Diazoxide prevents reactive oxygen species and mitochondrial
damage, leading to anti-hypertrophic effects
Aline M. Lucas
a, Francisco R. Caldas
a, Amanda P. da Silva
a, Maximiano M. Ventura
a,
Iago M. Leite
a, Ana B. Filgueiras
a, Claúdio G.L. Silva
a, Alicia J. Kowaltowski
b,
Heberty T. Facundo
a,*aFaculdade de Medicina, Universidade Federal do Cariri, Barbalha, CE, Brazil bDepartamento de Bioquímica, Instituto de Química, Universidade de S~ao Paulo, Brazil
a r t i c l e
i n f o
Article history:
Received 22 September 2016 Received in revised form 25 October 2016
Accepted 16 November 2016 Available online 17 November 2016
Keywords: Mitochondria Hypertrophy Oxidative stress Free radicals Calcium
a b s t r a c t
Pathological cardiac hypertrophy is characterized by wall thickening or chamber enlargement of the heart in response to pressure or volume overload, respectively. This condition will, initially, improve the organ contractile function, but if sustained will render dysfunctional mitochondria and oxidative stress. Mitochondrial ATP-sensitive Kþchannels (mitoKATP) modulate the redox status of the cell and protect
against several cardiac insults. Here, we tested the hypothesis that mitoKATP opening (using diazoxide) will avoid isoproterenol-induced cardiac hypertrophyin vivoby decreasing reactive oxygen species (ROS) production and mitochondrial Ca2þ-induced swelling. To induce cardiac hypertrophy, Swiss mice were
treated intraperitoneally with isoproterenol (30 mg/kg/day) for 8 days. Diazoxide (5 mg/kg/day) was used to open mitoKATP and 5-hydroxydecanoate (5 mg/kg/day) was administrated as a mitoKATP blocker. Isoproterenol-treated mice had elevated heart weight/tibia length ratios and increased myocyte cross-sectional areas. Additionally, hypertrophic hearts produced higher levels of H2O2and had lower glutathione peroxidase activity. In contrast, mitoKATP opening with diazoxide blocked all isoproterenol effects in a manner reversed by 5-hydroxydecanoate. Isolated mitochondria from Isoproterenol-induced hypertrophic hearts had increased susceptibility to Ca2þ
-induced swelling secondary to mitochondrial permeability transition pore opening. MitokATP opening was accompanied by lower Ca2þ
-induced mitochondrial swelling, an effect blocked by 5-hydroxydecanoate. Our results suggest that mitoKATP opening negatively regulates cardiac hypertrophy by avoiding oxidative impairment and mitochondrial damage.
©2016 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
Cardiac hypertrophy is initially an adaptive response in which the cardiac tissue grows and changes to accommodate hemody-namic overload [1]. Even though the human heart is a dynamic organ, the maintenance of the hemodynamic insult results in pro-gressive tissue deterioration and is an important risk factor for the development of heart failure [2]. Cardiac hypertrophy is also a major contributing factor to sudden cardiac death and morbidity in the Western world[3].
There is increasing evidence for the involvement of oxidative
imbalance in cardiac hypertrophy and heart failure[4e13]. What-ever the mechanism for increased oxidative imbalance,finding new tools to prevent it would be desirable. Thefirst evidence for the role of oxidative imbalance in cardiac hypertrophy emerged from a study reporting that angiotensin II induced reactive oxygen species (ROS) generation in cardiac myocytes [9]. This hypothesis was confirmed by other groups showing that hypertrophy can be abrogated by antioxidants[4,5,12,13]. Conversely, ROS are capable of inducing hypertrophic features in cardiomyocytes[14,15]. In this line, we have recently shown that cardiac hypertrophy suppressed the activity of antioxidant enzymes catalase and superoxide dis-mutase. Additionally, the opening of the mitochondrial ATP-sensitive potassium channel (mitoKATP) reversed the impairment of superoxide dismutase activity[16].
The transition of hypertrophy to heart failure is accompanied by *Corresponding author. Universidade Federal do Cariri, Rua Divino Salvador, 284,
Barbalha, Ceara, 63180-000, Brazil.
E-mail address:[email protected](H.T. Facundo).
