34
INOVAÇÃO NA TERAPÊUTICA ANTICOAGULANTE
Resumo
Introdução: A trombose arterial e a trombose venosa são ainda a principal causa de morbilidade e
mortali-dade. A terapêutica anticoagulante é fundamental para diminuir esta morbimortalidade, permitindo a não ocorrência ou o aumento de trombos.
Objetivo: Descrever alguns dos novos fármacos anticoagulantes que surgem na tentativa de colmatar as
limitações da atual terapêutica (heparinas, varfarina e acenocumarol).
Métodos: Revisão narrativa de artigos científicos relevantes publicados entre os anos de 2006 e 2012. Resultados: A estratégia destes novos fármacos antitrombóticos baseia-se na inibição seletiva de um
fa-tor de coagulação específico, obtendo assim inibidores seletivos com potencial para serem mais eficazes, seguros e fáceis de usar. Esta nova classe tem como principais alvos a trombina (bivalirudina, desirudina, lepirudina, argatrobana e dabigatrano) e o FXa (fondaparinux, rivaroxabano, o apixabano, edoxabano), verificando-se, no entanto, também o desenvolvimento de um novo antagonista da vitamina K, a tecarfarina, sem as limitações dos que estão atualmente comercializados, o que se deve essencialmente ao metabolismo não ocorrer via citocromo P450.
Conclusões: A terapêutica anticoagulante está a ser alvo de uma intensa investigação, de modo a que a sua
utilização na prática clínica seja de facto inovadora e revolucionária. Atualmente, em Portugal, ainda só estão aprovados e comercializáveis o fondaparinux sódico, o dabigatrano etexilato e o rivaroxabano.
Palavras-chave: Anticoagulante, inibidor, FXa, trombina, vitamina K.
Abstract
Introdution: Atherothrombosis and venous thromboembolism are the leading causes of morbidity and
mortality. Anticoagulant therapy is crucial to decrease these factors by preventing the clotting of blood.
Objective: Describe some of the new anticoagulant drugs, introduced in an attempt to overcome the
limitations of traditional anticoagulant drugs (heparins, warfarin and acenocumarol).
Method: Narrative literature review of relevant scientific articles published between 2006 and 2012. Results: A new strategy for the design of new antithrombotic drugs is based on selective inhibition of a
specific coagulation factor, with potential to be more effective, safe and easy to use. These include direct thrombin inhibitors (dabigatran, bivalirudin, desirudin, lepirudin and argatroban) and factor Xa inhibitors (otamixaban, rivaroxaban, o apixaban, edoxaban and fondaparinux). Also, a new vitamin K antagonist has been developed, tecarfarin. This does not present the same limitations as the other, which is due essencially to the fact that is not metabolized by CYP450 enzymes.
Conclusion: Anticoagulant therapy has been the target of an intense review so that its use in the clinical
practice becomes indeed innovative and revolutionary. Currently, in Portugal, only fondaparinux, the dabigatran etexilate and rivaroxaban have been approved and commercialized.
Keywords: Anticoagulant, inhibitors, FXa, thrombin, vitamin K. Sofia Ramos
Mestrado integrado em Ciências Farmacêuticas. Faculdade de Farmácia da Universidade de Lisboa.
Sofia de Oliveira Martins
Doutoramento em Farmácia – Farmacoepidemiologia. Professora auxiliar da Faculdade de Farmácia da Universidade de Lisboa, iMed.UL – Research Institute for Medicines and Pharmaceutical Sciences.
35
Introdução
A terapêutica anticoagulante tem como objetivo a administração de um fármaco de modo a prevenir a formação de um coágulo ou o seu aumento sem com-plicações hemorrágicas graves.
A anticoagulação oral surgiu em 1941. Karl Paul Link, da Universidade de Wisconsin, conseguiu isolar cuma-rina a partir de dicumarol, molécula derivada de uma planta doce chamada «erva de bisonte», que provocava hemorragia nos bovinos que a comiam. Este compos-to foi denominado de warfarin (Wisconsin Alumni Research Foundation, yarin de coumarin), exercendo o seu efeito anticoagulante através da inibição da ativação da vitamina K, mecanismo descrito em 1978. Este antago-nista da vitamina K, inicialmente usado como raticida, tem sido o pilar da terapêutica anticoagulante desde 1954 até aos dias de hoje. Os antagonistas da vitamina K foram, durante muito tempo, os únicos anticoagulantes orais em uso clínico para a prevenção e tratamento das diferentes doenças trombóticas1-5. Estes fármacos têm limitações, nomeadamente as interações com alimen-tos e medicamenalimen-tos, mas a principal é a sua margem terapêutica estreita, que implica a monitorização pe-riódica para ajuste da dosagem, através do international normalized ratio (INR).1 Nos últimos 30 anos surgiram outros anticoagulantes, mas de administração endo-venosa e subcutânea, a heparina não fracionada e a heparina de baixo peso molecular4.
Com o intuito de ultrapassar as desvantagens da tera-pêutica anticoagulante em uso, desenvolveram-se novos anticoagulantes com farmacocinética e farmacodinâmi-ca mais previsíveis, maior margem terapêutifarmacodinâmi-ca e doses fixas, sem necessidade de monitorização periódica. Nesse sentido, a estratégia desenvolvida para o design de novos fármacos antitrombóticos baseou-se na inibição seletiva de um fator específico da coagulação, nomeada-mente a trombina e o fator X ativado (FXa).
Têm assim surgido vários fármacos, sendo que alguns estão ainda em fase de ensaios clínicos, mas outros já foram autorizados e são utilizados na prática clínica, continuando a ser testados para outras indicações. Este artigo tem como objetivo descrever os novos fármacos anticoagulantes que surgiram na tentativa de colmatar as limitações da terapêutica anticoagulante tradicional.
Métodos
A metodologia desta revisão narrativa consistiu numa análise pormenorizada de um conjunto de artigos ob-tidos por pesquisa nas bases de dados Google Scholar, Pubmed, B-on e consulta das bases de dados de medi-camentos da Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. (INFARMED), da European
Medicines Agency (EMA) e da Food and Drug Admi-nistration (FDA). Os artigos analisados foram publica-dos entre os anos de 2006 e 2012. Os selecionapublica-dos esta-vam escritos em inglês, castelhano, francês e português e foram utilizados os seguintes descritores: anticoagu-lants, coagulation, warfarin, dabigatran, heparin, ximelagatran, otamixaban, bivalirudin, fondaparinux, acenocoumarol, bivali-rudin, desibivali-rudin, lepibivali-rudin, argatroban, rivaroxaban, apixaban, edoxaban, idraparinux, tecarfarin. A seleção dos artigos nas várias pesquisas efetuadas teve em consideração o ano do artigo, nunca anterior a 2006, e o seu resumo. Após a seleção inicial, os artigos escolhidos foram analisados de modo a comprovar a sua relevância direta para esta revisão. O tipo de estudo não constituiu limitação à sua inclusão na presente análise, desde que os resultados apresentados fossem fiáveis e relevantes – foram assim incluídos artigos de revisão, estudos experimentais e observacionais. Foi dada preferência a artigos relaciona-dos com esturelaciona-dos efetuarelaciona-dos em seres humanos. Apenas se incluíram estudos realizados em animais na ausência de informação sobre estudos em seres humanos. Foram excluídos artigos com informação repetida, pouco rele-vante ou de metodologia pouco fiável.
Resultados
Fisiologia da Coagulação
A compreensão da patogénese da trombose é essencial para identificar potenciais alvos terapêuticos, com o consequente desenvolvimento de novos fármacos para prevenção e tratamento6.
