificação), sensibilidade, precisão e exactidão (Guia RELACRE 13, 2000). O processo de validação envolve o estudo de parâmetros por avaliação directa e avaliação indirecta.
3.2 Validação dos métodos de AAS-F e de ICP-AES
Seguidamente serão apresentados os resultados da validação relativos ao elemento cobre, sendo a validação dos elementos magnésio, sódio, cálcio e ferro apresentados, respectivamente, nos Anexos I, II, III e IV.
3.2.1 Avaliação indirecta
- Gama de trabalho
A gama de trabalho pode ser avaliada pelo teste de homogeneidade de variâncias. Isto garante que o método dos mínimos quadrados é aplicável. De acordo com a norma ISO 8466-1 recomendam-se dez pontos de calibração distribuídos de igual modo na gama de concentrações, não devendo este número ser inferior a cinco. O primeiro e o último
- 26 -
padrão são analisados em dez réplicas independentes e a gama de trabalho considera-se ajustada se a diferença de variâncias relativas aos dois padrões não for significativa (Guia RELACRE 13, 2000), isto é, se for igual ou inferior ao valor de F de Ficher para n-1 graus de liberdade. Assim, para este teste, efectuaram-se dez leituras de sinal para o primeiro e para o último padrão da gama, encontrando-se os resultados na Tabela 12.
Tabela 12: Medições de P0 e P6 para o cobre em AAS-F.
Neste caso, a homogeneidade de variâncias não se verificou uma vez que o valor de PG é superior ao valor tabelado. No caso da AAS-F foi possível obter novas gamas com homogeneidade de variâncias (Tabela 13), com a excepção do cálcio que precisou de padrões intermédios. As gamas escolhidas estão resumidas na Tabela 10.
Tabela 13: Medições de P2 e P7 para o cobre em AAS-F.
Padrões P2 P7 0.068 0.281 0.070 0.283 0.071 0.285 0.069 0.286 0.071 0.283 0.071 0.281 0.071 0.281 0.072 0.282 0.073 0.286 In te n si d ad es 0.071 0.283 Variância 2.01E-06 3.88E-06
PG 1.93 < 3.179 Padrões P0 P6 0.000 0.296 -0.001 0.300 -0.002 0.294 0.000 0.301 0.000 0.302 -0.001 0.302 -0.002 0.303 -0.001 0.299 -0.001 0.298 In te n si d ad es -0.001 0.296 Variância 6.11E-07 9.03E-06
- 27 -
Como se pode verificar, para a nova gama de concentrações o valor de PG já se mostrou superior ao valor de F, podendo considerar-se a gama de trabalho ajustada.
Para os resultados obtidos em ICP-AES, procedeu-se exactamente da mesma forma obtendo-se os seguintes resultados:
Tabela 14: Medições de P0 e P6 para o cobre em ICP-AES.
Padrões P0 P6 1272 187604 1197 188311 1175 187825 1227 188690 1227 187287 1272 183853 1231 193466 1245 190661 1140 192321 In te n si d ad es 1148 191494 Variância 2.21E+03 8.12E+06
PG 3.66E+03 > 3.179
Também para este método as gamas não estavam ajustadas, procedendo-se à preparação de novos padrões. Com excepção do cálcio, as gamas escolhidas apenas cumpriam o teste para quatro padrões, pelo que teve de se preparar novos padrões com concentrações intermédias a partir da solução-mãe para, como referido, as rectas terem no mínimo cinco padrões. As gamas encontradas apresentaram as variâncias observadas na Tabela 15.
- 28 -
Tabela 15: Medições dos padrões P2 e P7 para o cobre em ICP-AES.
Padrões P2 P7 17630 50858 17685 50445 17346 49518 17022 50234 17350 50043 17046 49574 16958 49665 17411 49956 16991 49468 In te n si d ad es 16883 49160 Variância 8.41E+04 2.66E+05
PG 3.16 < 3.179
Como se pode verificar, esta nova gama de concentrações já apresentou homogeneidade de variâncias. É, no entanto, de salientar que para que as gamas cumprissem o referido teste foi necessário estreitá-las demasiado, obrigando à diluição de grande parte das amostras posteriormente analisadas para a comparação dos métodos.
Com as gamas de calibração ajustadas, prosseguiu-se com a validação de ambos os métodos.