Contents lists available atScienceDirect
Chemico-Biological Interactions
j o u r n a l h o m e p a g e :w w w . e l s e v i e r . c o m / l o c a t e / c h e m b i o i n t
http://dx.doi.org/10.1016/j.cbi.2016.11.012
mitochondrial damage [17]. Perturbed mitochondrial function would impair the ability of this organelle to produce ATP efficiently. It is important to point out that mitochondria produce 90% of all intracellular ATP[18]. Additionally, cardiac Ca2þ
participates as an obligatory signaling molecule resulting in increased calcium-dependent activation of intracellular hypertrophic factors [19]. Interestingly, stimulating mitochondrial potassiumflux, through mitoKATP opening triggers cardioprotection by decreasing mito-chondrial[20]and cytosolic[21] Ca2þ
accumulation. Cardiac hy-pertrophy induces mitochondrial Ca2þ overload and oxidative
imbalance triggering the opening of the mitochondrial perme-ability transition pore (mPTP)[22].
The aims of the present study were to investigate whether the opening of the mitoKATP, using diazoxide, triggers anti-hypertrophic effects by changing the cellular redox status and to determine if diazoxide avoids Ca2þ-induced mPTP opening during
cardiac hypertrophy as part of its mechanism of action. Ourfindings suggest that diazoxide may protect against cardiac hypertrophy through attenuation of ROS and mPTP formation.
2. Materials and methods
2.1. Animals
All animals were used in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The protocol was approved by the institutional Animal Experimentation Ethics Committee. All mice used were 60-day-old Swiss Male weighting between 25 and 30 g.
2.2. Hypertrophy induction
The animals were treated daily (for thefirst four days) with intraperitoneal (i.p.) injections of saline (0.9%econtrol group) or isoproterenol (30 mg/kg/dayeISO group). From that point on, all drugs were administered with a solution containing saline (0.9%) and DMSO (2%) and mice receiving isoproterenol were randomly divided into 3 groups and treated for four more days. In summary, one group received isoproterenol alone (30 mg/kg/day e ISO group), another received isoproterenol (30 mg/kg/day) plus diaz-oxide (5 mg/kg/daye DZX group) and the third group received isoproterenol (30 mg/kg/day) plus diazoxide (5 mg/kg/day) plus 5-hydroxydecanoate (5 mg/kg/day e 5-HD group). 5-hydroxydecanoate was administered 20 min before diazoxide in order to inhibit the mitoKATP. A solution containing saline (0.9%) and DMSO (2%) was administered to the control group. A summary of the treatment is depicted onTable 1.
2.3. Histology and image analysis
Hearts were collected and rapidlyfixed in 10% buffered formalin, and subsequently embedded in paraffin. Serial 5-
m
m heart sectionsfrom each group were cut transversely and stained with H&E for histological analysis. Only myocytes cut transversely with both a visible nucleus and unbroken cellular membrane were used for cross-sectional area measurement. We used the Image J software (from National Institutes of Health) to trace the outer border of the myocytes.
2.4. H2O2measurements
H2O2production was measured using Amplex Red (Molecular Probes) according to the manufacturer's instructions. In brief, left ventricles were cut into blocks (50 mg) and incubated (protected from light) with Amplex Red (50
m
mol/L) and horseradish peroxi-dase (1 U/mL) for 30 min at 37C in Krebs-Hepes buffer containing(in mM) 118 NaCl, 25 NaHCO3, 1.2 KH2PO4, 4.7 KCl, 1.2 MgSO4, 1.25 CaCl2, 10 glucose, and 10 Hepes, pH 7.4. The tubes were centrifuged at 5000gfor 2 min, the supernatant was transferred to a cuvette, and the absorbance measured at 560 nm. Background absorbance was determined by incubating Amplex red and horseradish peroxidase without the sample. H2O2release was calculated using H2O2standards to create a calibration curve (Fig. 2a) and expressed as
m
mol/50 mg tissue.2.5. Glutathione peroxidase activity assay
The left ventricle tissue was homogenized in ice-cold buffer containing 10 mM Tris, 1 mM EDTA and 20% Sucrose, pH 7.4 at 4C.
The homogenates were centrifuged at 12,000gfor 30 min at 4C.
The resulting supernatant was used. Glutathione peroxidase ac-tivity was determined by the decrease in NADPH concentrations. We used 1 mM cumene hydroperoxide (Sigma Aldrich) as a sub-strate. First the cellular extract was added to a buffer containing 50 mM potassium phosphate, 0.5 mM EDTA, 0.2 mM NADPH (Sigma Aldrich),1 mM GSH (Sigma Aldrich) and 0.2 U/mL Glutha-tione Reductase purified forS. cerevisiae(Sigma Aldrich), pH 7.0. After 5 min incubation, cumene hydroperoxide was added to initiate the reaction. We followed the disappearance of the co-substrate NADPH at 340 nm for 5 min. Glutathione peroxidase ac-tivity is expressed as Units/mg protein.