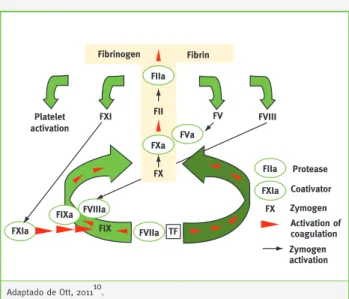
A cascata da coagulação é formada por duas vias, a in-trínseca ou de ativação de contacto e a exin-trínseca ou do fator tecidual (FT). No entanto, não há distinção clara entre as duas vias, uma vez que atuam de modo altamente interativo in vivo. Estas duas vias convergem, formando uma via comum, da qual resulta a ativação do fator X6. O FXa interage com o seu cofator V ativado na superfície de membranas contendo fosfolípidos anióni-cos (plaquetas), formando o complexo protrombinase e produzindo pequenas quantidades de trombina (IIa), que atua sobre o fibrinogénio para a formação do coágu-lo de fibrina. Em condições fisiológicas, as reações resul-tam numa produção equilibrada de quantidades apro-priadas de trombina e do coágulo de fibrina, em resposta adequada e proporcional ao dano vascular existente1,7-9. O início do processo de coagulação depende da expo-sição do sangue a componentes que, normalmente, não estão presentes no interior dos vasos, mas que surgem devido a danos vasculares ou alterações bioquímicas, como por exemplo a libertação de citocinas. Qualquer que seja o evento desencadeante, o início da coagula-ção ocorre mediante a expressão da sua componente
36
crítica, o FT e a sua exposição ao espaço intravascu-lar6,10. Após a exposição do FT aos fatores de coagulação forma-se um complexo com o fator VII ativado (FVIIa). O complexo FT-FVII é convertido num complexo en-zimático ativo FT-FVIIa pelo FXa, ou autocatálise pelo TF-FVIIa. A atividade deste complexo é influenciada pela composição da membrana e pela atividade pro-teolítica que requer um aumento da concentração de fosfolípidos aniónicos. O complexo TF-FVII/FVIIa ativa subsequentemente o FX em FXa e o FIX em FIXa (via extrínseca). O FXa produzido origina a conversão de pequenas quantidades de protrombina em trombina, quantidade suficiente para ativar o FVIII, FV e FXI. A trombina produzida pelo FXa ativa plaquetas, expõe fosfolípidos carregados negativamente, induzindo uma «explosão» de trombina. O FX também pode ser ativado por uma via diferente: o complexo de FIXa com o seu cofator FVIIIa (complexo intrínseco Xase) em que FIX é ativado pelo FXIa através da via intrínseca. Assim, o FIXa e o FXa representam pontos de convergência para as vias intrínseca e extrínseca. O FXa e o fator V ativado (FVa) formam o complexo protrombinase, ativando a protrombina em trombina, resultando na formação do coágulo de fibrina. A trombina serve também para amplificar a coagulação, pela ativação dos cofatores FV e FVIII e FXI na via intrínseca10.
Além disto, a trombina ativa as plaquetas, ativação ne-cessária para a formação do tampão hemostático. Re-sultando num equilíbrio regulado entre a formação de coágulos normais, dissolução (hemostase) e a formação patogénica de coágulos (trombose)10.
Em suma, pode dizer-se que a trombina tem um papel central na produção de trombos. Isto porque a
trombi-na ativa os fatores V, VIII e XI, que estão envolvidos trombi-na produção de mais trombina, e assim ativando também o fator XIII. Esta proteína está envolvida no cross-linking da fibrina e na estabilização do coágulo. A principal função da trombina é a conversão do fibrinogénio solúvel em fibrina insolúvel, enquanto é estimulada a ativação das plaquetas. A trombina pode ser inibida direta ou indire-tamente pela ligação de fármacos a um ou dois dos três domínios da trombina, sendo eles o sítio ativo, o exosítio um e dois. O exosítio um funciona como ligação para a fibrina e o exosítio dois funciona como domínio para a ligação da heparina11.
As reações bioquímicas da coagulação do sangue devem ser estritamente reguladas, de modo a evitar ativação excessiva do sistema, formação inadequada de fibrina e oclusão vascular. Assim, a atividade das proteases que participam na ativação da coagulação é regulada por nu-merosas proteínas inibitórias, que atuam como anticoa-gulantes naturais. Como principais inibidores fisiológi-cos da coagulação temos: o inibidor da via FT, a proteína C e a proteína S e a antitrombina.
O inibidor da via do FT é um inibidor de protea-se que regula a via extrínprotea-seca através da inibição do TF/FVIIa na iniciação da coagulação6.
A proteína C é uma glicoproteína plasmática ativada pela trombina, que inativa os fatores Va e VIIIa. Esta rea-ção é potenciada pela proteína S, que atua como cofator não enzimático nas reações de inativação6.
A antitrombina é o inibidor primário da trombina e exerce, também, um efeito inibitório sobre outras enzi-mas da coagulação, incluindo os fatores IXa, Xa, e XIa. Adicionalmente, a antitrombina acelera a dissociação do complexo FVIIa/FT e impede sua reassociação6.
Qualquer trombina que escapa aos efeitos inibitórios dos anticoagulantes naturais converte o fibrinogénio solúvel em fibrina insolúvel e ativa o fator XIII que esta-biliza a fibrina do coágulo6.
A fibrinólise é a degradação da fibrina, mediada pela plas-mina. O sistema fibrinolítico endógeno é ativado, de modo a limpar a fibrina e manter a permeabilidade da circulação. A plasmina é a protease major, que é ativada para digerir os produtos da degradação da fibrina. A regulação fisiológica da fibrinólise ocorre principalmente via ativador do plas-minogénio e antiplasmina que inibe a plasmina6.
Patologias com Indicações para Anticoagulação
A hemostase é um processo dinâmico cujo objetivo é a manutenção da integridade dos vasos sanguíneos e a circulação do sangue em fase líquida. Uma ativação pa-tológica origina uma produção excessiva de trombina, iniciando o fenómeno de trombose1,12,13.
As principais indicações da tromboprofilaxia são trom-boembolismo venoso (TEV), síndrome
pós-trombró-Fibrinogen Fibrin Platelet activation FXI FV FVIII FIIa FXa FXIa FIXa FVIIIa FVIIa FVa FIIa FXIa FX FX TF FIX Protease Coativator Zymogen Activation of coagulation Zymogen activation FII Adaptado de Ott, 201110.
Figura 1 – Ativação da cascata da coagulação dependente do FT
37
tica, hipertensão pulmonar tromboembolica crónica, fibrilação auricular (FA), síndrome coronária aguda e portadores de válvulas cardíacas12.
Tromboembolismo venoso (TEV)
O TEV, que compreende a trombose venosa profunda e o embolismo pulmonar, é uma das principais causas de morbimortalidade. Nos Estados Unidos, o trombo-embolismo pulmonar causa aproximadamente 250 mil mortes por ano14. Na União Europeia, a incidência anual de TEV é acima de um milhão, resultando em mais de 500 mil mortes por ano15, cerca de 12 por cento das mor-tes que ocorrem anualmente16.
A doença tromboembólica venosa constitui a princi-pal causa de morbimortalidade prevenível em doentes hospitalizados, podendo ocorrer complicações graves e com risco de vida, como a embolia pulmonar1,12, e a se-gunda causa de morte em doentes com cancro1,17. Além destas causas existem condições trombofílicas here-ditárias, como a mutação do fator V de Leiden, o alelo pró-trombina G20210A, bem como as deficiências em antitrombina, proteína C e S, que aumentam o risco de trombose, quer espontânea quer secundária1. O TEV deve ser considerado uma doença crónica, e não uma doença aguda3.
Os doentes com um primeiro episódio de TEV apresen-tam maior risco de recorrência. A incidência cumulativa de recorrência é de aproximadamente 18 por cento após dois anos, 25 por cento depois de cinco anos e 30 por cento após oito anos3.
Uma trombose venosa profunda recorrente está asso-ciada ao desenvolvimento de síndrome pós-trombótica. Esta síndrome é uma complicação a longo prazo da trombose venosa profunda, caracterizada por dor cró-nica, persistente, inchaço, descoloração da pele, poden-do mesmo resultar numa ulceração venosa no membro afetado3. Na maioria dos casos, a síndrome pós-trom-bótica desenvolve-se um a dois anos após trombose venosa profunda18.
A embolia pulmonar predispõe os pacientes a hiperten-são pulmonar crónica19.
Fibrilação auricular (FA)
A FA, que é a arritmia cardíaca mais comum com signi-ficado clínico, aumenta o risco de enfarte e morte16,20-24. Apresenta uma prevalência de aproximadamente 1 por cento para a população em geral e 10 por cento na popu-lação com mais de 80 anos16,23. É um problema crescente de saúde pública associado a morbidade e mortalidade significativas24.
A presença de FA é um importante fator de risco para acidente vascular cerebral (AVC) isquémico e outros
eventos tromboembólicos24. A FA é o principal fator de risco de AVC cardioembólico16,23 e o risco de AVC is-quémico aumenta entre quatro e cinco vezes nos pacien-tes com FA1,3, e está associado ao embolismo sistémico e morte1,25. A terapêutica anticoagulante a longo prazo pode reduzir o risco de AVC em cerca de 60 por cento dos pacientes com FA não valvular21,24,25 e em mais de 90 por cento o risco de enfarte em indivíduos com FA21. Fármacos Anticoagulantes Tradicionais
A terapêutica anticoagulante tradicional inclui a hepari-na não fraciohepari-nada e a heparihepari-na de baixo peso molecular, anticoagulantes parentéricos e os antagonistas da vita-mina K, de administração oral. Estes fármacos atuam em vários fatores de coagulação.