- Curva de calibração
Em análises quantitativas, a calibração é um processo pelo qual a resposta do equipamento se relacionada com uma concentração conhecida. Caso se utilize o método dos mínimos quadrados, o eixo das ordenadas representa a resposta instrumental e o eixo das abcissas representa as concentrações dos padrões. Para avaliar uma calibração analítica, o coeficiente de correlação pode ser usado, devendo ter valores superiores a 0.995 (Guia RELACRE 13, 2000).
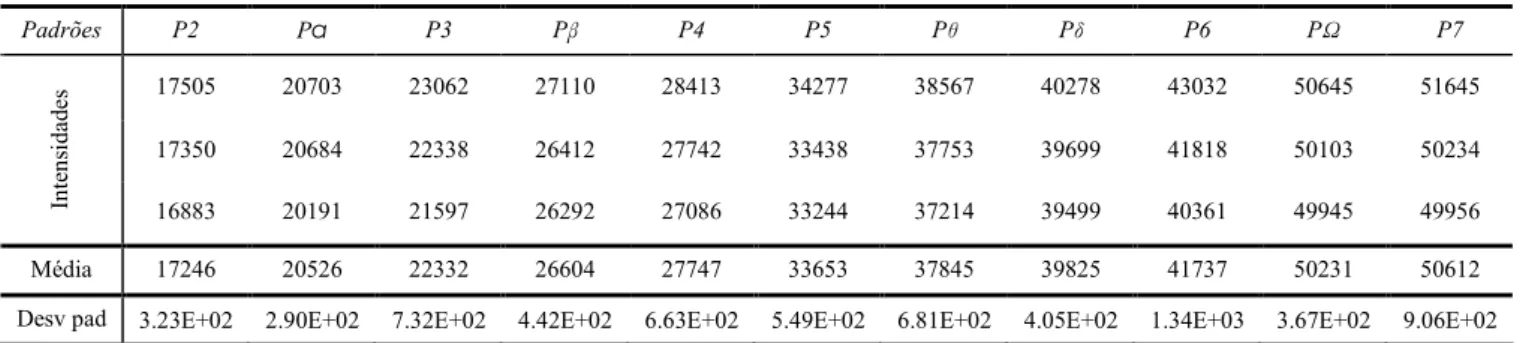
As intensidades obtidas para cada padrão de calibração de cobre das gamas escolhidas, assim como a respectiva média e desvio padrão, apresentam-se nas Tabelas 16 e 17.
- 29 -
Tabela 16: Intensidades obtidas para os padrões de calibração de cobre em AAS-F.
Padrões P2 P3 P4 P5 P6 P7 0.069 0.113 0.155 0.200 0.239 0.282 0.068 0.111 0.156 0.201 0.240 0.281 0.070 0.111 0.155 0.201 0.240 0.283 0.071 0.113 0.156 0.199 0.242 0.285 In te n si d ad es 0.069 0.114 0.156 0.201 0.243 0.286 Média 0.069 0.112 0.156 0.200 0.241 0.283 Desv pad 1.1E-03 1.34E-03 5.48E-04 8.94E-04 1.64E-03 2.07E-03
Tabela 17: Intensidades obtidas para os padrões de calibração de cobre em ICP-AES.
Padrões P2 Pα P3 Pβ P4 P5 Pθ Pδ P6 PΩ P7 17505 20703 23062 27110 28413 34277 38567 40278 43032 50645 51645 17350 20684 22338 26412 27742 33438 37753 39699 41818 50103 50234 In te n si d ad es 16883 20191 21597 26292 27086 33244 37214 39499 40361 49945 49956 Média 17246 20526 22332 26604 27747 33653 37845 39825 41737 50231 50612 Desv pad 3.23E+02 2.90E+02 7.32E+02 4.42E+02 6.63E+02 5.49E+02 6.81E+02 4.05E+02 1.34E+03 3.67E+02 9.06E+02
A partir destes valores calculou-se então a curva de calibração do cobre para cada método, segundo o método dos mínimos quadrados:
Tabela 18: Curva de calibração para o cobre em AAS-F.
