departamento de f´ısica te´
orica e experimental
programa de p´
os-graduac
¸˜
ao em f´ısica
transporte eletrˆ
onico e propriedades
termodinˆ
amicas de nanobiomol´
eculas
leonardo mafra bezerril
natal-rn
transporte eletrˆ
onico e propriedades
termodinˆ
amicas de nanobiomol´
eculas
Tese de doutorado apresentada ao
Departamento de F´ısica Te´orica e
Experimental da Universidade Federal do
Rio Grande do Norte como requisito parcial `a obten¸c˜ao do grau de Doutor em F´ISICA.
Orientador: Prof. Dr. EUDENILSON LINS
DE ALBUQUERQUE
Natal-RN
Minha esposa
Emanuella L. F. Vidal Bezerrile
meus pais Leˆoncio S. de Bezerril e Jeanete Mafra Bezerril.
Lembre-se de que a resposta de ontem pode n˜ao ter nada a ver com o problema de hoje
(Don Ward).
Em primeiro lugar a Deus por me aben¸coar nessa ´ardua jornada, e em todas outras que me aguardam;
Aos meus pais pelo suporte, carinho, amor e aten¸c˜ao, sem medidas, que me s˜ao dados desde crian¸ca;
`
A minha esposa, pelo amor e paciˆencia dedicada ao longo desses dois anos de doutora-mento;
Ao professor Eudenilson Lins de Albuquerque pela orienta¸c˜ao, apoio, e excelentes dis-ciplinas ministradas, que me foram ´uteis na concretiza¸c˜ao deste trabalho;
Ao professor Umberto Laino pelo suporte e agrad´aveis discuss˜oes sobre f´ısica;
Aos professores Ananias Monteiro Mariz, Luciano Rodrigues, Fernando Dantas Nobre, Dory H´elio, Rui Tertuliano, Jos´e Wilson e Artur Carri¸co, pela forma¸c˜ao profissional;
Aos meus colegas, pelo suporte e companherismo, em especial a Darlan Ara´ujo Mor-eira, Antonio de Macedo Filho, Ricardo Ricardo Gondim Sarmento, Rodolfo Bezerra da Silva, Gustavo de Oliveira G. Rebou¸cas, Gabriel Alves Mendes e Carlos Alexandre Amaral Ara´ujo.
Aos funcion´arios do Programa de P´os-Gradua¸c˜ao em F´ısica PPGF, em especial, `a Celina, pelos servi¸cos e amizada;
Ao CNPQ pelo apoio financeiro.
da diferen¸ca de potencial, no contexto da liga¸c˜ao forte, em seq¨uˆencias de dupla fita do DNA. Com o intuito de investigar a relevˆancia das correla¸c˜oes subjacentes nas distribui¸c˜oes dos nucleot´ıdeos, comparamos os resultados de uma seq¨uˆencia genˆomica do DNA com duas seq¨uˆencias artificiais (Fibonacci e Rudin-Shapiro, que apresentam correla¸c˜ao de longo alcance) e uma seq¨uˆencia aleat´oria, prot´otipo de sistemas de correla¸c˜ao de curto alcance. A seq¨uˆencia aleat´oria utilizada apresenta a mesma correla¸c˜ao de pares de primeiros vizinhos que a seq¨uˆencia do DNA humano. Observamos que a caracter´ıstica de correla¸c˜ao de longo alcance ´e importante para o espectro de transmissividade, apesar das curvas I×V serem mais influenciadas por correla¸c˜oes de curto alcance.
Neste trabalho, analisamos tamb´em as propriedades t´ermicas e eletrˆonicas de uma sequˆencia α-h´elice, obtida de um pept´ıdeo α3, o qual apresenta a seguinte sequˆencia unidi-mensional (Leu-Glu-Thr-Leu-Ala-Lys-Ala)3 (estrutura prim´aria). C´alculos ab initio quˆanticos
s˜ao utilizados para obter as energias dos orbitais moleculares mais altos (HOMO, highest occu-pied molecular orbital), bem como suas integrais de transferˆencias de cargas quando a sequˆencia
α-h´elice forma uma estrutura fibrosa (variante 5Q) e n˜ao fibrosa (variante 7Q), as quais podem ser observadas atrav´es de microscopia eletrˆonica de transmiss˜ao. A diferen¸ca entre as duas estruturas ´e que a estrutura 5Q (7Q) apresenta a substitui¸c˜ao Ala → Gln na 5a (7a) posi¸c˜ao,
respectivamente. N´os estimamos, teoricamente, a densidade de estado bem como o espectro de transmiss˜ao eletrˆonico dos pept´ıdeos, utilizando um Hamiltoniano no formalismo da liga¸c˜ao-forte juntamente com a equa¸c˜ao de Dyson. Al´em disso, n´os resolvemos a equa¸c˜ao de Schr¨odinger dependente do tempo para obter o espalhamento de um pacote de onda inicialmente localizado.
que as importantes diferen¸cas observadas no estudo das propriedades eletrˆonicas de transporte nos encorajam a sugerir este m´etodo como uma ferramenta de diagn´ostico molecular.
voltage (I×V) characteristics of sequences of double-strand DNA molecules. In order to reveal the relevance of the underlying correlations in the nucleotides distribution, we compare the results for the genomic DNA sequence with those of artificial sequences (the long-range corre-lated Fibonacci and RudinShapiro one) and a random sequence, which is a kind of prototype of a short-range correlated system. The random sequence is presented here with the same first neighbors pair correlations of the human DNA sequence. We found that the long-range charac-ter of the correlations is important to the transmissivity spectra, although the I×V curves seem to be mostly influenced by the short-range correlations.
We also analyze in this work the electronic and thermal properties along an α-helix sequence obtained from an α3 peptide which has the uni-dimensional sequence
(Leu-Glu-Thr-Leu-Ala-Lys-Ala)3. An ab initio quantum chemical calculation procedure is used to obtain the
highest occupied molecular orbital (HOMO) as well as their charge transfer integrals, when the
α-helix sequence forms two different variants with (the so-called 5Q variant) and without (the 7Q variant) fibrous assemblies that can be observed by transmission electron microscopy. The difference between the two structures is that the 5Q (7Q) structure have Ala→Gln substitution at the 5th (7th) position, respectively. We estimate theoretically the density of states as well as the electronic transmission spectra for the peptides using a tight-binding Hamiltonian model together with the Dyson’s equation. Besides, we solve the time dependent Schr¨odinger equation to compute the spread of an initially localized wave-packet. We also compute the localization length in the finite α-helix segment and the quantum especific heat. Keeping in mind that fibrous protein can be associated with diseases, the important differences observed in the present
Resumo iv
Abstract vi
Lista de Figuras xiii
Lista de Tabelas xiv
1 Introdu¸c˜ao 1
2 Modelos Te´oricos 4
2.1 Introdu¸c˜ao . . . 4
2.2 O Modelo Tight-binding . . . 5
2.2.1 Formalismo da primeira quantiza¸c˜ao ou da fun¸c˜ao de onda . . . 5
2.2.2 Formalismo da segunda quantiza¸c˜ao ou do n´umero de ocupa¸c˜ao . . . 7