Apesar de alcançar reduções significativas na incidên-cia de complicações tromboembólicas, esta terapêutica utilizada na prevenção e tratamento de TEV apresenta inúmeras limitações26.
Heparinas
A heparina não fracionada é um anticoagulante indire-to que se liga à antitrombina III, aumentando a sua ca-pacidade para inibir o FXa, a trombina e outros fatores de coagulação. No entanto, o coágulo ligado à trombina está relativamente protegido da inibição por heparina-antitrombina, possivelmente porque o local de ligação à heparina está inacessível quando a trombina se encontra ligada à fibrina3.
A heparina não fracionada liga-se a um número de pro-teínas plasmáticas, que contribuem para a sua resposta anticoagulante variável. Além disso, a heparina não fra-cionada está associada ao risco de desenvolvimento de trombocitopenia induzida por heparina e osteoporose. Consequentemente, é necessária uma monitorização ri-gorosa e frequente da coagulação3.
A heparina de baixo peso molecular resulta da despoli-merização química ou enzimática da heparina não fra-cionada. Apresenta uma anticoagulação mais previsível, podendo ser administrada em doses fixas e sem moni-torização periódica da coagulação1,3. No entanto, tanto a heparina não fracionada como a heparina de baixo peso molecular requerem administração parentérica, o que limita a sua utilização em ambulatório30. Contudo, as heparinas de baixo peso molecular também podem causar trombocitopenia induzida por heparina, apesar de este risco ser menor em comparação com a heparina não fracionada1,3.
Antagonistas da vitamina K
A vitamina K na sua forma reduzida (hidroquinona) é um cofator da enzima hepática gama-glutamil-carboxilase,
38
que catalisa a carboxilação de vários resíduos de gluta-mato a ácido gama-carboxiglutámico, nas regiões ter-minais NH das proteínas dependentes da vitamina K, sendo elas os fatores II, VII, IX e X e as proteínas C e S. A carboxilação é um passo necessário a uma alteração conformacional dependente de cálcio, que ocorre nas proteínas de coagulação, levando à ligação de cofatores nas superfícies fosfolipídicas do local de lesão vascular. Assim, o mecanismo dos resíduos Gla confere ativida-de biológica aos fatores ativida-depenativida-dentes da vitamina K. O processo de carboxilação oxida a vitamina K na sua forma epóxido, que se reduz voltando à sua forma ati-va (hidroquinona) pela epoxirredutase da vitamina K (VKORC). Como tal, a ação dos anticoagulantes orais antagonistas da vitamina K é inibir a enzima epoxirredutase da vitamina K1,31.
No entanto, a vitamina K exógena (proveniente da die-ta, por exemplo) pode contornar o antagonismo da varfarina, conseguindo produzir o cofator (vitamina KH2) necessário à carboxilação das proteínas da coagula-ção dependentes da vitamina K, através duma via menos sensível à varfarina (a da enzima vitamina K redutase)31. Os inibidores da vitamina K são anticoagulantes efetivos, com uma boa absorção e baixo custo. Existem métodos para reverter o efeito do excesso de anticoagulantes. As principais desvantagens são as interações com fárma-cos, produtos naturais e alimentos, que modificam o seu metabolismo, podendo causar aumento ou inibição do efeito anticoagulante, e a variabilidade dose-resposta de pessoa para pessoa1. As doenças intercorrentes, o início ou paragem de tratamentos com outros fármacos, mu-danças na dieta ou na função intestinal podem causar alterações no efeito anticoagulante21,24,25. Como tal, esta terapêutica exige uma monitorização periódica através da realização do INR, medida normalizada a nível in-ternacional para minimizar as variações nos valores do tempo de protrombina entre diferentes laboratórios1. No entanto, existem situações em que estes anticoagu-lantes estão contraindicados ou são impraticáveis, como na gravidez e em indivíduos que vivem em locais geogra-ficamente inacessíveis29.
Para a manutenção de um nível de anticoagulação tera-pêutica adequada é necessário um bom entendimento da farmacocinética e farmacodinâmica, dado serem im-previsíveis, tal como é fundamental uma boa comunica-ção médico-doente3,32,33.
A varfarina é o anticoagulante oral mais utilizado a ní-vel mundial, quer na prática clínica quer em estudos clínicos1. A varfarina inibe a síntese dos pró-fatores II, VII, IX e X, tal como acontece com os anticoagulantes endógenos, proteínas C e S31,34. Este fármaco é uma
mistura de enantiómeros, composta pelo R-varfarina e S-varfarina, sendo que a atividade do enantiómero-S é cinco vezes mais potente que o R, sendo por isso o enantiómero-S o principal responsável pela ativi-dade da varfarina. São ambos metabolizados no fíga-do, mas por vias diferentes. O tempo de semivida é de 35 a 45 horas1.
A metabolização da varfarina ocorre por múltiplas en-zimas hepáticas do citocromo P450 (CYP450), o que origina a sua elevada suscetibilidade a interações com fármacos e alimentos. Esta também é influenciada pelas variações genéticas destas enzimas. O polimorfismo da enzima epoxirredutase da vitamina K contribui ainda mais para esta variabilidade33,35. Como tal, as doses e os valores de INR não são previsíveis. No entanto, estão a ser desenvolvidos algoritmos para propor as doses indi-cadas a cada indivíduo28,36.
A varfarina é usada na prevenção de eventos tromboem-bólicos associados a várias condições médicas, como FA, doença valvular cardíaca e trombose venosa profunda5,33. O acenocumarol é um derivado da cumarina e apresenta um tempo de semivida mais curto, entre oito e 24 horas, o que pode conferir uma maior instabilidade terapêutica1. O estabelecimento de dose terapêutica apropriada das cumarinas é difícil, devido à sua intravariabilidade ou in-tervariabilidade, associada a numerosos fatores genéticos e não genéticos, à farmacocinética, à farmacodinâmica e à estreita janela terapêutica. Apesar de serem muito seme-lhantes, para a varfarina e o acenocumarol as doses reco-mendadas são diferentes, uma vez que a farmacocinética e a farmacodinâmica de ambas são diferentes e também devido à influência dos fatores genéticos. O efeito de va-riantes defeituosas de CYP2C9 é mais pronunciado para a varfarina do que para o acenocumarol, podendo dizer-se que a variabilidade para a varfarina é de 10 a 15 por cen-to e apenas de 5 por cencen-to para o acenocumarol28,36. A dose é inicialmente definida por critérios clínicos, sen-do depois controlada pela determinação individual sen-do INR. Critérios a ter em conta na determinação da dose são o género, a idade, o peso, a altura, tal como variações nos genes VKORC (alvo cumarínico) e CYP2C9, que expressa o citocromo P450 2C9 (enzima responsável pelo metabolismo da cumarina). Dada a elevada taxa de efeitos adversos (incluindo hemorragias fatais), devido a doses incorretamente calculadas, é necessário desen-volver novas estratégias para a determinação da dose se-manal apropriada de acenocumarol, tal como algoritmos farmacogenéticos para prever uma dose de acenocumarol apropriada foram propostos36-38. Tem sido demonstrada a importância da pré-genotipagem dos polimorfismos VKORC1, CYP4F2, CYP2C9*2 e *3 em doentes que ne-cessitam de doses extremas de acenocumarol28.
39
O acenocumarol é altamente eficaz no tratamento de doenças tromboembólicas, como tromboembolismo ve-noso profundo e embolia profunda, na FA e portadores de válvulas cardíacas artificiais36.
Novos Fármacos Anticoagulantes
O desenvolvimento dos novos fármacos anticoagulantes tem como objetivo a seleção de um fármaco que reúna o máximo das características possíveis do anticoagulante ideal. Essas características são: eficácia no tratamento da doença arterial e venosa; segurança; risco mínimo de efeitos adversos; reversibilidade fácil com ou sem antídoto; propriedades farmacocinéticas e farmacodi-nâmicas, como rápido início de ação, dose-resposta e farmacocinética previsível, ampla margem terapêutica com variabilidade intra e interindividual mínima, ad-ministração oral e sem ajuste de dose, ausência de inte-ração com alimentos e fármacos e a não necessidade de monitorização de rotina15,39.