Padrões xi xi2 xi-xméd (xi-xméd)2 yi yi-yméd (yi-yméd)2 (xi-xméd)(yi-yméd) yest |yi-yest| (yi-yest)2
P2 255.9 6.55E+04 -3.90E+02 1.52E+05 0.069 -1.08E-01 1.16E-02 4.19E+01 6.96E-02 2.00E-04 4.00E-08 P3 412.1 1.70E+05 -2.33E+02 5.44E+04 0.112 -6.46E-02 4.17E-03 1.51E+01 1.13E-01 2.69E-04 7.22E-08 P4 565.3 3.20E+05 -8.01E+01 6.42E+03 0.156 -2.14E-02 4.58E-04 1.72E+00 1.55E-01 6.98E-04 4.87E-07 P5 730.6 5.34E+05 8.51E+01 7.25E+03 0.200 2.34E-02 5.48E-04 1.99E+00 2.00E-01 7.46E-05 5.57E-09 P6 876.4 7.68E+05 2.31E+02 5.33E+04 0.241 6.38E-02 4.07E-03 1.47E+01 2.41E-01 1.19E-04 1.41E-08 P7 1032 1.07E+06 3.87E+02 1.50E+05 0.283 1.06E-01 1.13E-02 4.12E+01 2.84E-01 2.73E-04 7.46E-08 média 645.4 4.87E+05 1.89E-14 7.05E+04 0.177 -9.25E-18 5.36E-03 1.94E+01 1.77E-01 2.72E-04 1.16E-07 soma 3873 2.92E+06 0.00E+00 4.23E+05 1.062 0.00E+00 3.21E-02 1.17E+02 1.06E+00 1.63E-03 6.93E-07
Obteve-se:
- 30 -
Tabela 19: Parâmetros da curva de calibração.
Parâmetro Valor
r 1.000
declive (b) 2.76E-04 ord.origem (a) - 9.62E-04
sy/x 3.72E-04
sb 5.73E-07
sa 1.12E-04
Com base nestes parâmetros foi possível obter o gráfico da curva de calibração (Figura 8) e também dos intervalos de confiança (Figura 9), que se apresentam de seguida.
Figura 8: Curva de calibração para o cobre em AAS-F.
- 31 - Tabela 20: Curva de calibração para o cobre em ICP-AES.
Padrões xi xi2 xi-xméd (xi-xméd)2 yi yi-yméd (yi-yméd)2 (xi-xméd)(yi-yméd) yest |yi-yest| (yi-yest)2 P2 82.82 6.86E+03 -7.85E+01 6.17E+03 17246 -1.62E+04 2.64E+08 1.28E+06 1.75E+04 2.64E+02 6.97E+04 Pα 95.81 9.18E+03 -6.55E+01 4.29E+03 20526 -1.30E+04 1.68E+08 8.49E+05 2.02E+04 3.72E+02 1.38E+05 P3 108.7 1.18E+04 -5.27E+01 2.77E+03 22332 -1.12E+04 1.24E+08 5.88E+05 2.28E+04 4.37E+02 1.91E+05 Pβ 125.4 1.57E+04 -3.59E+01 1.29E+03 26604 -6.88E+03 4.74E+07 2.47E+05 2.62E+04 4.23E+02 1.79E+05 P4 135.6 1.84E+04 -2.57E+01 6.61E+02 27747 -5.74E+03 3.29E+07 1.48E+05 2.83E+04 5.08E+02 2.58E+05 P5 164.0 2.69E+04 2.61E+00 6.79E+00 33653 1.66E+02 2.75E+04 4.32E+02 3.40E+04 3.64E+02 1.33E+05 Pθ 178.3 3.18E+04 1.69E+01 2.87E+02 37845 4.36E+03 1.90E+07 7.38E+04 3.69E+04 9.12E+02 8.31E+05 Pδ 188.0 3.53E+04 2.66E+01 7.09E+02 39825 6.34E+03 4.02E+07 1.69E+05 3.89E+04 9.19E+02 8.44E+05 P6 207.4 4.30E+04 4.60E+01 2.12E+03 41737 8.25E+03 6.81E+07 3.80E+05 4.28E+04 1.11E+03 1.24E+06 PΩ 239.0 5.71E+04 7.76E+01 6.02E+03 50231 1.67E+04 2.80E+08 1.30E+06 4.93E+04 9.53E+02 9.08E+05 P7 249.9 6.24E+04 8.85E+01 7.84E+03 50612 1.71E+04 2.93E+08 1.52E+06 5.15E+04 8.92E+02 7.96E+05 média 161.3 2.90E+04 3.36E-14 2.92E+03 33487 -3.97E-12 1.22E+08 5.95E+05 3.35E+04 6.51E+02 5.08E+05 soma 1775 3.19E+05 3.69E-13 3.22E+04 368357 -4.37E-11 1.34E+09 6.55E+06 3.68E+05 7.16E+03 5.59E+06
Obteve-se:
y = (20.3 ± 1.33)E+01 x + (6.59 ± 28.4)E+02
Tabela 21: Parâmetros da curva de calibração.