2.3 O Modelo de Anderson . . . 10
2.3.1 Introdu¸c˜ao ao modelo de Anderson . . . 11
2.4 A Mol´ecula do DNA . . . 13
2.5 A mol´ecula α-h´elice e suas variantes 5Q e 7Q . . . 18
2.6.1 Aproxima¸c˜ao de Born-Oppenheimer . . . 21
2.6.2 Teoria do Funcional da Densidade (DFT) . . . 23
2.6.3 O M´etodo de Kohn-Sham . . . 25
2.7 Conclus˜oes. . . 28
3 Propriedades de Transporte Eletrˆonico da Mol´ecula do DNA 29 3.1 Introdu¸c˜ao. . . 29
3.2 As Seq¨uˆencias. . . 31
3.3 Transmitˆancia . . . 35
3.3.1 Resultados . . . 46
3.4 Caracter´ısticas I×V. . . 47
3.4.1 Resultados . . . 50
3.5 Conclus˜oes . . . 52
4 Propriedades de Transporte Eletrˆonico das Variantes 5Q e 7Q da Mol´ecula α3 54 4.1 Introdu¸c˜ao . . . 54
4.2 Densidade de Estado . . . 54
4.2.1 A Equa¸c˜ao de Dyson . . . 60
4.2.2 C´alculo das Energias de Ioniza¸c˜ao . . . 68
4.2.3 Resultados . . . 70
4.3 Transmitˆancia . . . 72
4.3.1 Resultados . . . 73
4.4 Caract´eristicas I×V . . . 76
4.4.1 Resultados . . . 77
4.5 Dinˆamica da Fun¸c˜ao de Onda . . . 79
4.5.1 Resultados . . . 81
6 Conclus˜oes e Perspectivas 96
Apˆendices 98
A 98
A.1 O M´etodo de Euler . . . 98
A.2 O M´etodo de Runge-Kutta de quarta ordem . . . 99
B 102
B.1 C´alculo do n´ıveis de energia dos α3-pept´ıdeos . . . 102
C 107
C.1 Trabalhos publicados e submetidos . . . 107
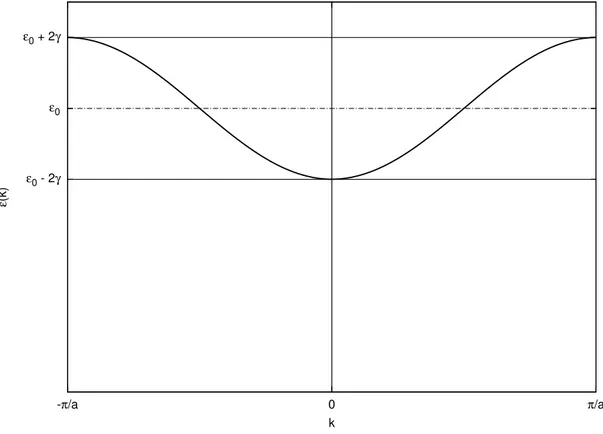
2.1 Curva de dispers˜aoǫ(k) versusk na primeira zona de Brillouin|k| ≤π/a.. . . 8
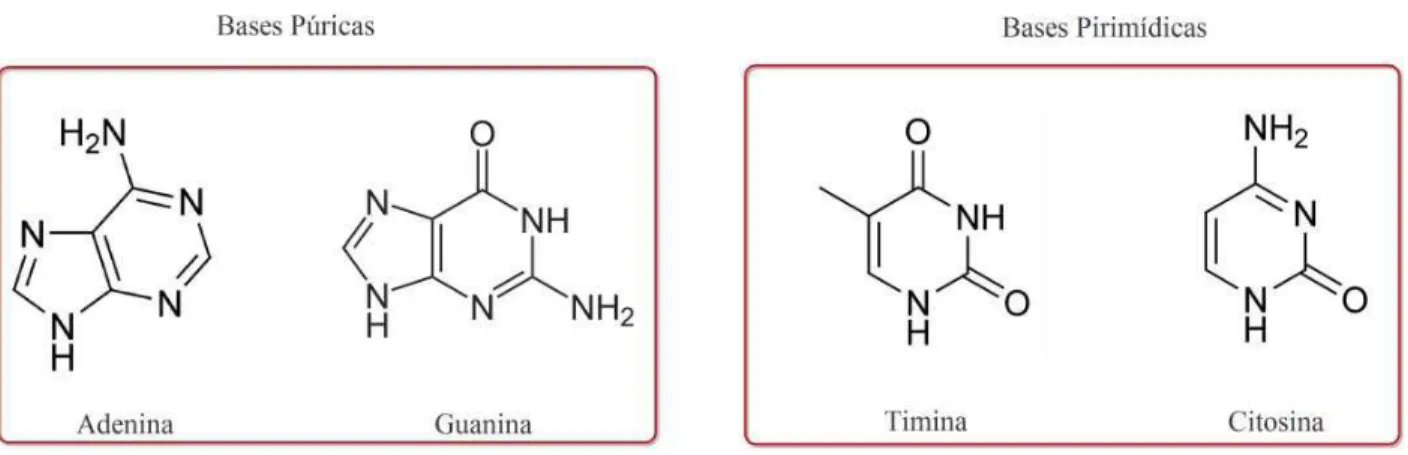
2.2 Bases nitrogenadas que comp˜oem o DNA. . . 15
2.3 Componentes estruturais do esqueleto do DNA. . . 16
2.4 Parte de uma seq¨uˆencia do DNA. Esqueleto do DNA em destaque. . . 16
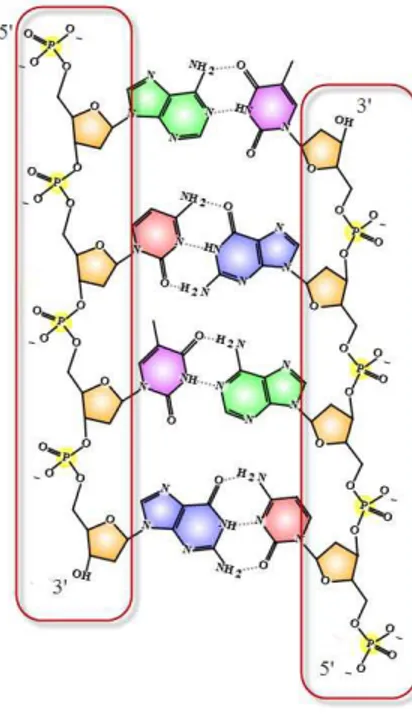
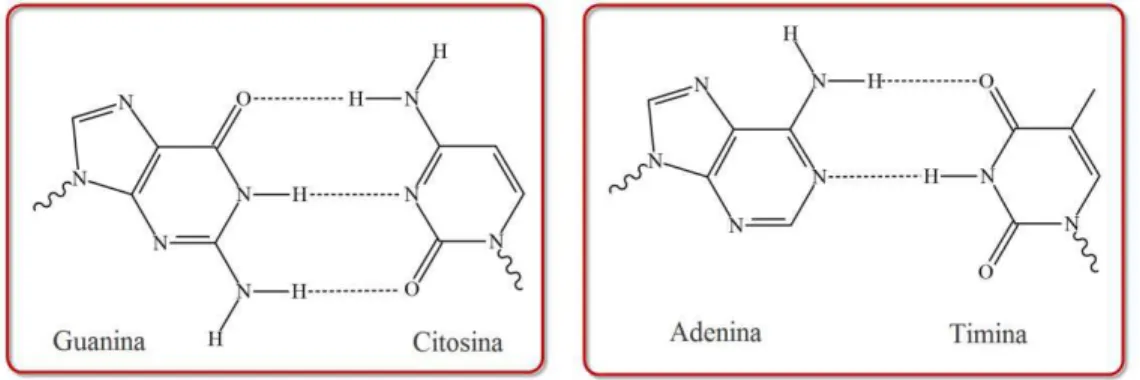
2.5 Pontes de hidrogˆenio entre as bases que formam o DNA. . . 17
2.6 Tipos de conforma¸c˜oes do DNA. Da esquerda para a direita, DNA tipo A, B e Z.. . . 18
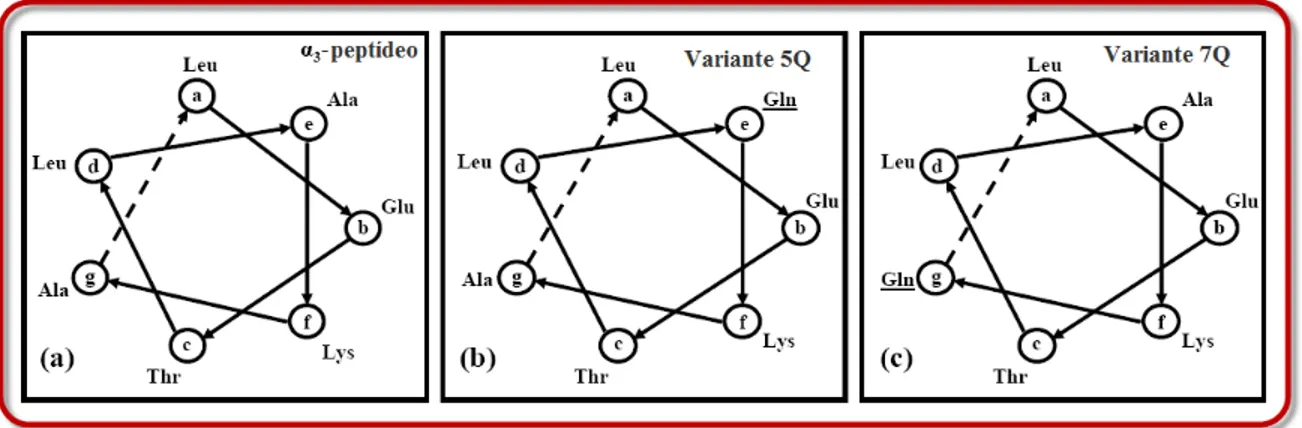
2.7 a) Representa¸c˜ao do polipet´ıdeo α3. A seq¨uˆencia com 7 res´ıduos Leu-Glu-Thr-Leu-Ala-Lys-Ala ´e repetida 3 vezes. b) Variante 5Q produzida pela subistitui¸c˜ao do res´ıduo Ala por Gln na posi¸c˜ao e (5a) na seq¨uˆencia α 3-pept´ıdeo. c) Variante 7Q produzida pela subistitui¸c˜ao do res´ıduo Ala por Gln na posi¸c˜aog (7a) na seq¨uˆencia α 3-pept´ıdeo. 19 2.8 Representa¸c˜ao do grupo carboxila (em destaque). . . 20
2.9 Representa¸c˜ao do grupo amina (em destaque). Amina prim´aria.. . . 20
2.10 Esquema da rea¸c˜ao de forma¸c˜ao de um amino´acido. . . 20
3.1 Primeiras gera¸c˜oes da seq¨uˆencia de Fibonacci. . . 33
3.2 Primeiras gera¸c˜oes da seq¨uˆencia de Rudin-Shapiro. . . 34
3.3 Representa¸c˜ao diagram´atica da mol´ecula do DNA planificada (modelo de escada). . . 35
3.4 Representa¸c˜ao do modelo de dupla fita do DNA.. . . 36
seq¨uˆencias de: (a) Fibonacci; (b) Rudin-Shapiro; (c) aleat´oria; (d) cromossomo humano Ch22, respectivamente. . . 53
4.1 Mol´ecula do pept´ıdeoα3 entre dois eletrodos. . . 63
4.2 Cadeia linear composta apenas pelos elementos do eletrodo. . . 65
4.3 Cadeia linear composta pelos elementos do eletrodo e por um elemento distinto. Na figura temos A0 =A2 =S. . . 66
4.4 Cadeia linear composta pelos elementos do eletrodo e por dois elementos distintos. Na figura temos A0 =A3 =S. . . 67
4.5 Densidade de estado eletrˆonica como fun¸c˜ao da energia, em eV, para as variantes 5Q (linha cheia) e 7Q (linha pontilhada). . . 71
4.6 Coeficiente de transmissividade TN(E) como fun¸c˜ao da energia, em eV, para as
vari-antes 5Q (linha cheia) e 7Q (linha pontilhada). . . 74
4.7 Expoente de Lyapunov γ(E) como fun¸c˜ao da energia E, em unidades de eV, para as duas configura¸c˜oes 5Q (linha cheia) e 7Q (linha pontilhada). Para a variante 5Q podemos observar a presen¸ca de 5 singularidades, o que sugere desordem efetiva infinita (comprimento de localiza¸c˜ao infinito). . . 75
4.8 Aproxima¸c˜ao da integral 4.77. . . 77
4.9 Caracter´ıstica da corrente como fun¸c˜ao da diferen¸ca de potencial para as duas con-figura¸c˜oes 5Q (linha cheia) e 7Q (linha pontilhada). Observe um comportamento de retificador para ambas variantes na forma de altas correntes a voltagens negativas quando comparado com as positivas. . . 78
4.10 Espalhamento da fun¸c˜ao de onda, definido pela raiz quadrada do desvio quadr´atico m´edio, como fun¸c˜ao do tempo, para as duas variantes 5Q e 7Q. . . 83
5.2 . . . 91
5.3 Potencial qu´ımico para as variantes 5Q (linha cheia) e 7Q (linha tracejada); (a) fra¸c˜ao de ocupa¸c˜aoNe/N = 2/7; (b) fra¸c˜ao de ocupa¸c˜aoNe/N = 4/7; (c) fra¸c˜ao de ocupa¸c˜ao
Ne/N = 5/6; (d) fra¸c˜ao de ocupa¸c˜ao Ne/N = 6/7. . . 92
5.4 Calor espec´ıfico fermiˆonico para a variante 5Q como fun¸c˜ao da temperatura. Note que o n´umero de oscila¸c˜oes no perfil do calor espec´ıfico aumenta com o aumento da fra¸c˜ao de ocupa¸c˜ao. . . 93
5.5 Calor espec´ıfico fermiˆonico para a variante 7Q como fun¸c˜ao da temperatura. Aqui vemos o mesmo comportamento exibido pela variante 5Q, isto ´e, aumento do n´umero de oscila¸c˜oes com o aumento da fra¸c˜ao de ocupa¸c˜ao. . . 94
5.6 Calor espec´ıfico fermiˆonico para as variantes 5Q (linha cheia) e 7Q (linha tracejada); (a) fra¸c˜ao de ocupa¸c˜ao Ne/N = 2/7; (b) fra¸c˜ao de ocupa¸c˜ao Ne/N = 4/7; (c) fra¸c˜ao
de ocupa¸c˜aoNe/N = 5/6; (d) fra¸c˜ao de ocupa¸c˜ao Ne/N = 6/7. . . 95
pept´ıdeos 5Q e 7Q α-h´elice (todas energias expressas em eV). . . 69
Cap´ıtulo
1
Introdu¸c˜
ao
O estudo te´orico de mol´eculas biologicamente ativas vem ganhando grande destaque atualmente. Devido aos importantes avan¸cos nos m´etodos da Mecˆanica Quˆantica e algoritmos computacionais, conduzido por um n´umero crescente de estudos computacionais devotados ao comportamento estrutural e conformacional dessas biomol´eculas, este tema vem se tornando, hoje em dia, um campo de pesquisa promissor (para uma lista atualizada de trabalhos ver Refs. [1–3]). As principais dificuldades encontradas nesta ´area s˜ao de natureza topol´ogica, bem como o alto grau de precis˜ao requerido na caracteriza¸c˜ao de tais biomol´eculas. Al´em disso, elas quase sempre apresentam um alto n´ıvel de complexidade, e devem, portanto, ser tratados por meio de m´etodos aproximativos [4; 5].