Inibidores diretos da trombina
Os inibidores diretos da trombina ligam-se diretamen-te à trombina, sem a necessidade de um cofator como a antitrombina para exercerem o seu efeito. Estes inibem tanto a trombina solúvel como a trombina ligada à fibri-na. Outra das vantagens deste grupo farmacológico é o facto de permitir um efeito anticoagulante mais previsí-vel que as heparinas, devido à sua não ligação a outras proteínas plasmáticas, um efeito antiplaquetário e a au-sência de trombocitopenia11.
Tanto os inibidores diretos da trombina orais como os pa-rentéricos têm sido investigados para a profilaxia e o trata-mento de tromboembolismo venoso, prevenção de compli-cações tromboembólicas em doentes com trombocitopenia induzida por heparina ou em risco de desenvolver trombo-citopenia induzida por heparina ou sujeitos a intervenção coronária percutânea, síndrome coronária aguda, com ou sem angioplastia coronária transluminal percutânea (PTCA), prevenção secundária de eventos coronários após síndromes coronárias agudas e FA não valvular11.
Inibidores diretos da trombina orais
Estes inibidores podem ligar-se de modo reversível ou irreversível ao sítio ativo do fator IIa4. Os inibidores FIIa orais representam a nova era da anticoagulação na pre-venção e tratamento de tromboembolismo venoso e arte-rial seletivo. O primeiro a ser desenvolvido e aprovado na Europa, nunca tendo sido aprovado nos Estados Unidos, foi o ximelagatrano, pró-fármaco do melagatrano. No en-tanto, foi retirado 20 meses após a sua aprovação devido ao risco de hepatoxicidade demonstrada em tratamentos superiores a 35 dias11,22,40.
Estes são antagonistas competitivos da trombina, capa-zes de inibir a sua função tanto na fração solúvel como unida à fibrina. Esta propriedade de inibir a trombina unida à fibrina diferencia-os das heparinas. Além disto, estes novos fármacos atuam na ativação por trombina dos fatores V, VIII e XI e a sua propriedade de agonis-ta plaquetário. Como principais vanagonis-tagens apresenagonis-tam o seu uso em dose fixa e sem necessidade de monitori-zação dos níveis de anticoagulação, devido ao seu perfil farmacocinético previsível1,41.
O dabigatrano etexilato é um pró-fármaco do dabigatrano, conversão que ocorre rapidamente, via hidrólise39. O dabigatrano é um pequeno peptidomimético, molécula de baixo peso molecular, que inibe diretamente a trom-bina através ligação ao sítio ativo da tromtrom-bina por inte-rações iónicas4,11. É um inibidor reversível, competitivo e direto da trombina42. Inibe rápida e reversivelmente quer a fração livre quer a ligada ao coágulo, dependen-te da concentração com uma constandependen-te de inibição (Ki) de 4,5nmol-14,11. Apresenta uma elevada especificidade para a trombina sob outras proteases da serina. Como molécula carregada e altamente polar, o dabigatrano apresenta uma baixa absorção intestinal e deixa de estar biodisponível após absorção oral. A conversão do gru-po carboxilato do dabigatrano num grugru-po éster e do grugru-po fortemente básico da benzamidina num éster carbamato lipofílico levou ao desenvolvimento do dabigatrano etexilato, um pró-fármaco altamente lipofílico e absor-vido pelo sistema gastrointestinal. Após a absorção, o dabigatrano etexilato é rapidamente convertido no dabigatrano activo e em dois intermédios, o BIBR 951 e BIBR 1087, após clivagem de ambos os grupos lipofílicos pelas esterases da serina11,42,43.
Para que haja absorção é necessário um pH ácido, pelo que se recomendava que a sua toma não fosse concomitante com um inibidor da bomba de pro-tões1. No entanto, a nova formulação do dabigatrano etexilato foi desenvolvida de modo a obter o ambien-te ácido ideal para a sua absorção, conseguido através de cápsulas com pequenos pellets contendo um núcleo de ácido tartárico revestido. Assim, as variações de pH gástrico intrínseco não afetam significativamente a absorção deste, mesmo na presença de inibidores da
bomba de protões11. A conversão deste pró-fármaco
na sua forma ativa ocorre a nível hepático, sem utili-zar o citocromo P4501, mas estando as esterases ubi-quitárias presentes no plasma envolvidas nesta con-versão. Sendo por isso as suas principais vantagens o rápido início de ação e a não interação com o citocro-mo P450, fazendo com que a interação com alimen-tos e fármacos seja mínima; apresenta um excelente perfil de segurança, não necessita de monitorização
40
de rotina, tem uma ampla janela terapêutica e a uma dose de administração fixa11,42.
Após a absorção do dabigatrano etexilato, a bioconver-são a dabigatrano ocorre nos enterócitos, hepatócitos e na veia portal. O correspondente a cerca de 20 por cen-to do dabigatrano é conjugado com ácido glucurónico para produzir conjugados de glucuronido farmacologi-camente ativos. Uma vez metabolizado em dabigatrano, o pró-fármaco e os seus metabolitos intermediários são detetáveis no plasma de indivíduos saudáveis aproxi-madamente duas horas após a ingestão. Os resultados de vários estudos demonstram que a insuficiência he-pática moderada não afeta a eficácia ou o perfil de segu-rança do dabigatrano, não sendo necessário o ajuste de dose em situações de insuficiência hepática11.
Este fármaco apresenta uma baixa ligação às proteínas plasmáticas: aproximadamente 35 por cento. Em si-tuações de overdose ou hemorragia severa, em que seja necessário um efeito anticoagulante reversivo rápido, a hemodiálise pode ser efetiva ao acelerar a clearance plas-mática do dabigatrano, especialmente em doentes com falha renal4. Apresenta uma biodisponibilidade de 7,2 por cento, atingindo a concentração plasmática máxi-ma entre umáxi-ma hora e meia e três horas e umáxi-ma semivida de 12 a 17 horas1. Após a absorção ocorre uma fase rápida de distribuição, à qual se segue um tempo de eliminação prolongado. O volume de distribuição do dabigatrano é moderado (60-70L)11. A sua excreção é maioritaria-mente renal (80 por cento), pelo que doentes com insu-ficiência renal têm sido excluídos desta terapêutica1. A restante excreção ocorre através da bílis4. Os alimentos podem prolongar o tempo para a concentração plasmá-tica em duas horas, mas sem efeito na extensão da ab-sorção do dabigatrano11.
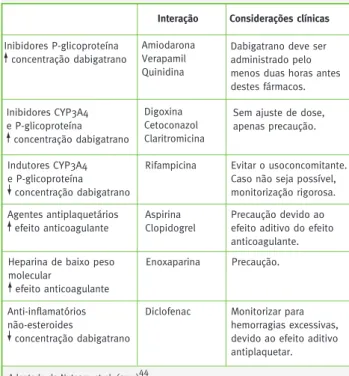
As principais interações do dabigatrano com outros fár-macos, tal como algumas considerações clínicas, resu-mem-se na Tabela I.
Tanto o tempo trombina como o tempo de coagulação do ecarin são testes altamente sensíveis para quantificar os efeitos anticoagulantes do dabigatrano. O INR, pro-longado pelo dabigatrano, e o tempo trombina não são sensíveis para avaliação do efeito anticoagulante deste fármaco41. O tempo de tromboplastina parcial ativado (TTPa) é igualmente prolongado, mas não de uma for-ma dose-dependente. Assim, este teste pode ser usado como teste qualitativo, dado que é menos sensível a concentrações subterapêuticas do dabigatrano4. Não há agentes específicos para reverter o efeito do dabigatrano4.
Este fármaco foi aprovado no Canadá e na Europa em 2008 para a prevenção de tromboembolismo venoso após cirurgia de substituição da anca ou joelho,
sen-Adaptado de Nutescu et al. (2011)44
Inibidores P-glicoproteína concentração dabigatrano
Tabela 1 – Interações do dabigatrano com outros fármacos Interação Considerações clínicas Amiodarona
Verapamil Quinidina
Dabigatrano deve ser administrado pelo menos duas horas antes destes fármacos. Inibidores CYP3A4 e P-glicoproteína concentração dabigatrano Digoxina Cetoconazol Claritromicina
Sem ajuste de dose, apenas precaução. Indutores CYP3A4
e P-glicoproteína concentração dabigatrano
Rifampicina Evitar o usoconcomitante. Caso não seja possível, monitorização rigorosa. Agentes antiplaquetários efeito anticoagulante Aspirina Clopidogrel Precaução devido ao efeito aditivo do efeito anticoagulante. Heparina de baixo peso
molecular efeito anticoagulante Enoxaparina Precaução. Anti-inflamatórios não-esteroides concentração dabigatrano
Diclofenac Monitorizar para hemorragias excessivas, devido ao efeito aditivo antiplaquetar.
do também uma das opções farmacológicas indica-das para esta situação na Norma da Direção-Geral da Saúde 026/2012, relativa à profilaxia do tromboembo-lismo venoso em ortopedia, com nível de evidência A, grau de recomendação I, estando referido que o fármaco deve ser iniciado apenas após a cirurgia.