Parâmetro Valor
r 0.9979
declive (b) 2.03E+02 ord.origem (a) 6.59E+02 sy/x 1.06E+03
sb 5.89E+00
sa 1.26E+03
- 32 -
Figura 11: Intervalo de confiança para os padrões de cobre em ICP-AES.
Como o coeficiente de correlação de todas as rectas obtidas foi superior a 0.995 para a totalidade dos metais estudados, considerou-se que as mesmas eram uma boa representação das intensidades obtidas em função da concentração dos padrões. Isto pode também ser verificado com gráficos de resíduos (Figuras 12 e 13).
Figura 12: Resíduos para o cobre em AAS-F.
- 33 -
Como se pode visualizar em ambos os gráficos e para todos os metais analisados (com excepção do sódio em ICP-AES), os resíduos tanto são positivos como negativos, podendo considerar-se os dois métodos livres de tendências.
- Sensibilidade
Pode ser definida como o quociente entre o acréscimo do valor lido e a variação da concentração correspondente àquele acréscimo. Esta característica avalia a capacidade de um método discriminar entre pequenas diferenças de concentração de analito (Guia RELACRE 13, 2000; Coimbra, 2007). A definição quantitativa aceite pela IUPAC é sensibilidade de calibração - declive da curva de calibração que é constante na gama de trabalho (Guia RELACRE 13, 2000). As sensibilidades obtidas para o cobre são apresentadas na Tabela 22.
Tabela 22: Sensibilidade dos dois métodos para o cobre.
Método
Parâmetro AAS-F ICP-AES
Sensibilidade (µg L-1) 2.76E-04 2.03E+02
Como se pode verificar, a sensibilidade para o cobre foi maior no método de ICP-AES. Isto aconteceu para todos os metais estudados, sendo a diferença entre os dois métodos de seis ordens de grandeza para o cobre, o magnésio e o ferro e de cinco ordens de grandeza para o sódio e o cálcio. Assim, pode-se concluir que para estes metais, o ICP-AES é bastante mais sensível do que o AAS-F.
- Limiares analíticos
Todos os métodos instrumentais têm um grau de ruído associado à medida, que limita a quantidade de analito que pode ser detectada (Christian, 1994). Uma medida torna-se credível quando é maior do que a sua incerteza associada. O ponto a partir do qual isso ocorre é designado limite de detecção (LD) (Taylor, 1988), ou seja, trata-se do teor mínimo medido, a partir do qual é possível detectar a presença do analito com uma certeza estatística razoável. Em termos qualitativos, este conceito corresponde à concentração mínima distinguível do branco (Miller & Miller, 1993; DL 236/98; Guia RELACRE 13,
- 34 -
2000). O mínimo sinal analítico distinguível é tomado como a soma da média dos sinais do branco, Sbl, mais um múltiplo k do desvio padrão do branco, sb, que segundo o Guia da
RELACRE 13 será 3.3 para um nível de confiança de 99.7%.
O nível mais baixo onde as medidas se tornam quantitativamente significantes é chamado limite de quantificação (LQ) (Taylor, 1988; Skoog et al., 1998). Assim sendo, o LQ é a menor concentração medida a partir da qual é possível a quantificação do analito com uma dada precisão e exactidão (Miller & Miller, 1993; Guia RELACRE 13, 2000). Para medidas quantitativas, as concentrações devem ser pelo menos 10 vezes o limite de detecção (Christian, 1994) pelo que este limite é geralmente tomado como sendo igual a 10 vezes o desvio padrão de repetitivas medidas do branco (Miller & Miller, 1993; Skoog et al., 1998; Guia RELACRE 13, 2000).
Para além de poderem ser calculados a partir das medidas das intensidades dos brancos estes limites podem também ser determinados através da curva calibração. Assim, neste trabalho procedeu-se à análise de dez brancos independentes e comparou-se os limites obtidos com os determinados pela calibração considerando-se o valor maior.
Tabela 23: Limiares analíticos para o cobre.