Atualmente, m´etodos computacionais tˆem obtido n˜ao apenas um conjuto de dados para valida¸c˜ao e parametriza¸c˜ao dos campos de for¸cas, largamente aplicados em simula¸c˜ao de dinˆamica molecular, mas tamb´em insights sobre efeitos qu´ımicos respons´aveis pela estabilidade de importantes biomol´eculas. Dentre eles, m´etodos ab initio baseados na solu¸c˜ao quˆantica do problema de intera¸c˜ao el´etron-´ıon, vˆem se revelando ´otimos candidatos para lidar com estes tipos de problemas. Contudo, na pr´atica, devido ao grande volume computacional e outros tipos de limita¸c˜oes, m´etodos ab initio tradicionais como Hartree-Fock e da fun¸c˜ao de onda correlacionada, s˜ao restritos a pequenas mol´eculas provendo, assim, um banco de dados limitado para o ajuste de parˆametros de potenciais emp´ıricos [6]. Felizmente, o desenvolvimento de poderosos programas computacionais vem superando este entrave, possibilitando o seu uso numa vasta gama de simula¸c˜oes de dinˆamica molecular (para artigos de revis˜ao ver Ref. [7]). Para ser mais preciso, m´etodos baseados na teoria do funcional da densidade de
O entendimento do mecanismo e da dinˆamica do transporte eletrˆonico dos portadores de carga (el´etrons e buracos) [10] no DNA, ´e de crucial importˆancia para a engenharia de disposi-tivos nanoeletrˆonicos baseados no DNA. Diferentemente das prote´ınas, a quest˜ao do transporte eletrˆonico na mol´ecula do DNA n˜ao ´e simplesmente um problema de transferˆencia el´etron-buraco. A raz˜ao para isto baseia-se no mecanismo propriamente dito. Ele falha ao explicar a persistˆencia da eficiˆencia dos transportadores de carga quando as taxas de transferˆencia n˜ao diminuem rapidamente com a distˆancia [11]. Assim sua aplicabilidade como um potencial dis-positivo nanoeletrˆonico molecular n˜ao pode depender apenas da transferˆencia de longo alcance de el´etrons e buracos atrav´es da mol´ecula. No entanto, seu arrranjo π dos pares da base, aqui considerados como uma seq¨uˆencia simb´olica das quatro letras do alfabeto representando os quatros diferentes nucleot´ıdeos, realmente nos d´a o apropriado caminho para o transporte de carga de longo alcance [12; 13], embora o mecanismo de transporte de longo alcance e o da transferˆencia de curto alcance possam ser completamente diferentes [14; 15].
Outros biopol´ımeros igualmente importante para os seres vivos s˜ao as prote´ınas. A conforma¸c˜ao estrutural das prote´ınas, bem como sua composi¸c˜ao, est´a diretamente ligada `a fun¸c˜ao que as mesmas exercem nos organismos dos seres vivos. Mudan¸cas na conforma¸c˜ao estrutural de uma dada prote´ına podem fazer com que ela assuma uma fun¸c˜ao prejudicial em nosso organismo, acarretando doen¸cas neuro-degenerativas como Alzheimer, Parkinson e v´arios tipos de cˆancer. Recentemente, foi sintetizada [16; 17] atrav´es do m´etodo de Novo (s´ıntese de mol´eculas complexas partindo de mol´eculas mais simples como amino´acidos e a¸c´ucares, em vez da degrada¸c˜ao parcial de mol´eculas maiores) um polipept´ıdeo α3. Este polipept´ıdeo ´e composto de 6 tipos de amino´acidos, a saber; a leucina (Leu), ´acido glutˆamico (Glu), Treonina (Thr), alanina (Ala), lisina (Lys) e glutamina (Gln). Estes amino´acidos s˜ao dispostos em cadeias de 7 elementos que se repetem trˆes vezes, formando um polipept´ıdeo com 21 amino´acidos. Tal polipept´ıdeo apresenta caracter´ısticas bastante distintas dependendo do amino´acido que ocupe a quinta e s´etima posi¸c˜oes da cadeia que se repete (presen¸ca, ou n˜ao, de estruturas fibrosas, que podem acarretar doen¸cas como as citadas anteriormente).
ioniza¸c˜ao e das integrais de transferˆencia), bem como o m´etodo de Dyson em conjunto com a t´ecnica da matriz de transferˆencia com um Hamiltoniano no contexto da liga¸c˜ao forte, adequado para descrever o movimento eletrˆonico numa cadeia polipept´ıdica. Dessa forma, obtemos a densidade de estado eletrˆonico (DOS) e a transmissividade eletrˆonica em cada variante do polipept´ıdeo α3. Al´em disso, resolvemos a equa¸c˜ao de Schr¨ondiger dependente do tempo para descrever a dinˆamica de um pacote de onda, inicialmente localizado, em um dado amino´acido da cadeia pept´ıdica.
Pretendemos tamb´em estudar as propriedades eletrˆonicas da mol´ecula de DNA pro-pondo, inicialmente, um modelo quasi-peri´odico da seq¨uˆencia de nucleot´ıdeos do DNA e com-parando este resultado com as propriedades f´ısicas de uma seq¨uˆencia de DNA encontrada na natureza, o cromossomo 22 do genoma humano Ch22. A quasi-periodicidade garante o
trans-porte de longo alcance devido a sua principal caracter´ıstica f´ısica, a autosimilaridade de sua estrutura.
No Cap´ıtulo 2 fazemos uma breve revis˜ao da mol´eucla do DNA e do polipept´ıdeo α3
em estudo, expondo suas variantes 5Q e 7Q. Revisamos tamb´em o modelo aplicado no estudo de tais mol´eculas, o modelo da liga¸c˜ao forte (Tight-binding) e o modelo de localiza¸c˜ao eletrˆonica de Anderson. E, por fim, uma revis˜ao superficial do m´etodo ab initio.
No Cap´ıtulo 3 introduzimos os tipos de seq¨uˆencias empregadas para o estudo das pror-piedades eletrˆonicas do DNA, a saber as seq¨uˆencias quasi-peri´odicas de Fibonacci e Rudin-Shapiro. Em seguinda abordamos o c´alculo do coeficiente de transmissividade juntamente com a t´ecnica da matriz de transferˆencia. E por fim, a c´elebre f´ormula de Landauer-B¨uttiker, que ´e utilizada para obter o perfil I×V da cadeia do DNA.
No Cap´ıtulo 4 calculamos as propriedades eletrˆonicas de transporte (como a desisdade de estado, DOS), das variantes 5Q e 7Q, atrav´es da equa¸c˜ao de Dyson, bem como o expoente de Lyapunov (que est´a ligado diretamente ao comprimeto de localiza¸c˜ao do paconte de onda). Em seguinda, atrav´es da t´ecnica da matriz de transferˆencia, obtemos tamb´em o coeficiente de transmissividade e a curva corrente como fun¸c˜ao da diferen¸ca de potencial. Por ´ultimo, resolvendo a equa¸c˜ao de Schr¨odinger dependente do tempo, analisamos o espalhamento de um pacote de onda incialmente localizado.
No Cap´ıtulo 5 investigamos as propriedades t´ermicas das varintes 5Q e 7Q, utilizando para isto a estat´ıstica de Fermi-Dirac. Obtemos assim, o calor espec´ıfico quˆantico, bem como o potencial qu´ımico.
2.1
Introdu¸c˜
ao
Neste cap´ıtulo faremos uma breve revis˜ao do modelo da liga¸c˜ao forte (tight-binding model), o qual nos permite a obten¸c˜ao da estrutura de banda de um cristal (ou mol´ecula), energia do estado fundamental, entre outros parˆametros. Veremos ainda que ao contr´ario do modelo do el´etron livre (onde assume-se que as fun¸c˜oes de onda s˜ao delocalizadas de seus ´ıons, formando assim o que chamamos de n´uvem eletrˆonica), este modelo descreve estados eletrˆonicos no limite onde admite-se orbitais atˆomicos isolados. Em seguida, faremos uma breve descri¸c˜ao do modelo de Anderson uni-dimensional em que os el´etrons s˜ao as entidades m´oveis do sistema. Neste modelo, de sistema desordenado, a dinˆamica quˆantico-mecˆanica de certas entidades do sistema (como spins ou el´etrons, por exemplo), que eram descritas em termos da temperatura, passam, agora, a ser descritas atrav´es de saltos quˆanticos entre os s´ıtios da rede.
Neste cap´ıtulo anida discutiremos, brevemente, uma das mais importantes estruturas biol´ogicas, a saber a mol´ecula do DNA. Esta mol´ecula, que cont´em toda informa¸c˜ao gen´etica em trechos de sua cadeia chamados de genes, ´e um pol´ımero composto por unidades monom´ericas, os nucleot´ıdeos, ligadas a uma estrutura do tipo a¸c´ucar-fosfato. E fecharemos o cap´ıtulo com outro tipo de pol´ımero igualmente importante, as prote´ınas (tamb´em conhecidas como polipept´ıdeos), que s˜ao compostas por cadeias de amino´acidos unidos por liga¸c˜oes pept´ıdicas.
2.2
O Modelo Tight-binding
2.2.1
Formalismo da primeira quantiza¸c˜
ao ou da fun¸c˜
ao de onda
O modelo da liga¸c˜ao forte (tight-binding model) ´e um m´etodo aproximativo que inicia com fun¸c˜oes de ondas de ´atomos livres, tamb´em conhecido como combina¸c˜ao linear dos orbitais atˆomicos LCAO (linear combination of atomic orbitals) [18; 19]. Este modelo nos possibilita o c´alculo da estrutura de bandas, bem como a energia do estado fundamental utilizando um conjunto de fun¸c˜oes de ondas aproximativas, baseado na superposi¸c˜ao das fun¸c˜oes de ondas dos ´atomos isolados. O Modelo da liga¸c˜ao forte ´e comumente aplicado a materiais cristalinos onde as posi¸c˜oes atˆomicas est˜ao bem localizadas, isto ´e, os ´atomos est˜ao distribu´ıdos em s´ıtios que pertencem a uma rede com espa¸camento peri´odico, por´em ´e poss´ıvel tamb´em aplicar tal modelo a materias n˜ao cristalinos cujas posi¸c˜oes atˆomicas s˜ao determindas a posteriori. Assim, em primeira aproxima¸c˜ao, o Hamiltoniano do sistema ´e considerado como a soma do Hamiltoniano atˆomico de cada ´atomo da rede (modelo do el´etron livre), juntamente com as intera¸c˜oes entre diferentes s´ıtios atˆomicos que s˜ao tratadas como uma perturba¸c˜ao.