Foi também aprovado em 2010 pela FDA na prevenção de enfarte em indivíduos com FA11. Foram desenvolvi-dos inúmeros estudesenvolvi-dos para demonstrar a eficácia clíni-ca do dabigatrano nas seguintes situações: prevenção de tromboembolismo em cirurgias de substituição da anca e joelho – alguns dos ensaios clínicos realizados nesta indicação foram: RE-NOVATE, RE-MOBILIZE e
RE-MODEL39; tratamento e prevenção secundária de
tromboembolismo venoso (RE-MEDY, RE-COVER, RE-SONATE); prevenção de enfarte em indivíduos com FA sem prótese valvular (RE-LY)39. Potenciais indicações para o uso de dabigatrano são: preven-ção secundária de eventos coronários após síndrome coronária aguda; prevenção de trombose em interven-ção coronária percutânea eletiva e preveninterven-ção de trom-bos nas válvulas mecânicas cardíacas5,11.
Inibidores diretos da trombina parentéricos
Nos Estados Unidos, quatro inibidores diretos da trombina parentéricos estão aprovados para se-rem usados como anticoagulantes: a bivalirudina, a desirudina, a lepirudina e o argatrobano11. Em Por-tugal, apenas a argatrobana não está autorizada pelo
41
INFARMED, apesar de a lepirudina ser a única co-mercializada.
A lepirudina e a desirudina são derivados da hirudina, um péptido originalmente isolado a par-tir das glândulas salivares de sanguessugas medici-nais desenvolvidas por tecnologia recombinante em Saccharomyces cerevisiae. Ambas as hirudinas recom-binantes são inibidores diretos da trombina bivalen-tes que se ligam simultaneamente ao sítio da enzima ativa e ao exosítio domínio um da trombina, uma característica que aumenta a sua especificidade. Es-tes também apresentam uma elevada afinidade para a trombina, formando rapidamente complexos irre-versíveis. Comparativamente às hirudinas, a afinida-de afinida-destes dois fármacos para a trombina é afinida-dez vezes mais fraca; no entanto, eles são considerados os inibi-dores da trombina mais potentes11.
A lepirudina tem como indicação a profilaxia ou tratamento de complicações trombóticas causadas por trombocitopenia induzida por heparina45. A sua afinidade para a trombina é elevada (Ki=0,23 pM), formando um complexo irreversível. A sua via de administração pode ser endovenosa ou subcutânea e a dose depende do peso. A semivida plasmática é de aproximadamente 80 minutos. A excreção deste fármaco é renal; como tal, é necessário um ajuste de dose em indivíduos com insuficiência renal. As prin-cipais desvantagens da lepirudina são a estreita jane-la terapêutica e o potencial aumento de eventos he-morrágicos. Além disto, há a formação de anticorpos anti-hirudina após o tratamento de trombocitopenia induzida por heparina em 40 por cento dos indiví-duos. Estes complexos imunogénicos podem atra-sar a excreção renal da lepirudina, provocando a sua acumulação. Por isso, é necessário um ajustamento da dose baseado no TTPa durante o tratamento11,45. Embora suceda raramente, pode ocorrer choque ana-filático em indivíduos com anticorpos anti-hirudina quando re-expostos à hirudina. Atualmente, não existe antídoto específico para reverter os efeitos das hirudinas recombinantes ou qualquer outro inibidor direto da trombina11.
A desirudina é o único inibidor direto da trombina administrado subcutaneamente com dose fixa apro-vado pela FDA para a prevenção pós-operatória de tromboembolismo venoso sujeito a cirurgia eletiva de substituição da anca, e encontra-se disponível desde 2010 nos Estados Unidos11. A sua afinidade para a trombina é elevada (Ki=0,26 pM) e a via de adminis-tração pode ser endovenosa ou subcutânea, apresen-tando uma semivida plasmática de 60 minutos para administração endovenosa e de 120 minutos para a
administração subcutânea. O metabolismo e a clearance são renais, e como tal é necessário ajuste de doses nos casos de insuficiência renal severa, tal como o seu controlo por TTPa. Tal é, no entanto, desnecessário na insuficiência renal moderada, de acordo com es-tudos farmacocinéticos mais recentes. As vantagens deste fármaco são o facto de não precisar de cálculos de dose tendo em conta o peso e de não precisar de monitorização periódica11.
Nos ensaios HELVETICA e GUSTO-IIb, comparan-do as heparinas com a desirudina, concluiu-se que o efeito anticoagulante da desirudina é superior e que o perfil de segurança é semelhante. Atualmente, está a decorrer um estudo PREVENT-HIT sobre a potencialidade deste anticoagulante em indivíduos com trombocitopenia induzida por heparina, com ou sem trombose11.
A bivalirudina é um aminoácido sintético, bivalente análogo da hirudina, com uma atividade inibidora da trombina, aproximadamente 800 vezes mais fraco que a hirudina. Ao contrário das hirudinas recombi-nantes, a ligação da bivalirudina à trombina é rever-sível, pois uma vez formada a ligação, esta é clivada pela trombina. Como tal, a atividade da trombina é inibida apenas transitoriamente e a atividade enzi-mática da trombina é restaurada. Esta reversibilida-de na ligação entre a bivalirudina e a trombina poreversibilida-de contribuir para diminuir o risco de hemorragia e me-lhorar o perfil de segurança, quando comparada com as hirudinas recombinantes11. A sua afinidade para a trombina é intermédia, apresentando um Ki=1,9 nM. A bivalirudina é essencialmente eliminada por clivagem proteolítica e metabolismo hepático, apresentando no entanto uma excreção renal de 20 por cento, sendo assim necessário um ajuste de dose em insuficientes renais moderados e contraindicado na insuficiência renal severa11.
A bivalirudina é administrada por via endovenosa e tem um início de ação rápido, aproximadamente cin-co minutos, e uma semivida plasmática de aproxima-damente 25 minutos. A via de administração pode ser uma das suas desvantagens, pois é incómoda para ser utilizada num tratamento crónico11,26.
Este inibidor parentérico da trombina, tem-se mostrado eficaz e seguro em pacientes com síndrome coronária agu-da que têm um stent implantado26, sendo também indica-do em indivíduos com angina instável sujeitos a PTCA, em intervenções coronárias percutâneas com uso provisório de inibidor de glicoproteína, em indivíduos com ou em ris-co de trombocitopenia induzida por heparina ou trombo-citopenia induzida por heparina com síndrome trombóti-ca sujeitos a intervenção coronária percutânea11.
42
A bivalirudina está a ser extensamente investigada em vários ensaios clínicos sobre a sua eficácia na redução da morte, enfarte do miocárdio ou revascularizações em doentes com síndrome coronária aguda que realizem intervenções coronárias percutâneas. Os estudos como o REPLACE-2 mostram uma melhor ou igual eficácia deste fármaco comparado com as heparinas e uma sig-nificativa redução da hemorragia, tendo sido por isso aprovado pela FDA em 2000 como alternativa à hepa-rina em doentes sujeitos a PTCA. Foi alargado em 2005 para o uso concomitante de glicoproteína IIb/inibidor IIIa para doentes sujeitos a PCI eletiva ou urgente11. O argatrobano é um derivado sintético da arginina. Este inibidor direto da trombina é pequeno e univalente, li-gando-se de forma não-covalente e reversível ao sítio ati-vo da trombina. A sua afinidade para a trombina é baixa (Ki=5-38nM). A sua via de administração é endovenosa e apresenta uma semivida plasmática de 45 minutos. O metabolismo é hepático e é predominantemente excre-tado pelo sistema biliar; como tal, é necessário ajuste de dose em indivíduos com insuficiência hepática modera-da e contraindicado em insuficiência hepática severa, o que não acontece na insuficiência renal11,45.