Método
Parâmetro AAS-F ICP-AES
LD (µg L-1) 18.48 ± 4.106 17.14 ± 15.45
LQ (µg L-1) 39.94 ± 3.998 51.95 ± 14.20
O método de ICP-AES apresentou menor limite de detecção para o cobre, mas é a AAS-F que tem menor limite de quantificação. Para o sódio e o ferro a AAS-F apresentou menores limiares analíticos nas condições em que este trabalho foi realizado, tendo o ICP-AES menores limiares para magnésio e cálcio.
A legislação de referência (DL 306/2007) impõe que os métodos utilizados apresentem, no mínimo, um limite de detecção de 10% do valor paramétrico, o que foi cumprido para o cobre e também para os restantes elementos em ambos os métodos.
- Coeficiente de variação
O desvio padrão de um método, e respectivo coeficiente de variação, permitem ao analista avaliar a qualidade do seu trabalho (Guia RELACRE 13). Eles são calculados da seguinte forma:
- 35 - Tabela 24: Coeficientes de variação para o cobre.
Método
Parâmetro AAS-F ICP-AES
Sm 1.35 5.19
CVm (%) 0.209 3.22
Os valores de coeficiente de variação obtidos para o cobre e para os outros elementos podem ser considerados baixos. O ICP-AES apresentou menores valores de coeficiente de variação para magnésio, sódio e cálcio e a AAS-F teve menores valores para cobre e ferro. Em todo o caso o CVm nunca ultrapassa os 5%, o que se pode considerar um valor bastante
razoável.
- Ensaios de recuperação
A capacidade de um método identificar e distinguir um analito, em particular numa mistura complexa sem interferência dos outros componentes denomina-se selectividade. Já discriminar o analito relativamente a outras substâncias eventualmente presentes na amostra a analisar, oferecendo garantias de que a grandeza medida provém apenas do analito designa-se especificidade (Guia RELACRE 13, 2000; Coimbra, 2007). Geralmente utiliza-se o método da adição de padrão para avaliar as interferências (Guia RELACRE 13, 2000). O método envolve adicionar um ou mais incrementos de padrão (spikes) a alíquotas iguais de amostra e diluí-las ao mesmo volume final (Loon, 1980; Miller & Miller, 1993; Skoog et al., 1998). O padrão é portanto sujeito à mesma matriz da amostra, tal como o analito, sendo o aumento do sinal devido ao padrão adicionado e o sinal original devido ao analito (Christian, 1994). Um método analítico é considerado específico e selectivo quando após a realização destes testes se verificar que a taxa de recuperação é próxima de 100% (Guia RELACRE 13, 2000; Coimbra, 2007).
Para este teste escolheram-se duas amostras por cada metal estudado e fizeram-se três spikes em cada amostra. Os resultados obtidos para o cobre estão nas Tabelas 25 e 26.
declive y/x S Sm = 100 x S méd m × = m CV
- 36 - Tabela 25: Ensaios de recuperação para o cobre em AAS-F.
Amostra C amostra (µg L -1 ) C spike (µg L -1 ) C am+spike (µg L -1 ) Recuperação (%) 326.4 342.5 97.76 612.0 595.1 93.42 19 23.37 918.0 912.1 96.82 346.8 479.8 97.96 530.4 696.0 104.8 30 140.1 734.4 889.3 102.0
Tabela 26: Ensaios de recuperação para o cobre em ICP-AES.
Amostra C amostra (µg L -1 ) C spike (µg L -1 ) C am+spike (µg L -1 ) Recuperação (%) 61.80 149.5 80.26 164.8 285.9 112.8 7 99.94 267.8 393.9 109.8 82.40 205.0 85.08 185.4 314.8 97.05 37 134.9 247.2 346.0 85.42
Os valores obtidos estão dentro do intervalo 80 a 120%, o que indicou não haver influência da matriz na detecção do analito. Isto ocorreu para todos os metais em ambos os métodos. Assim, podem considerar-se os dois métodos específicos e selectivos, não sendo as interferências relevantes.
- Precisão
A precisão dos dados analíticos é o grau de concordância mútua numa série de medidas obtidas da mesma forma. Fornece uma medida dos erros aleatórios de uma análise (Loon, 1980; Christian, 1994; Dean, 1997; Skoog et al., 1998; Vogel, 2002). A precisão avalia a dispersão de resultados entre ensaios independentes repetidos sobre uma mesma amostra, amostras semelhantes ou padrões, em condições definidas. Existem duas medidas extremas para avaliar esta dispersão: a repetibilidade e reprodutibilidade. Entre elas existe a precisão intermédia ou variabilidade intralaboratorial (Guia RELACRE 13, 2000).