O Hamiltoniano do sistema, no contexto da liga¸c˜ao forte, ´e dado ent˜ao por
H(r) = X
j
Hat(r−rj) + ∆U(r), (2.1)
onde Hat(r−rj) ´e o Hamiltoniano do ´atomo num dado s´ıtio j da rede, e ∆U(r) ´e a corre¸c˜ao
adicionada devido `a presen¸ca dos orbitais vizinhos. Se considerarmos que a influˆencia de um ´atomo sobre seu vizinho ´e pequena, obtemos uma fun¸c˜ao de onda aproximada para um el´etron do sistema atrav´es da seguinte equa¸c˜ao
ψk(r) =
X
j
Ckjφ(r−rj), (2.2)
onde k´e o vetor de onda, φ(r−rj) ´e o orbital atˆomico centrado emrj e o somat´orio se estende
sobre todos os pontos do sistema. No caso da presen¸ca de potenciais peri´odicos, do teorema de Bloch vemos que os coeficientes Ckj devem ser dados por Ckj = N−
1
os seguintes elementos de matrizes
hk|H|ki=N−1X
j
X
m
exp [ik·(rj −rm)]hφm|H|φji, (2.4)
onde φm ≡φ(r−rm). Temos ainda que:
hφm|H|φji=
Z
d3rφ∗(r−rm)Hφ(r−rj). (2.5)
Fazendo R=r−rj e ρmj =rm−rj, a equa¸c˜ao 2.5 assume a seguinte forma:
hφm|H|φji=
Z
d3Rφ∗(R−(rm−rj))Hφ(R) =
Z
d3Rφ∗(R−ρmj)Hφ(R) (2.6)
Logo a equa¸c˜ao 2.4 ´e dada por:
hk|H|ki = N−1X
mj
exp (−ik·ρmj)
Z
d3Rφ∗(R−ρmj)Hφ(R) =
= X
ρmj
exp (−ik·ρmj)
Z
d3Rφ∗(R−ρmj)Hφ(R), (2.7)
onde as soma de N2 termos nos ´ındices j e m foi substitu´ıda por um soma de N termos na
Logo chegamos a seguinte rela¸c˜ao de dispers˜ao:
ǫ(k) = ǫ0−
X
ρjm6=0
exp (−ik·ρmj)γ(ρmj), (2.8)
onde ǫ0 e γ(ρmj) s˜ao dadas por:
ǫ0 = Z
d3Rφ∗(R)Hφ(R) (2.9)
γ(ρmj) = −
Z
d3Rφ∗(R−ρmj)Hφ(R). (2.10)
No caso de um sistema unidimensional, temos que os primeiros vizinhos do s´ıtio ms˜ao os s´ıtios m±1. Se o parˆametro de rede do sistema em quest˜ao for a, temos que ρmm±1 =∓a,
e sendo o sistema peri´odico γ = γ(ρmm+1) = γ(ρmm−1), temos que a rela¸c˜ao de dispers˜ao 2.8
pode ser expressa como
ǫ(k) = ǫ0−γ[exp (ika) + exp (−ika)] = ǫ0−2γcos (ka). (2.11)
O comportamento da rela¸c˜ao de dispers˜ao 2.11 na primeira zona de Brillouin, isto ´e |k| ≤π/a, encontra-se na figura 2.1.
2.2.2
Formalismo da segunda quantiza¸c˜
ao ou do n´
umero de ocupa¸c˜
ao
O Hamiltoniano do modelo da liga¸c˜ao forte, equa¸c˜ao 2.1, pode ser expresso em ter-mos dos operadoes de cria¸c˜ao e aniquila¸c˜ao, formalismo este conhecido como segunda quan-tiza¸c˜ao. Neste formalismo, ´e frequentemente poss´ıvel, atrav´es de considera¸c˜oes f´ısicas, reduzir o Hamiltoniano para uma forma quadr´atica que pode ser ent˜ao diagonalizada por meio de uma transforma¸c˜ao canˆonica. Assim, dado o seguinte operador gen´erico
J =X
j
J(rj), (2.12)
ε0 - 2γ
-π/a 0 π/a
ε
(k)
k
Figura 2.1: Curva de dispers˜aoǫ(k) versuskna primeira zona de Brillouin |k| ≤π/a.
b
J =
Z
d3rψb†(r)J(r)ψb(r)
= X
kk′ Z
d3rψ∗(r)
kJ(r)ψk′(r)c†kck′
= X
kk′
hk|J|k′ic†kck′ (2.13)
onde ψb†(r) e ψb(r) s˜ao os operadores de campo definidos em termos dos operadores de cria¸c˜ao e aniquila¸c˜ao, c† e c, respectivamente por
b
ψ†(r) =X
k
ψ∗k(r)c
†
e
b
ψ(r) = X
k
ψk(r)ck. (2.15)
Nas equa¸c˜oes 2.14 e 2.15 a vari´avel k refere-se ao conjunto de n´umeros quˆanticos nescess´arios para descrever o sistema em estudo. Assim, para o Hamiltoniano 2.1 temos a seguinte representa¸c˜ao
b
H =X
kk′
hk|H|k′ic†kck′, (2.16)
onde o elemento de matriz hk|H|k′i´e dado por:
hk|H|k′i= Z
d3r Z
d3r′hk|rihr|H|r′ihr′|k′i= Z
d3r Z
d3r′ψ∗k(r)hr|H|r′iψk′(r′). (2.17)
Utilizando a defini¸c˜ao do Hamiltoniano 2.1, o elemento de matriz hr|H|r′i assume a seguinte forma na representa¸c˜ao da posi¸c˜ao:
hr|H|r′i = hr|X j
Hat(r−rj) + ∆U(r)|r′i=
= X
j
hr|Hat(r−rj)|r′i+ ∆U(r)hr|r′i=
= X
j
Hat(r−rj)δ(r−r′) + ∆U(r)δ(r−r′), (2.18)
assim a equa¸c˜ao 2.17 pode ser escrita como:
hk|H|k′i= = X
j
Z
d3rψk∗(r)Hat(r−rj)ψk′(r) + Z
d3rψk∗(r)∆U(r)ψk′(r)
= X
j
b
H = X
kk′ X
j
ǫj(k)δkk′c†kck′ + X
kk′
tkk′c†kck’
= X
k
(ǫk+tk)nbk+
X
k6=k′
tkk′c†kck′, (2.21)
comǫk=Pjǫj(k),tk =tkk ebnk =c†kck′, chamado de operador n´umero de part´ıculas. Notemos que o somat´orio em k pode ser incorporado ao potencial qu´ımico no formalismo do ensemble grande canˆonico, de forma que o Hamiltonaino efetivo para o sistema em quest˜ao fica
b
Hef f =
X
k6=k′
tkk′c†kck′. (2.22)
Novamente, no caso de um sistema unidimensional, levando em conta apenas intera¸c˜ao do tipo primeiros vizinhos temos que:
b
Hef f =−t
X
j
(c†jcj+1+c†j+1cj) = −t
X
j
(c†jcj+1+h.c), (2.23)
onde assumimos quet =−tjj+1 =−tjj−1eh.cdenota o conjugado hermitiano do termo anterior
no somat´orio.
2.3
O Modelo de Anderson
por Anderson, considerava-se o movimento eletrˆonico apenas sob a influˆencia de um potencial aleat´orio devido `a desordem no meio, por´em, atualmente, estudos recentes motivam `a inclus˜ao de correla¸c˜oes na desordem. No regime de baixas temperaturas, T →0, Anderson mostrou que h´a a ocorrˆencia de uma transi¸c˜ao met´alica para uma fase isolante (transi¸c˜ao metal-isolante in-duzinda por desordem) conhecida como transi¸c˜ao de Anderson [21–25]. O modelo de Anderson prevˆe que este tipo de transi¸c˜ao n˜ao ocorre em dimens˜ao (para correla¸c˜ao de curto alcance)
D ≤ 2, onde os estados estacion´arios eletrˆonicos s˜ao exponecialmente localizados, isto ´e, a fun¸c˜ao de onda eletrˆonica apresenta o seguinte comportamento
ψ(r)∼exp (r−r0
λ ), (2.24)
onde λ ´e chamado de comprimento de localiza¸c˜ao eletrˆonico. Por outro lado, se levarmos em conta intera¸c˜oes de longo alcance, a transi¸c˜ao de um estado eletrˆonico localizado para um delocalizado pode ocorrer mesmo em sistemas unidimensionais desordenados.
2.3.1
Introdu¸c˜
ao ao modelo de Anderson
Consideremos um sistema unidimensional composto porN s´ıtios, destribu´ıdos uniforme-mente ou aleatoriauniforme-mente, no qual os el´etrons podem se locomover. Cada el´etron pode ocupar um dado s´ıtioj, apresentando assim, energiaǫj. A desordem nesta cadeia linear ´e caracterizada
pelo fato das energias ǫj’s estarem destribu´ıdas aleatoreamente pelos s´ıtios da rede dentro do
intervalo [−W/2, W/2], onde W ´e chamada de largura da desordem. Neste modelo, a dinˆamica eletrˆonica se d´a atrav´es de saltos eletrˆonicos (hopping) entre os s´ıtios i e j (n˜ao nescessaria-mente primeiros vizinhos), que ´e dado pela integral de transferˆencia 2.20. O Hamiltoniano de Anderson pode ent˜ao ser escrito como
H =X
n
ǫn|nihn|+
X
m6=n
tmn|mihn|, (2.25)
0 0 0 · · · ǫn−2 tn−2n−1 0
0 0 0 · · · tn−1n−2 ǫn−1 tn−1n
0 0 0 · · · 0 tnn−1 ǫn
Como antes, |ni representa o orbital atˆomico centrado no s´ıtio n, isto ´e, hr|ni ´e a fun¸c˜ao de onda de um el´etron localizado no s´ıtio n. O conjunto formado por {|ni} representa, no contexto do modelo da liga¸c˜ao forte, uma base ortonornal hn|mi=δnm. Assim, expandindo
2.25 nesta base temos
Eψn =ǫnψn+t(ψn+1+ψn−1), (2.27)
onde considerou-se hoppings iguais entre os primeiros vizinhos. No caso de um s´olido cristalino (W = 0), isto ´e, onde todas as energias ǫn s˜ao iguais (e sem perda de generalidade podem ser
feitas iguais a zero) temos que
Eψn=t(ψn+1+ψn−1). (2.28)
Temos tamb´em que ψn pode ser escrita como (do teorema de Bloch)
ψn =ψ0exp (ink), (2.29)
de modo que a equa¸c˜ao 2.28 se escreve
Eψ0exp (ink) = t[ψ0exp (ink) exp (ik) +ψ0exp (ink) exp (−ik)]
Da equa¸c˜ao 2.30 vemos que as energias acess´ıveis ao sistema se encontram na regi˜ao onde −2t ≤ E ≤ 2t. Dessa maneira temos que a largura da banda cristalina ´e B = 4t. De forma geral, para um sistema com z primeiros vizinhos (n´umero de coordena¸c˜ao z) e hopping
constante entre tais vizinhos, a largura da banda para um sistema de dimens˜ao D pode ser escrita como B = 2zt.