O efeito anticoagulante é monitorizado pelo TTPa e pelo tempo de coagulação ativado e a dose é ajustada de modo a que o TTPa seja 1,5 a 3,0 vezes o valor inicial11,45. O argatrobano prolonga o tempo de protrombina e o INR11. Nos Estados Unidos, a sua indicação é a profilaxia ou tratamento de trombocitopenia induzida por heparina e indivíduos com ou sem risco de trombocitopenia induzida por heparina sujeitos a intervenção percu-tânea coronária11,45.
Inibidores do fator X
O FXa é um bom alvo para novos anticoagulantes, uma vez que tem um papel crucial na ativação da coagulação, sendo capaz de ativar a protrombina após a sua ligação ao FVa, e está demonstrado que quando ativada uma molécula de FXa pode originar mais de mil moléculas de trombina. Os fármacos podem inibir o FXa de modo direto ou indireto via antitrombina. A via indireta precisa que se verifique a formação de um complexo fármaco-an-titrombina, complexo de elevado peso molecular capaz de inibir o FXa em circulação mas incapaz de bloquear a atividade do coágulo. Por outro lado, os fármacos capa-zes de inibir diretamente o FXa são moléculas pequenas com a capacidade de entrar no trombo e de inibir tanto o FXa em circulação como o FXa ligado ao trombo46. Inibidores diretos do fator X ativado
Os inibidores diretos do FXa atuam numa etapa inicial da cascata da coagulação. Inibem o FXa livre, unido ao
complexo trombinase (junto ao cofator V), e o FXa asso-ciado à trombina. A sua dose terapêutica é fixa e não re-quer uma monitorização dos níveis de anticoagulação1. Os inibidores diretos do FXa podem ser de nistração parentérica, o otamixabano, ou de admi-nistração oral, como o rivaroxabano, o apixabano e o edoxabano. Os inibidores diretos do FXa orais são inibidores pequenos, moléculas ativas oralmen-te, capazes de se ligar reversivelmente ao sítio ati-vo do FXa. O único autorizado e comercializado em Portugal é o rivaroxabano, aprovado em 2008 na Europa e no Canadá39.
O otamixabano é um inibidor direto e reversível do FXa47. Este inibidor parentérico tem um início de ação rápido, uma semivida curta, de apenas 30 mi-nutos48 e uma excreção renal inferior a 25 por cen-to. Apresenta um efeito anticoagulante previsível, o que dispensa uma monitorização da coagulação de rotina. Ao contrário da heparina não fracionada e da heparina de baixo peso molecular, este fármaco não causa trombocitopenia induzida por heparina. Nos es-tudos SEPIA-PCI e SEPIA-ACS, este inibidor seletivo mostrou ser eficaz e seguro48. Como tal, o otamixabano é um bom candidato a substituir a heparina não fracio-nada e a heparina de baixo peso molecular em indiví-duos com síndrome coronária aguda46. As vantagens do otamixabano são a sua simplicidade de uso e a fá-cil transição em intervenção coronária percutânea de emergência, com uma redução significativa de morte e eventos isquémicos49.
O rivaroxabano é um inibidor altamente seletivo, re-versível e direto do FXa. Este fármaco tem um início de ação rápido e é absorvido através do sistema gas-trointestinal, com uma biodisponibilidade de 80 por cento46,50. A concentração plasmática máxima ocor-re entocor-re as duas e as quatro horas1,3,50. A ligação às proteínas plasmáticas é de 92 a 95 por cento, sendo a albumina sérica a principal3, e o seu volume de dis-tribuição é de 50 litros39. A semivida é de três a nove horas. Três aspetos relevantes da farmacodinâmica do rivaroxabano são: a inibição dependente da concen-tração do FXa com elevada potência e seletividade, a inibição da trombina a partir da protrombina e uma inibição dose-dependente do FT4.
O rivaroxabano é excretado por várias vias: um terço é excretado de forma inalterada via renal, um terço é metabolizado no fígado, via oxidativa CYP3A4 e CYP2J2 no citocromo P4504, e eliminado pelas fezes e outro terço é metabolizado no fígado em metabo-litos inativos, sendo depois eliminados via renal50. Este fármaco é um substrato para o transporte da P-glicoproteína.
As principais interações do rivaroxabano com outros fármacos, tal como algumas considerações clínicas, resumem-se na Tabela II.
Adaptado de Nutescu et al. (2011)44
Absorção
Tabela 2 – Interações do rivaroxabano com outros fármacos Interação Considerações clínicas Antiácidos
Antagonistas recetor H2
Sem efeito significativo.
Inibidores CYP3A4 e P-glicoproteína concentração rivaroxabano Cetoconazol Itraconazol Ritonavir Claritromicina Contraindicado o uso concomitante. Precaução. Contraindicado. Precaução. Indutores CYP3A4 e P-glicoproteína concentração rivaroxabano Rifampicina Fenitoina Carbamazepina Fenobarbital
Evitar o uso concomitante. Caso não seja possível monitorização rigorosa. Precaução. Agentes antiplaquetários efeito antitrombótico Aspirina Clopidogrel Precaução devido à potenciação do efeito antitrombótico. Anticoagulantes efeito antitrombótico Enoxaparina Precaução. Anti-inflamatórios não-esteroides efeito antiplaquetar Naproxeno Precaução.
O perfil farmacológico deste fármaco não se mostrou afetado pelo excesso de peso corporal, idade ou sexo4. O ribaroxano prolonga o INR com sensibilidade do reagente utilizado4. Não existe agente específico para reverter os efeitos deste fármaco4. O rivaroxabano está indicado para a profilaxia e tratamento de tromboem-bolismo venoso40, prevenção de enfarte na FA e no tra-tamento de síndromes coronárias agudas46.
O rivaroxabano está indicado para a profilaxia e
trata-mento de tromboembolismo venoso40, sendo uma das
opções farmacológicas para a profilaxia da cirurgia ele-tiva da artroplastia da anca e joelho constantes da Nor-ma da Direção-Geral da Saúde 026/2012 – Profilaxia do Tromboembolismo Venoso em Ortopedia com nível de evidência A, grau de recomendação I. Com o mesmo ní-vel de evidência e grau de recomendação, é indicado que a terapêutica só deve ser iniciada depois da cirur-gia. É indicado, igualmente, na prevenção de enfarte na FA e no tratamento de síndromes coronárias agu-das46.
O apixabano é outro inibidor oral, direto, altamente seletivo e reversível do FXa51, com a capacidade de inibir tanto o FXa em circulação como o FXa ligado ao complexo da protrombinase33.
Este fármaco é rapidamente absorvido pelo sistema gastrointestinal, com uma concentração plasmática máxima, três horas após a administração1,3,46 e uma semivida entre as oito e as 15 horas3,4. A sua ligação às proteínas plasmáticas é de 87 por cento e o volume de distribuição é baixo39.
O apixabano é eliminado por múltiplas vias, incluindo o metabolismo oxidativo, excreção renal e intestinal. O metabolismo oxidativo ocorre via CYP3A4; como tal, o tratamento concomitante com inibidores do CYP3A4 está contraindicado nestes doentes46.
A sua excreção é maioritariamente fecal (56 por cento), sendo apenas entre 22 e 25 por cento por via renal1,3. O composto pai é o principal componente no plasma, uri-na e fezes. Existem, no entanto, outros metabolitos que perfazem aproximadamente um terço da dose recupe-rada, sendo o o-dimetilsulfato de apixabano o principal destes compostos3.
O apixabano é um substrato de transporte da proteína P-glicoproteína que funciona como uma bomba de efluxo para prevenir a absorção ou aumento na se-creção renal de certas drogas, como os substratos de P-glicoproteína4.
As principais interações do apixabano com outros fár-macos, tal como algumas considerações clínicas, resu-mem-se na Tabela III.
O apixabano tem um impacto mínimo sobre o INR e o TTPa em concentrações terapêuticas, mas o fator de inibição Xa parece sensível para detetar a sua presença4. Não há nenhum agente específico para reverter o efei-to anticoagulante deste fármaco4. A sua indicação é na profilaxia e tratamento de tromboembolismo venoso51, prevenção de enfarte na FA e no tratamento de síndro-mes coronárias agudas46.