Repetibilidade:
A repetibilidade exprime a precisão de um método efectuado em condições idênticas, isto é, refere-se a ensaios efectuados sobre uma mesma amostra em condições tão estáveis quanto possível em curtos intervalos de tempo. O limite de repetibilidade (r) é o valor
- 37 -
abaixo do qual se deve situar, com uma probabilidade de 95%, a diferença absoluta entre dois resultados obtidos nas condições referidas: |xi - xi-1| ≤ r (Guia RELACRE 13, 2000).
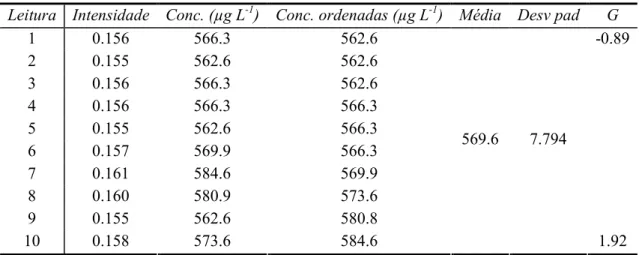
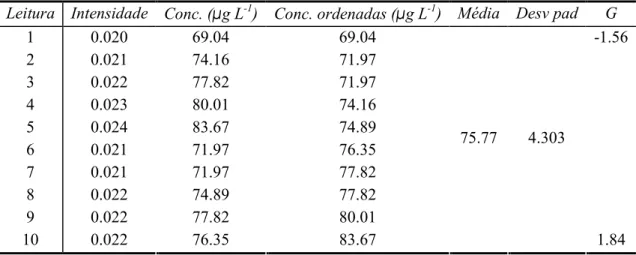
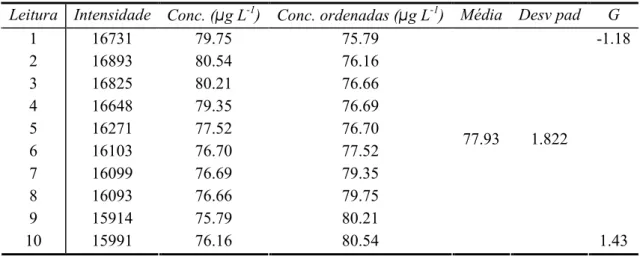
Para esta avaliação analisou-se em replicado uma amostra, três padrões da calibração e material de referência. Primeiramente testaram-se os resultados para valores aberrantes aplicando-se o teste de Grubbs. Neste teste ordenam-se os resultados obtidos e calcula-se a média e o desvio padrão, testando-se o valor menor e o maior:
Caso o teste indique a existência de valores aberrantes retira-se esse valor e volta-se a fazer o teste, caso contrário pode-se calcular a repetibilidade. Os resultados obtidos são apresentados nas Tabelas 27 a 36.
Tabela 27: Teste de Grubbs para o padrão de concentração mais baixa de cobre em AAS-F.
Leitura Intensidade Conc.(µg L-1) Conc. ordenadas (µg L-1) Média Desv pad G
1 0.068 244.5 244.5 -1.90 2 0.070 251.9 248.2 3 0.071 255.5 251.9 4 0.069 248.2 255.5 5 0.071 255.5 255.5 6 0.071 255.5 255.5 7 0.071 255.5 255.5 8 0.072 259.2 255.5 9 0.073 262.8 259.2 10 0.071 255.5 262.8 254.4 5.185 1.62
Tabela 28: Teste de Grubbs para o padrão de concentração mais baixa de cobre em ICP-AES.
Leitura Intensidade Conc. (µg L-1) Conc. ordenadas (µg L-1) Média Desv pad G
1 20859 99.79 96.55 -1.43 2 20401 97.56 96.88 3 20688 98.96 96.99 4 20260 96.88 97.56 5 20684 98.93 98.00 6 20770 99.35 98.93 7 20685 98.94 98.94 8 20490 98.00 98.96 9 20283 96.99 99.35 10 20191 96.55 99.79 98.19 1.150 1.38 padrão desvio média -suspeito valor = G