A propriedade mais importante do modelo de Anderson para um s´olido desordenado encontra-se na raz˜ao W/B entre a largura de destribui¸c˜ao dos potencias e a banda de en-ergias permitidas para um el´etron no s´olido. Dessa forma, se a raz˜ao W/B for suficiente-mente grande, todos os estados eletrˆonicos s˜ao exponencialsuficiente-mente localizados, isto ´e, W > B ⇒
localiza¸c˜ao eletrˆonica. Por´em, se W ≈ B ⇒ desordem intermedi´aria, isto ´e, estados estendi-dos e localizaestendi-dos podem existir simultaneamente no s´olido. Para entendermos como os estaestendi-dos eletrˆonicos s˜ao suscept´ıveis `a localiza¸c˜ao na presen¸ca de um potencial desordenado, basta ana-lisarmos o movimento eletrˆonico entre dois s´ıtios vizinhos. Se a diferen¸ca de energia entre dois s´ıtios vizinhos i e j for, no m´aximo, da ordem da largura da banda, na verdade da ordem de
B/z, o el´etron pode mover-se entre os s´ıtio i e j. Por outro lado, se a diferen¸ca for maior que
B/z, n˜ao haver´a transporte eletrˆonico entre tais s´ıtios, de forma que tudo se passa como se tais s´ıtios fossem desacoplados.
2.4
A Mol´
ecula do DNA
Desde os prim´ordios da hist´oria humana, o homem desejava saber como os tra¸cos s˜ao herdados de uma gera¸c˜ao para outra. Apesar de crian¸cas parecerem mais com um dos pais do que com o outro, as caracter´ısticas herdadas parecem ser um conjunto herdado de ambos os pais. Centenas de anos de reprodu¸c˜ao de plantas e animais dom´esticos mostraram que certos tra¸cos ´
nenhuma fun¸c˜ao celular espec´ıfica. Enquanto que com rela¸c˜ao as prote´ınas, j´a se conheciam suas fun¸c˜oes enzim´aticas e estruturais nas c´eluas vivas. Tamb´em era de conhecimento que as prote´ınas, conhecidas como polipept´ıdeos, eram compostas por in´umeros amino´acidos, sendo 20 os mais importantes para o ser humano. Assim, era de se esperar que o alfabeto dos 20 amino´acidos fosse o respos´avel pela transmiss˜ao da informa¸c˜ao gen´etica, comparado com o al-fabeto de quatro letras do DNA.
Na d´ecada de 20 do s´eculo XX, experimentos mostraram que uma certa esp´ecie de bact´eria inofensiva poderia se tornar infecciosa quando misturada a uma esp´ecie de bact´eria (morta) extremamente agressiva. A bact´eria morta aparentemente provinha algum suporte qu´ımico que transformava a bact´eria inofensiva em infecciosa. Este, ent˜ao chamado, princ´ıpio da transforma¸c˜ao [31] parecia ser um gene. Um grupo de cientistas liderados por Oswald Avery doRockefeller Institute [32], seguiram de forma rigorosa este tipo de experimento na d´ecada de 40. E, em seguida, mostraram que um gene ´e composto pelo DNA, apesar da grande relutˆancia por parte de muitos cientistas, esta era uma prova de que o DNA, e n˜ao as prote´ınas, era a mol´ecula gen´etica.
Figura 2.2: Bases nitrogenadas que comp˜oem o DNA.
Na tentativa de se obter a conforma¸c˜ao estrutural da mol´ecula do DNA, uma s´erie de experimentos mostraram que a raz˜ao de adeninas para timinas, e a raz˜ao de guaninas para citosinas eram aproximadamente constantes em todos os seres vivos. Em seguida, experimentos de raio X proveram o caminho final para a estrutura de dupla fita torcida do DNA. Em 1953, a corrida para determinar como as pe¸cas deste quebra-cabe¸ca se encaixavam em uma estrutura tridimensional, culminou com a descoberta de James Watson e Francis Crick do Cavendish Laboratory em Cambridge [33–35]. Eles apresentaram um modelo de escada torcida, onde os corrim˜oes da escada eram compostos pelas mol´eculas da desoxirribose e fosfato, e os degraus da escada eram formados por pares de bases nitrogenadas.
A intera¸c˜ao do grupamento fosfato com o ´acido fosf´orico (H3PO4), d´a origem a
Figura 2.3: Componentes estruturais do esqueleto do DNA.
Figura 2.4: Parte de uma seq¨uˆencia do DNA. Esqueleto do DNA em destaque.
A liga¸c˜ao da base nitrogenada com o esqueleto a¸c´ucar-fosfato ´e muito mais forte que a liga¸c˜ao entre as duas fitas do DNA (liga¸c˜ao covalente), que ´e realizada via ponte de hidrogˆenio. Assim sendo, quando se aumenta a temperatura da regi˜ao onde se encontra a mol´ecula, as pontes de hidrogˆenio v˜ao se desfazendo muito antes da estrutura molecular come¸car a se desfazer (como um zipper se abrindo). Dessa forma, existir´a uma temperatura cr´ıtica TC para a qual todas
Figura 2.5: Pontes de hidrogˆenio entre as bases que formam o DNA.
Figura 2.6: Tipos de conforma¸c˜oes do DNA. Da esquerda para a direita, DNA tipo A, B e Z.
2.5
A mol´
ecula
α
-h´
elice e suas variantes 5Q e 7Q
Um tipo importante de conforma¸c˜ao estrutural em prote´ınas ´e conhecido comoα-h´elice, freq¨uentemente detectada em sua topologia tridimensional, que apresenta um papel fundamen-tal na estabilidade e dobragem (folding) das prote´ınas. A rela¸c˜ao entre a estabilidade e o tipo de seq¨uˆencia que comp˜oe as prote´ınas, bem como outras intera¸c˜oes entre cadeias laterais e a tendˆencia intr´ıseca dos amino´acidos em formarem h´elices, vem sendo estudada exaustiva-mente [47]. Este tipo de conforma¸c˜ao estrutural foi primeiraexaustiva-mente proposta por Pauling [48] e, em seguida, confirmada para diversas prote´ınas atrav´es da determina¸c˜ao de suas estruturas tridimensionais por raio X [49].
Recentemente foi sintetizado, por meio de engenharia gen´etica, e caracterizado, um polipept´ıdeo α3 com 21 res´ıduos, formado pela repeti¸c˜ao de sete amino´acidos, cuja seq6uˆencia ´e a seguinte: Leu-Glu-Thr-Leu-Ala-Lys-Ala. Este polipept´ıdeo apresenta um comportamento anfip´atico, isto ´e, uma regi˜ao hidrof´obica (neste caso a superf´ıcie hidrof´obica da leucina), e uma regi˜ao hidrof´ılica (devido `as superf´ıcies hidrof´ılicas da lisina e do ´acido glutˆamico). A forma¸c˜ao da α-h´elice foi confirmada pela an´alise de infravermelho por transformada de Fourier (FTIR).
glutamina (Glu), nas posi¸c˜oese(5a) eg(7a) noα3-pept´ıdeo (ver Fig. 2.7 ), mostram importantes
diferen¸cas na forma¸c˜ao da α-h´elice: o 5Qα3-pept´ıdeo (onde Q ´e um s´ımbolo alternativo para a glutamina) obtido pela substitui¸c˜ao Ala→Glu na posi¸c˜ao edoα3-pept´ıdeo original, mostrou a
presen¸ca de pequenas fibras de formato el´ıptico alongado, enquanto que o 7Qα3-pept´ıdeo com a substitui¸c˜ao Ala → Glu na posi¸c˜aog perdeu a habilidade de forma¸c˜ao de tal tipo de estrutura, apesar de ambos serem α-h´elices. De agora em diante chamaremos tais varia¸c˜oes de variante 5Q e 7Q. Estes resultados indicam que a presen¸ca da Ala na posi¸c˜ao g ´e a chave fundamental para a forma¸c˜ao de estruturas fibrosas [17].
Nos ´ultimos anos, foram identificadas v´arias patologias humanas e animais que resul-tam de folding incorreto ou de baixa estabilidade de certas prote´ınas. Desse modo, o problema dofolding e estabilidade prot´eica deixou de ser um campo de interesse restrito aos bioqu´ımicos para se tornar um t´opico essencial na compreens˜ao dos mecanismos moleculares de diversas patologias. Entre as doen¸cas mais conhecidas causadas por folding incorreto ou agrega¸c˜ao de prote´ınas encontram-se a doen¸ca de Alzheimer e a doen¸ca de Parkinson [50]. O caso mais re-portado de altera¸c˜oes fibrosas anormais de polipept´ıdeos ´e o do pr´ıon (agregado supramolecular acelulado) causador de v´arias doen¸cas, como a encefalopatia espongiforme bovina (vulgarmente conhecida como ”doen¸ca da vaca louca”) [51].
Figura 2.7: a) Representa¸c˜ao do polipet´ıdeoα3. A seq¨uˆencia com 7 res´ıduos Leu-Glu-Thr-Leu-Ala-Lys-Ala ´e repetida 3 vezes. b) Variante 5Q produzida pela subistitui¸c˜ao do res´ıduo Ala por Gln na posi¸c˜ao e(5a) na seq¨uˆencia α
3-pept´ıdeo. c) Variante 7Q produzida pela subistitui¸c˜ao do res´ıduo Ala por Gln na posi¸c˜aog (7a) na seq¨uˆencia α
3-pept´ıdeo.
Figura 2.8: Representa¸c˜ao do grupo carboxila (em destaque).
Figura 2.9: Representa¸c˜ao do grupo amina (em destaque). Amina prim´aria.
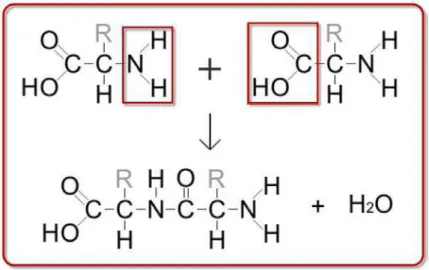
ainda, ativamente em mecanimos imunol´ogicos de defesa (anticorpos). Do ponto de vista qu´ımico, as prote´ınas s˜ao compostas por α-amino´acidos ligados entre si por liga¸c˜oes pept´ıdicas, isto ´e, uma liga¸c˜ao qu´ımica que ocorre entre duas mol´eculas quando o grupo carboxila (Fig. 2.8) de uma mol´ecula reage com o grupo amina (Fig. 2.9) de outra mol´ecula, liberando uma mol´ecula de ´agua H2O. Trata-se assim de uma rea¸c˜ao de desidrata¸c˜ao que ocorre entre mol´eculas de
amino´acidos (Fig. 2.9).