O edoxabano é um inibidor direto, específico e oral do FXa. Apresenta uma elevada seletividade para o FXa. Este fármaco ativo é rapidamente absorvido pelo sis-tema gastrointestinal, com a concentração máxima plasmática a ocorrer uma a duas horas após a admi-nistração3,46, regressando aos níveis basais 12 horas após a administração. A inibição do FXa levou a uma redução correspondente na produção de trombina e no prolongamento do tempo de coagulação. Num estudo com múltiplas doses, o edoxabano prolongou o TTPa e o tempo de protrombina de uma forma dose-depen-dente3. Apresenta uma semivida de nove a 11 horas46 e a taxa de ligação às proteínas plasmáticas é de 40 a 59
por cento3. O edoxabano apresenta um mecanismo de
eliminação duplo: um terço é excretado via renal, en-quanto o restante é excretado através das fezes46. Estudos efetuados com este fármaco, como HOSUSAI e ENGAGE, têm como alvo a profilaxia de tromboembolismo venoso e a prevenção de enfarte em indivíduos com FA15,46.
Adaptado de Nutescu et al. (2011)44
Absorção
Tabela 3 – Interações do apixabano com outros fármacos Interação Considerações clínicas Antiácidos
Pantoprazol
Sem efeito significativo. Inibidores CYP3A4
e P-glicoproteína concentração apixabano
Cetoconazol Precaução na administração concomitante.
Indutores CYP3A4 e P-glicoproteína
concentração apixabano
Rifampicina Evitar o uso concomitante, não foram efetuados estudos específicos. Agentes antiplaquetários efeito antitrombótico Aspirina Clopidogrel Monitorização rigorosa devido à potenciação do efeito antitrombótico. Anticoagulantes efeito antitrombótico Enoxaparina Heparina Potenciação do efeito anticoagulante, não foram efetuados estudos. Anti-inflamatórios não-esteroides efeito antiplaquetar Ibuprofeno Naproxeno Efeito antiplaquetar potenciado. Monitorizar para excessivas hemorragias, sem estudos específicos.
Inibidores indiretos do fator X ativado
Tal como a heparina não fracionada e a heparina de bai-xo peso molecular, esta classe de fármacos precisa da antitrombina, de modo a exercer a sua ação farmaco-lógica, que consiste na inibição da actividade do FXa e consequentemente na produção de trombina46.
Nesta classe incluem-se o fondaparinux sódico, o idraparinux e idrabioparinux. No entanto, apenas o pri-meiro está autorizado e comercializado em Portugal. O fondaparinux sódico foi o primeiro agente sintético da nova classe de antitrombóticos inibidores do FXa. É um análogo sintético da antitrombina de ligação. Este pentassacarídeo é idêntico à sequência presente na mo-lécula nativa de heparina, que se liga à antitrombina com elevada afinidade. Apresenta uma estrutura de-masiado curta para permitir a ponte entre a antitrom-bina e a tromantitrom-bina, sendo este o mecanismo de inibição específico para o FXa46. Assim, o fondaparinux poten-cia seletivamente a atividade do antifator Xa da anti-trombina e não tem nenhum efeito sobre a anti-trombina. O fondaparinux produz uma resposta anticoagulante pre-visível que dispensa a monitorização de coagulação de rotina, o ajuste de dose e não causa trombocitopenia in-duzida por heparina. A sua biodisponibilidade é de 100 por cento e a semivida de 17 horas30.
Este pentassacárideo liga-se à antitrombina, inibin-do assim indireta e seletivamente o FXa. A admi-nistração ocorre por via subcutânea3 e não existe
antídoto para reverter o seu efeito30. No entanto, o fondaparinux está contraindicado em pacientes com insuficiência renal (clearance da creatinina menor que 30 mL/min), porque a sua excreção é renal30. O fondaparinux tem demonstrado a sua maior eficácia em relação à heparina de baixo peso molecular em ensaios clínicos randomizados. Está aprovado pela FDA para o tratamento e prevenção de tromboembolismo veno-so em indivíduos sujeitos a cirurgias ortopédicas ou gerais45. Este fármaco é considerado uma das opções farmacológicas, a iniciar após a cirurgia, para a pro-filaxia da cirurgia eletiva da artroplastia da anca e joelho e da cirurgia de fratura da extremidade proxi-mal do fémur, segundo a Norma da Direção-Geral da Saúde 026/2012 já referida, com nível de evidência A, grau de recomendação I.
Está também licenciado no tratamento de indivídu-os com tromboembolismo venindivídu-oso com síndrome coronária aguda com ou sem elevação do segmento ST. Vários estudos – EPHESUS, PENTATHLON, PENTHIFRA e PENTAMAKS – demonstraram que o fondaparinux é superior à enoxaparina em cirurgias ortopédicas de alto risco como fratura da anca, subs-tituição anca e do joelho46.
Idraparinux e idrabiotaparinux
O idraparinux pertence à segunda geração de pentassa-carídeos sintéticos, derivando de uma hipermetilação do fondaparinux com elevada afinidade para a antitrombi-na. Este fármaco é eliminado via renal e apresenta uma semivida plasmática longa de 80 a 130 horas, o que per-mite uma única administração subcutânea semanal. O principal problema do idraparinux é a eliminação retar-dada após prolongada administração, o que pode expli-car a elevada incidência de complicações hemorrágicas observadas nos ensaios clínicos13,46. Por este motivo, foi desenvolvido o idrabiotaparinux, pentassacarídeo reversível de longa ação30, de modo a obter uma rápida inibição da atividade anticoagulante. A diferença entre o idrabiotaparinux e o idraparinux é que o primeiro tem na sua estrutura uma fração de biotina, conferindo-lhe a capacidade de reverter o efeito anticoagulante com uma administração endovenosa de avidina. A avidina é uma proteína da clara do ovo, que se liga à fração de biotina com elevada afinidade, formando um complexo que é rapidamente excretado pelo rim46. A sua biodisponibi-lidade é de 100 por cento e a semivida de 80 horas. A ex-creção é renal e não apresenta a desvantagem de causar trombocitopenia induzida por heparina30.
Novos antagonistas da vitamina K
A tecarfarina é um antagonista oral da
se da vitamina K que apresenta uma semivida média de 119 horas, com um intervalo entre as 107 e as 140 horas. A sua estrutura é análoga à varfarina, com uma ligação às proteínas plasmáticas de aproximadamen-te 99 por cento. A sua metabolização é efetuada pelas carboxilesterases nos microssomas hepáticos, originan-do um único metabolito inativo, o ATI-5900. O meca-nismo de ação é semelhante ao da varfarina. A principal diferença com os outros antagonistas da vitamina K é o facto de a tecarfarina ser um único enantiómero, em vez de uma mistura, e não ser metabolizado pelo citocro-mo P450. Consequentemente, as possíveis interações com outros fármacos e alimentos são mínimas1,35. Este último aspeto foi o mais considerado na conceção deste novo fármaco para diminuir as possíveis interações com os inibidores ou indutores do CYP450. A biotransfor-mação da tecarfarina é completamente independente da NADPH, cofator necessário para o CYP45034. A monitorização deste fármaco é efetuada pela medição do INR, tal como os outros antagonistas da vitamina K35. A segurança, tolerabilidade e eficácia deste fármaco foram avaliadas num estudo open-label de fase IIA35. No primeiro estudo, Tecarfarin-CLN-504, realizado em indivíduos com FA, este fármaco foi bem tolerado, sem toxicidade aparente ou clínica e sem evidência bioquí-mica de acumulação do fármaco ou seu metabolito35. Dado que um anticoagulante oral com intervalo de tem-po terapêutico baixo está associado a piores resultados, e que a tecarfarina apresenta um melhor intervalo de tempo terapêutico, tal sugere que possa resultar em melhoria das taxas de eventos tromboembólicos ou he-morrágicos em indivíduos que necessitam de terapêuti-ca anticoagulante35.
A tecarfarina pode proporcionar uma especial vantagem sobre a varfarina em indivíduos com genótipos variantes CYP2C9, devido à ausência de interações de CYP2C9 com alimentos e fármacos ou outras interações farma-cogenomicas. Esta população em estudos com varfarina apresenta um intervalo de tempo terapêutico baixo e valores de INR supraterapêuticos elevados e uma ele-vada propensão para hemorragias. Relativamente aos indivíduos com variante no polimorfismo VKORC1, não apresenta especial vantagem sobre a varfarina, con-sistente com o facto de terem um mecanismo de ação semelhante35.
Conclusões
A aterotrombose e o tromboembolismo venoso são ain-da a principal causa de morbiliain-dade e de mortaliain-dade. A terapêutica anticoagulante é fundamental para dimi-nuir esta morbimortalidade dos indivíduos, pois permi-te a não ocorrência ou aumento de trombos. Ao longo
de mais de 40 anos, os únicos fármacos disponíveis para uso clínico eram os antagonistas da vitamina K e a hepa-rina não fracionada. Apesar de serem amplamente uti-lizados e eficazes, apresentam significativas desvanta-gens. A varfarina tem uma estreita margem terapêutica, uma imprevisibilidade de dose-resposta causada pelos fatores genéticos e inumeras interações com medica-mentos e alimedica-mentos, devido ao seu metabolismo ocorrer a nível do citocromo P450. A heparina não fracionada tem origem animal, é administrada via endovenosa e pode causar trombocitopenia induzida por heparina. Ambos necessitam de monitorização laboratorial para garantir a sua eficácia e segurança. Como tal, no senti-do de ultrapassar estas limitações, surgiu a heparina de baixo peso molecular, com uma resposta mais previsí-vel, permitindo uma dose fixa e a dispensa de monitori-zação. No entanto, continua a apresentar desvantagens, como a trombocitopenia induzida pela heparina, a ad-ministração parentérica e a excreção renal.
Nos últimos anos intensificou-se a investigação com o intuito de desenvolver novos fármacos anticoagulantes, que permitissem ultrapassar as limitações dos anticoa-gulantes tradicionais. A estratégia destes novos fárma-cos antitrombótifárma-cos baseia-se na inibição seletiva de um fator de coagulação específico, obtendo assim ini-bidores seletivos com potencial de serem mais eficazes, mais seguros e mais fáceis de usar. Esta nova classe tem como principais alvos a trombina e o FXa.
Os inibidores do FIIa oferecem as seguintes vanta-gens: inibição direta da trombina ou da trombina ligada ao coágulo, a não necessidade de cofatores, previsibilidade de resposta e ausência de trombocito-penia induzida por heparina. Estes inibidores podem ser parentéricos (bivalirudina, desirudina, lepirudina e argatrobano) ou orais, como o dabigatrano, que apre-senta como vantagens um perfil farmacocinético e far-macodinâmico previsível, ausência de interações devido ao citocromo P450, rápido início de ação, excelente per-fil de segurança, não necessitando de monitorização de rotina, ampla janela terapêutica, dose de administração fixa; todas estas qualidades de um anticoagulante ideal. Quanto aos inibidores FXa, temos o fondaparinux, pentassacárido sintético, que se liga indiretamente à antitrombina. Outros inibidores seletivos do FXa de administração oral são o rivaroxabano, o apixabano e o edoxabano.
Apesar das inúmeras vantagens desta nova classe de antitrombóticos seletivos, existem algumas limitações. Nenhum deles, com exceção do idrabiotaparinux, tem antídoto para a reversão do efeito anticoagulante. Outra das limitações comum a vários destes fármacos é a sua excreção renal, tendo a dose administrada de ser
46
da ou estando mesmo contraindicado em situações se-veras. Ao contrário da maioria, o argatrobano tem uma excreção hepática e, como tal, a sua contraindicação é a insuficiência hepática grave. Relativamente aos inibi-dores da trombina, temos ainda a estreita margem tera-pêutica da lepirudina, tal como a formação de anticorpos anti-hirudinas. Os inibidores do FXa, nomeadamente o apixabano e o rivaroxabano, têm como principal des-vantagem a sua potencial interação com inibidores ou indutores de CYP3A4 e P-glicoproteína.
Em Portugal, estão aprovados e comercializáveis o fondaparinux sódico, o dabigatrano etexilato e o rivaroxabano.
Estão em investigação vários inibidores diretos orais do FIIa, sendo o que apresenta maior potencialidade o AZD0837, um pró-fármaco rapidamente convertido na sua forma ativa com atividade farmacológica superior ao xime-legatrano, mas sem os seus problemas de toxicidade11. Além dos inibidores do FXa referidos neste traba-lho, existem outros em investigação, quer para admi-nistração oral quer parentérica. Entre eles, temos: a semuloparina e o M118, heparinas de ultrabaixo peso molecular, que funcionam como inibidores indiretos. O betrixabano é um inibidor competitivo e direto do FXa, capaz de bloquear tanto o FXa livre como a protrombinase ligada ao FXa. A sua excreção é quase exclusivamente biliar e a depuração renal baixa, o que se apresenta como uma vantagem para doentes com insuficiência renal, e a sua metabolização também não ocorre a nível do citocro-mo P450, sendo assim as interações com medicamentos e alimentos mínimas. Outros inibidores específicos, dire-tos e de administração oral do FXa, são o YM150 e o TAK-44246.
Além dos alvos já referidos, também o fator V está a ser alvo de novos anticoagulantes. O FVa tem um papel crucial na ativação da coagulação e produção de trom-bina; mais especificamente, atua como cofator do FXa e os dois juntamente com os fosfolípidos da membrana das plaquetas formam o complexo da protrombinase que converte a protrombina em trombina na mem-brana celular. Nesta classe inclui-se a drotrecogina alfa ativada e o ART-12346. A drotrecogina alfa ativa-da foi aprovaativa-da pela FDA para indivíduos com sepsis e elevado risco de morte52.
Referências Bibliográficas
1. Berkovits A, Aizman A, Zúñiga P, et al. Nuevos anticoagulantes orales. Rev Med Chile. 2011;139:1347-1355.
2. Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists. Chest. 2008;133:160S-198S.
3. Mavrakanas T, Bounameaux H. The potential role of new oral anticoagulants in the prevention and treatment of thromboembolism. Pharmacology & Therapeutics. 2011;130:46-58.
4. Galanis T, Thomson L, Palladino M, et al. New oral anticoagulants. J Thromb Thrombolysis. 2011;31:310-320.
5. Liem T, DeLoughery T. Direct thrombin inhibitors for the treatment of venous thromboembolism:analysis of the dabigatran versus warfarin clinical trial. Semin Vasc Surg.2011;24:157-161. 6. Vandita J, Chandravathi L. Brief overview of the coagulation cascade. Dis Mon. [revista em linha]. 2012 [citado em outubro de 2012];58:421-423. Disponível em http://dx.doi.org/10.1016/j. disamonth.2012.04.004.
7. Ma T, Yan B, Lam Y. Dabigatran etexilate versus warfarin as the oral anticoagulant of choice? A review of clinical data. Pharmacology & Therapeutics. 2011;129:185-194.
8. Adams R, Bird R. Review article: coagulation cascade and therapeutics update: relevance to nephrology. part 1: overview of coagulation, thrombophilias and history of anticoagulants. Nephrology. 2009;14:462-470.
9. Hoffman M, Monroe D. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007;21:1-11.
10. Ott I. Inhibitors of the initiation of coagulation. Br J Clin Pharmacol. 2011;72(4):547-552.
11. Lee C, Ansell J. Direct thrombin inhibitors. Br J Clin Pharmacol. 2011;72:581-592.
12. Maegdefessel L, Spin J, Azuma J, et al. New options with dabigatran etexilate in anticoagulant therapy. Vascular Health and Risk Management. 2010;6:339-349.
13. Franchini M, Mannucci P. A new era for anticoagulants. European Journal of Internal Medicine. 2009;20:562-568.
14. Cohen A, Agnelli G, Anderson F, et al. Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. 2007;98:756-764.
15. Brenner B, Hoffman R. Emerging options in the treatment of deep vein thrombosis and pulmonary embolism. Blood Reviews. 2011;25:215-221. 16. Becattini C, Vedovati M, Agnelli G. Old and new oral anticoagulants for venous thromboembolism and atrial fibrillation: A review of the literature. Thrombosis Research. 2012;129:392-400.
17. Zhu T, Martinez I, Emmerich J. Venous thromboembolism: Risk factors for recurrence. Arterioscler Thromb Vasc Biol. 2009;29:298-310. 18. Tapson V, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension from acute to chronic pulmonary embolism. Proc Am Thorac Soc. 2006;3:564-567. 19. Kearon C, Kahn R, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th Ed). Chest. 2008;133:454S-545S.
20. Walenga J, Adiguzel C. Drug and dietary interactions of the new and emerging oral anticoagulants. Int J Clin Pract. 2010;64:956-967. 21. Bereznicki L, Peterson G. New antithrombotics for atrial fibrillation. Cardiovascular Therapeutics. 2010;28:278-286. 22. Connolly S, Ezekowitz M, Yusuf S. Dabigatran versus warfarin in patients with atrial fibrillation. 2009;361(12):1139-1151. 23. Hirsh J, Bauer K, Donati M, et al. Parenteral anticoagulants: American College of Chest Physicians evidence-based clinical practice guidelines (8th Ed). Chest. 2008;133:141S-159S.