Figura 2.10: Esquema da rea¸c˜ao de forma¸c˜ao de um amino´acido.
um polipept´ıdeo.
Os polipept´ıdeos naturais s˜ao capazes de exercer suas fun¸c˜oes biol´ogicas gra¸cas `as seq¨uˆencias ordenadas dos amino´acidos que as comp˜oem, e de seu arranjo tridimensional bem determinado. Dessa forma, a primeira etapa no estudo de um (poli)pept´ıdeo, ou prote´ına, consiste na determina¸c˜ao da seq¨uˆencia de amino´acidos, a chamada estrutura prim´aria. Com o aparecimento de t´ecnicas mais sofisticadas, detalhes cada vez mais complicados puderam ser observadas. Tais detalhes incluem a natueza das rela¸c˜oes espaciais dos amino´acidos pr´oximos, a chamada estrutura secund´aria, e a disposi¸c˜ao espacial da cadeia pept´ıdica, estrutura terci´aria, bem como as rela¸c˜oes entre duas ou mais cadeias polipet´ıdicas, a estrutura quatern´aria.
2.6
O Modelo
ab initio
Nesta se¸c˜ao, faremos uma exposi¸c˜ao das principais teorias e conceitos que d˜ao suporte `as metodologias de c´alculo e c´odigos adotados no estudo dos α3-pept´ıdeos. A predi¸c˜ao de propriedades eletrˆonicas como energia de ioniza¸c˜ao e integrais de transferˆencia (hopping) a partir de c´alculos de primeiros princ´ıpios ´e um tema dominante na f´ısica do Estado S´olido.
2.6.1
Aproxima¸c˜
ao de Born-Oppenheimer
O Hamiltoniano de um sistema constitu´ıdo por Ne el´etrons e por Nn n´ucleos atˆomicos,
possui termos de intera¸c˜ao coulombiana el´etron-n´ucleo, el´etron-el´etron e n´ucleo-n´ucleo, como podemos observar na equa¸c˜ao 2.31
H =−~2 2 Ne P i=1 ∇2 i
me −
~2 2 Nn P I=1 ∇2 I
MI −
1 4πε0
Ne P i=1 Nn P I=1
e2ZI
|ri−RI|+
+4πε10
Ne
P
i=1
Ne
P
j=i+1
e2
|ri−rj|+
1 4πε0
Nn
P
I=1
Nn
P
J=I+1
e2Z IZJ
|RI−RJ|.
(2.31)
Te(r) =− ~2 2 Ne X i=1 ∇2 i me , (2.33)
Tn(R) =−
~2 2 Nn X I=1 ∇2 I MI , (2.34)
Ve,n(r, R) = −
1 4πε0 Ne X i=1 Nn X I=1 e2Z
I
|ri −RI|
, (2.35)
Ve,e(r) =
1 4πε0
Ne
X
i=1
Ne
X
j=i+1 e2
|ri−rj|
, (2.36)
Vn,n(R) =
1 4πε0 Nn X I=1 Nn X
J=I+1 e2Z
IZJ
|RI −RJ|
. (2.37)
Os n´ucleos possuem mais massa que os el´etrons de modo que o termo de energia cin´etica nuclear pode ser considerado pequeno se comparado com a energia cin´etica dos el´etrons. A aproxima¸c˜ao de Born-Oppenheimer consiste em considerar que o movimento dos n´ucleos n˜ao influencia no movimento dos el´etrons, o que equivale a dizer que os n´ucleos est˜ao em repouso, onde cada um deles comparece como uma carga positiva externa envolvida por uma nuvem eletrˆonica. Como conseq¨uˆencia, o termo de energia cin´etica nuclear n˜ao figura na equa¸c˜ao 2.31 e o termo de energia potencial nuclear se reduz a uma constante. O Hamiltoniano descrito na equa¸c˜ao 2.31 reduz-se a
2.6.2
Teoria do Funcional da Densidade (DFT)
Os principais m´etodos aplicados ao estudo de sistemas com muitos el´etrons pressup˜oem a determina¸c˜ao da fun¸c˜ao de onda |Ψi dos el´etrons constituintes como fundamento para a obten¸c˜ao de propriedades f´ısicas de interesse. No entanto, a fun¸c˜ao de onda de um sistema de muitos corpos composto por Ne el´etrons depender´a de 4Ne vari´aveis, 3Ne coordenadas espaciais
e Ne coordenadas de spin. Assim, a resolu¸c˜ao da equa¸c˜ao de autovalor para uma fun¸c˜ao de
onda com um n´umero t˜ao grande de vari´aveis ´e impratic´avel e, mesmo em alguns casos mais restritos, a interpreta¸c˜ao adequada dos processos f´ısicos ´e comprometida pela complexidade das equa¸c˜oes envolvidas.
Os tratamentos mais simples, que prescindem do c´alculo direto da fun¸c˜ao de onda, baseados em uma aproxima¸c˜ao de campo m´edio, onde os el´etrons se deslocam como part´ıculas independentes em um potencial efetivo criado por ´ıons e por outros el´etrons, fornecem uma solu¸c˜ao bastante satisfat´oria para o problema de muitas part´ıculas. Uma teoria, chamada Teoria do Funcional da Densidade (DFT), formalmente estabelecida por Hohenberg e Kohn [54] e depois desenvolvida por Kohn e Sham [55], tornou o tratamento do problema de muitos corpos menos dispendioso computacionalmente falando e com resultados de excelente qualidade.
A densidade eletrˆonica representa o n´umero de el´etrons que s˜ao encontrados num dado volume, sendo poss´ıvel obter a densidade de carga eletrˆonica a partir da densidade eletrˆonica multiplicando esta ´ultima grandeza pela carga do el´etron. Uma condi¸c˜ao necess´aria para a densidade eletrˆonica ´e que sua integral em todo o espa¸co deva ser igual ao n´umero de el´etrons do sistema. O fundamento da DFT ´e utilizar a densidade eletrˆonica expressa como fun¸c˜ao das trˆes coordenadas espaciais,ρe(r), para obter uma solu¸c˜ao da equa¸c˜ao de Schr¨odinger. Hohenberg
e Kohn propuseram dois teoremas que fundamentam a DFT, ambos envolvendo diretamente a densidade eletrˆonica do sistema. O primeiro teorema afirma:
• Primeiro teorema de Hohenberg-Kohn: O potencial externo Vext(r) ´e (a menos de
uma constante) um funcional ´unico de ρe(r); uma vez que V
ext(r) determina H, vemosˆ
que o estado fundamental completo de muitas part´ıculas ´e um funcional ´unico de ρe (r).
O primeiro teorema nos informa que o potencial externo Vext ´e especificado de modo
´
sistema em quest˜ao, isto ´e, s˜ao comuns a todos os sistemas independente do n´umero de el´etrons, das coordenadas e das cargas nucleares e s˜ao reunidos para formar o chamado funcional de Hohenberg-Kohn FHK.
FHK[ρe(r)] =Te[ρe(r)] +Ve,e[ρe(r)] (2.40)
A equa¸c˜ao 2.39 ´e reescrita na forma
E[ρe(r)] =F
HK[ρe(r)] +Ve,n(ρe(r)) (2.41)
onde ˆVe,n[ρe(r)] ´e um termo dependente do sistema. Quando o funcional de Hohenberg-Kohn
recebe uma densidade de carga arbitr´aria ρe(r) para operar, ele d´a como resultado o valor
esperado hΨ|Te+e,e|Ψi. Esta ´e a soma da energia cin´etica com o operador repuls˜ao
el´etron-el´etron para a fun¸c˜ao de onda do estado fundamental Ψ vinculada `a densidadeρe(r), de maneira
que Ψ, ´e dentre todas as fun¸c˜oes de onda, a que resulta no valor mais baixo para a energia. Isto ´e,
FHK[ρe(r)] =Te[ρe(r)] +Ve,e[ρe(r)] =hΨ|Te+Ve,e|Ψi. (2.42)
A determina¸c˜ao do funcional FHK ´e fundamental para a DFT. Se ele fosse conhecido
com exatid˜ao seria poss´ıvel resolver a equa¸c˜ao de Schr¨odinger para sistemas com poucos ou muitos ´atomos, uma vez que tal funcional independe do sistema. At´e o presente momento a forma exata de FHK n˜ao foi determinada.
O segundo teorema de Hohenberg-Kohn assegura que o funcional FHK aplicado `a
den-sidade eletrˆonica do estado fundamental, ρe
o, de um sistema fornecer´a a energia m´ınima deste.
• Segundo teorema de Hohenberg-Kohn: O funcional da energia do estado
Uma densidade eletrˆonica tentativa que satisfa¸ca as condi¸c˜oes de contorno do problema de muitos el´etrons e que est´a associada a um potencial externo fornecer´a um valor maior que a energia do estado fundamental E0. A energia ser´a igual a E0 somente se a densidade correta
para o estado fundamental for inserida na equa¸c˜ao 2.39.
2.6.3
O M´
etodo de Kohn-Sham
O teorema de Hohenberg-Kohn n˜ao diz como calcular a energia E0 a partir de ρe0(r),
pois o funcional FHK n˜ao est´a determinado e n˜ao mostra como encontrar ρe0(r) sem primeiro
encontrar a fun¸c˜ao de onda. Kohn e Sham [55] estabeleceram um m´etodo para calcular ρe
0(r)
e, em seguida, E0 a partir de ρe
0(r).
Kohn e Sham consideraram um sistema de referˆencia fict´ıcio, conhecido como sistema n˜ao-interagente, composto porNeel´etrons que se comportam totalmente independentes e
expe-rimentam a mesma energia potencialVs(ri). Ela ´e definida de forma que a densidade eletrˆonica
para o estado fundamentalρe
s(ri) do sistema de referˆencia seja igual `a densidade eletrˆonica para
o estado fundamental ρe
0(ri) do sistema real.
O Hamiltoniano para um sistema de el´etrons n˜ao interagentes ´e dado por
Hs = Ne
X
i=1
−2m~
e∇
2
i +Vs(ri)
≡
n
X
i=1
HiKS, (2.43)
onde HKS
i ´e o Hamiltoniano de um el´etron de Kohn-Sham. ´E poss´ıvel relacionar o sistema
fict´ıcio de referˆencia de Kohn-Sham ao sistema real escrevendo o Hamiltoniano
Hλ ≡Te+ Ne
X
i=1
Vλ(ri) +λVe,e, (2.44)
onde o parˆametro λ varia de 0 (sistema n˜ao interagente) at´e 1 (sistema real), e Vλ ´e o
po-tencial externo que definir´a a densidade eletrˆonica para o estado fundamental do sistema com Hamiltoniano Hλ igual a densidade para o estado fundamental do sistema real.
Kohn e Sham reescreveram a equa¸c˜ao de Hohenberg-Kohn a partir da defini¸c˜ao de uma quantidade ∆ ¯Te,s que ´e a diferen¸ca na energia cin´etica m´edia do estado fundamental entre a
∆ ¯Ve,e[ρe0(r)]≡V¯e,e[ρe0(r)]−
1 2
Z ρe
0(r)ρe0(r′)
|r−r′| d
3rd3r′, (2.46)
onde |r−r′|´e a distˆancia entre os pontos (x, y, z) e (x′, y′, z′), e a integral ´e a express˜ao cl´assica para a energia de repuls˜ao eletrost´atica entre dois el´etrons cujas cargas foram espalhadas segundo uma densidade de carga proporcional `a densidade eletrˆonica. Com estas defini¸c˜oes, a energia total ´e expressa por
EVEXT[ρ
e
0(r)] =
R
VEXT(r)ρe0(r)dr+ ¯Te,s[ρe0(r)] +
+12R ρe0(r)ρe0(r)
|r−r′| drdr
′ + ∆ ¯T
e,s[ρe0(r)] + ∆ ¯Ve,e[ρe0(r)].
(2.47)
Define-se o funcional de energia de troca e correla¸c˜ao, EXC[ρe(r)], pela express˜ao dada
a seguir, com os funcionais desconhecidos ∆ ¯Te,s e ∆ ¯Ve,e.
EXC[ρe(r)] = ∆ ¯Te,s[ρe(r)] + ∆ ¯Ve−e[ρe(r)]. (2.48)
Com isto, pode-se escrever a energia total do estado fundamental atrav´es da equa¸c˜ao
E0 = EVEXT [ρ
e
0(r)] =
Z
VEXT(r)ρe(r)dr+Te,s[ρe0(r)] +
+ 1
2 Z ρe
0(r)ρe0(r′)
|r−r′| drdr ′+E
XC[ρe0(r)]. (2.49)
real, ou seja, ρe
s =ρe0. Demonstra-se que [56]
ρe
0(r) = ρes(r) = Ne
X
i=1
θKS
i (ri)
2
, (2.50)
onde θKS
i s˜ao os orbitais de Kohn-Sham a serem determinados.
Os termos da equa¸c˜ao 2.47 guardam, ent˜ao, as seguintes rela¸c˜oes com a densidade eletrˆonica e os orbitais de Kohn-Sham:
Z
VEXT(r)ρe(r)dr=− Nn
X
I=1 ZI
Z ρe
0(r)
|r−RI|
dr. (2.51)
Te,s[ρe0(r)] =−
1 2hψs,0|
Ne
X
i=1
∇2
i |ψs,0i=−
1 2 Ne X i=1 θKS i (r)
∇2θKS i (r)
. (2.52)
Assim, a equa¸c˜ao 2.47 pode ser reescrita na forma
E0 = −
Nn X I=1 ZI Z ρe
0(r)
|r−RI|
dr− 1
2
Ne
X
i=1
θiKS(r)
∇2θKS i (r)
+
+ 1
2 Z ρe
0(r)ρe0(r′)
|r−r′| drdr ′+E
XC[ρe0(r)], (2.53)
de modo que o c´alculo de E0 a partir de ρe
0 pode ser efetuado quando conhecemos os orbitais
de Kohn-Sham θKS
i e o funcional EXC. O termo de energia que inclui a repuls˜ao nuclear,Vn,n,
deve ser acrescentado para levar em conta esta contribui¸c˜ao.
O segundo teorema de Kohn-Sham afirma que ´e poss´ıvel achar a energia do estado fundamental variando-se ρe, lembrando-se que ao mesmo imp˜oe o v´ınculo R ρe(r)dr = N
e, de
modo a minimizar o funcional E[ρe]. De forma equivalente, ´e poss´ıvel variar os orbitais de
Kohn-Sham θKS
i que determinam ρe como indica a equa¸c˜ao 2.48. Conseq¨uentemente, os orbitais de
onde VXC(r) ´e o potencial de troca e correla¸c˜ao obtido a partir da derivada funcional da energia
de troca e correla¸c˜ao, isto ´e,
VXC(r)≡
δEXC[ρe(r)]
δρe(r) , (2.55)
de modo que se conhecemos EXC sua derivada funcional pode ser calculada, e determina-se a
fun¸c˜ao VXC. No entanto, n˜ao se conhece o funcional correto EXC[ρe(r)] para se proceder com
o c´alculo de ρe e de E
0. Tal limita¸c˜ao conduz `a utiliza¸c˜ao de m´etodos aproximativos para a
determina¸c˜ao de EXC[ρe(r)].
2.7
Conclus˜
oes.
Cap´ıtulo
3
Propriedades de Transporte Eletrˆ
onico da
Mol´ecula do DNA
3.1
Introdu¸c˜
ao.
Devido `a possibilidade de aplica¸c˜oes em dispositivos eletrˆonicos, vem aumentando o interesse na s´ıntese, caracteriza¸c˜ao e estudo das propriedades eletrˆonicas de sistemas baseados na mol´ecula do DNA. Atrav´es de m´etodos bioqu´ımicos e f´ısicos, estudos vˆem comprovando que a dupla h´elice do DNA pode ser utilizada como um meio para o transporte eletrˆonico. Contudo, uma s´erie de estudos recentes sobre a condu¸c˜ao eletrˆonica em fibras de DNA mostraram con-clus˜oes bastante diversificadas. Algumas destas concon-clus˜oes sugerem alta mobilidade eletrˆonica atrav´es do DNA, enquanto outras apontam nenhuma condutividade. Pode-se encontrar at´e es-tudos que sugerem o DNA como um supercondutor. At´e o momento tais trabalhos n˜ao chegaram a um senso comum. A causa para tal variedade de resultados deve-se, provalvemente, `a maneira pela qual o DNA ´e conectado aos eletrodos, a sua integridade na ausˆencia de um meio aquoso e a exposi¸c˜ao `a altas diferen¸cas de potencial.
Dada a sua complexidade estrutural, quantificar o transporte eletrˆonico em cadeias de DNA tornou-se um experimento altamente tendencioso por causa da presen¸ca dos contatos (eletrodos), da intera¸c˜ao com algum substrato inorgˆanico e das condi¸c˜oes atmosf´ericas e de temperatura experimental. Portanto, o foco sobre a possibilidade do DNA ser capaz de mediar o transporte eletrˆonico mudou para como tal efeito acontence. Assim, estamos interessados
transporte eletrˆonico entre o doador e o aceptor d´a lugar, agora, a um novo paradigma onde as mol´eculas s˜ao vistas como os mediadores do transporte eletrˆonico, com observ´aveis como a taxa de transferˆencia eletrˆonica substitu´ıdo por rela¸c˜oes de corrente-voltagem, em jun¸c˜oes mo-leculares. De grande importˆancia ´e a nessecidade de entedermos a rela¸c˜ao entre as estruturas moleculares de tais jun¸c˜oes e suas fun¸c˜oes, isto ´e, suas propriedades de transmiss˜ao e a condu¸c˜ao. Tais investiga¸c˜oes, nas quais pequenas mol´eculas operam como condutores conectando compo-nentes eletrˆonicos tais como contatos met´alicos e semicondutores, constituem a maior parte do que hoje ´e conhecido como eletrˆonica molecular. A diversidade, versatilidade e viabilidade no controle e manipula¸c˜ao de tais dispositivos moleculares, os tornam componentes potencial-mente importantes na elebora¸c˜ao de dispositivos nanoeletrˆonicos. Neste contexto, um sistema eletrˆonico padr˜ao ´e formado por um doador, um aceptor e uma ponte molecular que os conec-tam. Nosso foco, neste cap´ıtulo, ´e a transferˆencia eletrˆonica entre os dois eletrodos atrav´es de um meio molecular. Tal desenvolvimento tem atra´ıdo bastante aten¸c˜ao da ind´ustria de semi-condutores, e grande interesse do ponto de vista aplicado na compreens˜ao da capacidade da condu¸c˜ao molecular. Ao mesmo tempo, este t´opico ´e de grande relevˆancia do ponto de vista da f´ısica b´asica existindo, assim, trˆes poss´ıveis mecanismos a serem considerados:
• Super troca (superexchange): as cargas tunelam do eletrodo doador (DN) para o receptor (AC) pela cadeia do DNA em um processo n˜ao adiab´atico. ´E previsto um decr´escimo exponencial na taxa de transporte de carga com o aumento do comprimento da cadeia do DNA.
• Saltos (hopping): as cargas presentes na cadeia do DNA atravessam a mol´ecula por meio de saltos entre os orbitais moleculares discretos do DNA.
• Saltos entre dom´ınios (Domain hopping): as cargas atravessam a cadeia do DNA delocal-izando sobre v´arias bases, formando assim um dom´ınio. Tal dom´ınio ent˜ao salta longas distˆancias atrav´es da mol´ecula do DNA, do eletrodo doador (DN) at´e o receptor (AC), at´e que haja localiza¸c˜ao eletrˆonica.
a mol´ecula do DNA unidas, as intera¸c˜oes entre os orbitais π das bases nitrogenadas ajudam a estabilizar a liga¸c˜ao entre as fitas. A configura¸cao eletrˆonica das bases que comp˜oem o DNA est˜ao descritas em detalhes na Ref. [57]. Dentre os v´arios modelos utilizados para se estudar a condu¸c˜ao de cargas de um ponto de vista te´orico, o que parece ser mais bem sucedido ´e o que considera a proposta feita em 1960 por Ladik de que os orbitais do tipo π presentes nas bases podem fornecer um caminho para as cargas. O transporte das cargas pode ser resultado de tunelamento coerente (superexchange) ou de saltos incorentes (envolvendo fˆonons).
Neste cap´ıtulo, discutiremos o c´alculo anal´ıtico e num´erico dos estados de um el´etrom em seguimentos de fita dupla do DNA. O modelo te´orico se baseia no Hamiltoniano no con-texto da liga¸c˜ao forte, juntamente com a t´ecnica da matriz de transferˆencia que nos permite simplificar a parte alg´ebrica. N´os consideramos um modelo no qual a mol´ecula do DNA ´e ”con-tida”entre os eletrodos (doador (DN) e receptor (AC), respectivamente), seguindo as seq¨uˆencias quasiperi´odicas de Fibonacci (FB) e Rudin-Shapiro (RS), e comparamos com seq¨uˆencias aleat´orias (RD) e com a parte da primeira seq¨uˆencia do cromossomo 22 do DNA humano (Ch22). A
seq¨uˆencia referente ao Ch22 foi obtida na p´agina do National Center of Biotechnology Informa-tion na internet. N´os investigamos a condu¸c˜ao eletrˆonica da mol´ecula do DNA por meio do seu coeficiente de transmiss˜ao e, resolvendo numericamente a equa¸c˜ao de Schr¨odinger indepen-dente do tempo, obtivemos tamb´em as caracter´ısticas de corrrente como fun¸c˜ao da diferen¸ca de potencial para todas as seq¨uˆencias de DNA acima citadas.
3.2
As Seq¨
uˆ
encias.
Conforme mencionado na se¸c˜ao 3.1, utilizamos seq¨uˆencias artificiais, a saber, as seq¨uˆencias de Fibonacci e Rudin-Shapiro. Estas duas foram escolhidas porque quando utilizadas em uma s´erie de outros sistemas f´ısicos foram as respons´aveis por apresentar espectros de energia alta-mente fragmentados, exibindo um padr˜ao autosimilar, pr´oprio de estruturas fractais, al´em de apresentarem correla¸c˜ao de longo-alcance.
Os n´umeros de Fibonacci formam uma seq¨uˆencia gerada de forma recursiva. Tal seq¨uˆencia foi introduzida por Leonardo de Pisa em 1202, tamb´em conhecido como Fibonacci (uma contra¸c˜ao de filius Bonaccio, filho de Bonaccio), em seu livro Liber Abaci. Por defini¸c˜ao os dois primeiros n´umeros de Fibonacci s˜ao 0 e 1. Dessa maneira todos os demais s˜ao obtidos destes pela seguinte rela¸c˜ao de recorrˆencia
Fn=Fn−1+Fn−2, (3.1)
com os seguintes valores
F0 = 0 e F1 = 1. (3.2)
Assim, utilizando a rela¸c˜ao de recorrˆencia 3.1 obt´em-se os seguintes n´umeros de Fi-bonacci: 0, 1, 1, 2, 3, 5, 8, 13, 21, 34, 55, 89, 144, 233, 377, 610, 987, 1597, 2584, 4181, 6765, 10946 ... No contexto de cria¸c˜ao da fita simples para o DNA, utilizaremos uma redefini¸c˜ao da rela¸c˜ao de recorrˆencia 3.1. A cadeia do DNA ´e ent˜ao crescida por um processo conhecido como
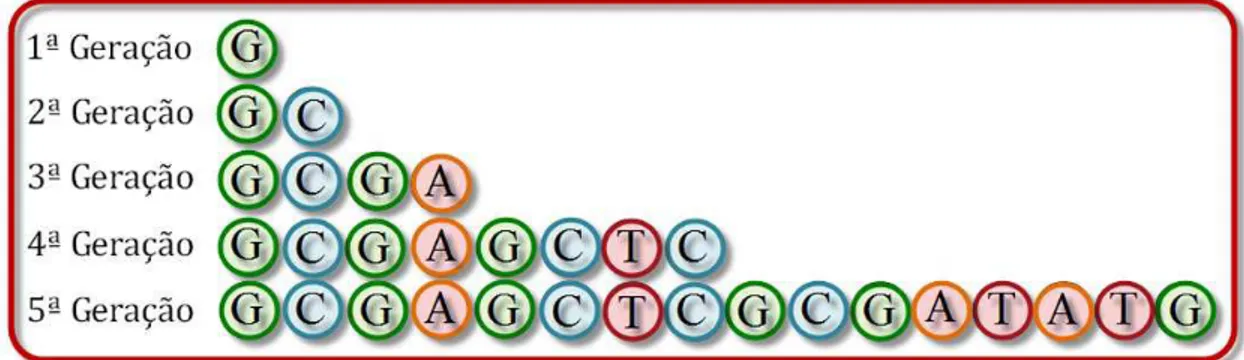
Infla¸c˜ao por Subistitui¸c˜ao de Palavras (ou em inglˆes,String Rewrite System). O processo ´e com-posto por duas partes: as regras (ou rela¸c˜oes de recorrˆencia) e a semente. Para a seq¨uˆencia de Fibonacci tem-se a seguinte regra: a guanina (G) deve ser substitu´ıda pelo par guanina-citosina (GC) e a citosina (C) deve ser substitu´ıda por uma guanina (G). A semente que utilizamos foi a guanina (G), isto ´e, a primeira gera¸c˜ao corresponde ao elemento G. A Fig 3.1. mostra algumas gera¸c˜oes obtidas desta maneira.
Figura 3.1: Primeiras gera¸c˜oes da seq¨uˆencia de Fibonacci.
para a mesma gera¸c˜ao, isto ´e, na quarta gera¸c˜ao de Fibonacci temos que F4 = 3 o qual ´e o mesmo n´umero de guaninas nesta gera¸c˜ao. Tem-se, ainda, que a raz˜ao do n´umero de guaninas e citosinas em cada gera¸c˜ao tende `a raz˜ao ´aurea definida como
xn =
Fn+1 Fn
= Fn+Fn−1
Fn
= 1 + Fn−1
Fn
= 1 + F1
n
Fn−1
= 1 + 1
xn−1
, (3.3)
como a seq¨uˆenciaxn ´e limitada e monotˆonica crescente ela ´e convergente, portanto,
lim
n→∞xn= limn→∞xn−1 =L, (3.4)
de onde obtemos a seguinte equa¸c˜ao quadr´atica, cuja raiz positva ´e chamada n´umero ´aureo, ou seja,
L2−L−1 = 0 =⇒φ= 1 +
√ 5
2 ≈1.61803. (3.5)
J´a a seq¨uˆencia de Rudin-Shapiro, tamb´em conheciada como seq¨uˆencia de Golay-Rudin-Shapiro, apresenta apenas dois poss´ıveis algarismos, a saber +1 e −1. O n-´esimo termo bn ´e
definido pelas seguintes regras
an=
X
i
ǫiǫi+1 e bn= (−1)an, (3.6)
onde ǫi s˜ao d´ıgitos da representa¸c˜ao bin´aria do ´ındice n de an. Assim, an conta o n´umero de
a6 =
3
X
i=1
ǫiǫi+1= 0·1 + 1·1 + 1·0 = 1, (3.8)
portanto, temos que b6 = (−1)1 =−1. Para o termo b7 ter´ıamos duas ocorrˆencias do par ”11”, de maneira que a7 = 2 e b7 = 1. Come¸cando com n = 0, os primeiros termos da seq¨uˆencia
an s˜ao: 0, 0, 0, 1, 0, 0, 1, 2, 0, 0, 0, 1, 1, 1, 2, 3 . . .. E a correspondente seq¨uˆencia bn fica
dada por: +1, +1, +1, -1, +1, +1, -1, +1, +1, +1, +1, -1, -1, -1, +1, -1, . . .. Assim, como no caso de Fibonacci, redefinimos as regras que formam a seq¨uˆencia afim de utilizarmos apenas as letras (os nucleot´ıdeos) que comp˜oem o DNA. Novamente temos um processo de Infla¸c˜ao por Subistitui¸c˜ao de Palavras. Desta vez a regra ´e a seguinte: a guanina (G) deve ser substitu´ıda pelo par (GC), a citosina (C) pelo par (GA), a adenosina (A) pelo par (TC), e a timina (T) pelo par (TA). A semente utilizada tamb´em foi a guanina. A Fig 3.2. mostra algumas gera¸c˜oes obtidas desta maneira.
Figura 3.3: Representa¸c˜ao diagram´atica da mol´ecula do DNA planificada (modelo de escada).
Uma vez criada uma das fitas do DNA, conforme dito anteriormente, a outra parte fica completamente determinada. Atrav´es de tal procedimento chegamos `a Fig. 3.3, onde cada nucleot´ıdeo ser´a representado por sua base nitrogenada no modelo em quest˜ao. O modelo consiste em duas cadeias de elementos dispostas paralelamente entre si. Tal modelo ´e conhecido como Ladder Model ou modelo de escada.
3.3
Transmitˆ
ancia
Figura 3.4: Representa¸c˜ao do modelo de dupla fita do DNA.
Na Fig. 3.4, a cada s´ıtio da dupla fita associamos um orbital quˆantico. Por´em, temos que destinguir em qual das duas fitas o s´ıtio se encontra. Para tal, vamos definir a seguinte base que gera o espa¸co das fun¸c˜oes de onda. Para a fita rotulada α temos a base {|αni}, onde
n rotula o s´ıtio na fita α. Para a fita β temos a base {|βmi}, de forma similar. Dessa forma, a base que gera a dupla fita ´e dada por {|ξki} (modelo da liga¸c˜ao forte), onde ξ pode assumir os valores α e β, e k varia de 1 a N (n´umero de elementos que comp˜oe a fita). Dessa maneira, temos a seguinte identidade
b1 =X
ξ
X
k
|ξkihkξ|, (3.9)
com a seguinte rela¸c˜ao de ortogonalidade
hξk|ζli=δξζδkl. (3.10)
No contexto da liga¸c˜ao forte, o Hamiltoniano da mol´ecula do DNA em um certo ponto da cadeia pode ser aproximado pelo Hamiltoniano Hnuc do nucleot´ıdeo naquele ponto, adicionado
de uma corre¸c˜ao ∆U devido `a presen¸ca dos orbitais vizinhos.
localizadas (fortemente ligadas aos nucleot´ıdeos), isto ´e, se ψξ
n(r) ´e uma fun¸c˜ao de onda tal que
Hnucψkξ =ǫ ξ kψ
ξ
k, (3.11)
onde ǫξk ´e a energia do orbital localizado no s´ıto k e na fita ξ, ent˜ao ψkξ(r) → 0 quando |r| for da ordem da distˆancia entre os nucleot´ıdeos. Dessa forma, o Hamiltoniano se escreve
H =Hnuc+ ∆U. (3.12)
Do modelo da liga¸c˜ao forte, temos ent˜ao que o orbital molecular do DNA pode ser escrito como combina¸c˜ao linear dos orbitais dos nucleot´ıdeos, ou seja, das autofun¸c˜oes de Hnuc
|φji=
X
ξ
X
k
Cjkξ |ψkξi=X
ξ
X
k
Cjkξ |ξki. (3.13)
O Hamiltoniano 3.12 pode ser escrito como:
H = X
γn
X
θm
|γnihγn|H|θmihmθ|
= X
γn
X
θm
Hnmγθ|γnihmθ|. (3.14)
O elemento de matriz hγn|H|θmi´e dado por
hγn|H|θmi = hγn|Hnuc+ ∆U|θmi=
= ǫθmδγθδnm+Unmγθ. (3.15)
Analisando a Fig. 3.4, podemos obter os elementos de matriz Uδθ
nm. Note que se γ 6=θ
(intera¸c˜ao entre elementos de fitas diferentes) s´o haver´a intera¸c˜ao se m = n. Na figura, essa intera¸c˜ao ´e representada pela letraw(hopping entre as fitas). Por outro lado, seγ =θ(intera¸c˜ao entre elementos da mesma fita), s´o haver´a intera¸c˜ao se m = n±1 (intera¸c˜ao do tipo primeiro vizinho). Na figura essa, a intera¸c˜ao ´e representada pela letrat(hopping entre orbitais dos s´ıtios vizinhos). Desta maneira, chegamos a seguinte representa¸c˜ao para os elementos de matriz Uδθ
nm
Uδθ
nm =
(
w, se γ 6=θ e m =n
t, se γ =θ e m=n±1. (3.17)
De modo que o segundo somat´orio da equa¸c˜ao 3.16 ´e escrito como:
X
γθ
X
mn
Unmδθ |γnihmθ| =
X
γ6=θ
X
n
Unnδθ|γnihnθ|+
X
γ=θ
X
n
X
δ=±1
Unnδθ+δ|γnih(n+δ)θ|=
= X
n
w[|αnihnβ+|βnihnα|] +
+ X
n
X
δ=±1
t[|αnih(n+δ)α|+|βnih(n+δ)β]. (3.18)