2.6. Mitochondrial isolation
Mitochondria were isolated by differential centrifugation. Briefly, mice hearts were removed and immediately washed in ice-cold buffer containing 300 mM sucrose, 10 mM Kþ
Hepes buffer, pH 7.2, and 1 mM Kþ
EGTA. The tissue was minced finely and then homogenized manually. Nuclei and cellular residues were pelleted by centrifugation at 600gfor 5 min. In order to obtain the mito-chondrial pellet the supernatant was recentrifuged at 9000g for 8 min. This pellet was washed to eliminate contaminating blood and resuspended in a minimal amount of buffer (~100
m
L). Samples were kept over ice and used within 1 h of isolation, to ensureTable 1
Treatment protocol.
Group Treatment
Control Saline (0.9%) for thefirst 4 days
Saline (0.9%) and DMSO (2%) for the following 4 days ISO 30 mg/kg/day isoproterenol in saline (0.9%) for thefirst 4 days
30 mg/kg/day isoproterenol in saline (0.9%) and DMSO (2%) for the following 4 days DZX 30 mg/kg/day isoproterenol in saline (0.9%) for thefirst 4 days
30 mg/kg/day isoproterenol and 5 mg/kg/day diazoxide in saline (0.9%) and DMSO (2%) for the following 4 days 5-HD 30 mg/kg/day isoproterenol in saline (0.9%) for thefirst 4 days.
mitoKATP activity and its pharmacological[23]regulation.
2.7. Mitochondrial swelling
Changes in light scattering due to Ca2þ uptake and swelling
were measured as a decrease over time in optical density at 520 nm. Mouse heart mitochondria (0.2 mg protein/mL) were incubated in buffer containing 100 mM KCl, 10 mM Hepes, 2 mM succinate, 2 mM MgCl2, 2 mM KH2PO4, 1
m
g/mL oligomycin, pH 7.2 (KOH). To induce opening of the mitochondrial permeability tran-sition pore, 2m
M Ca2þwas added.
2.8. Protein content
Total protein content in each sample was estimated using the Biuret method and bovine serum albumin as a standard.
2.9. Statistical analysis
Statistical analysis was conducted using Graphpad Prism soft-ware. Data are presented as mean±S.E.M. Student'st-test or one-way analysis of variance followed by a Tukey's post hoc test was performed where indicated. P<0.05 was considered statistically significant.
3. Results
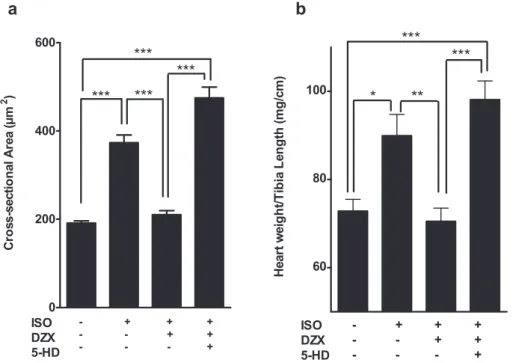
3.1. Diazoxide attenuates cardiac hypertrophy in vivo
In order to investigate the impact of mitoKATP opening on isoproterenol-induced cardiac hypertrophyin vivo, we pharmaco-logically induced mitoKATP opening using diazoxide (a mitoKATP opener). As expected, treating mice with isoproterenol for 8 days significantly increased myocyte cross-sectional areas (Fig. 1a, Supplementary Fig. 1) and heart weight/tibia length relationships (HW/TL eFig. 1b). Interestingly, mitoKATP opening significantly
prevented the elevation of both isoproterenol-induced hypertrophy gross markers. Strikingly, co-treatment with 5-hydroxydecanoate (a mitoKATP blocker) reversed the protective effect of mitoKATP opening.
3.2. Effect diazoxide on cardiac oxidative stress during isoproterenol-induced hypertrophy
We hypothesized that mitoKATP opening could protect myo-cytes from isoproterenol-induced hypertrophy by changing the intracellular redox state. To test this hypothesis, wefirst tested if the gross regression of cardiac hypertrophy was correlated with changes in oxidant generation. Using an Amplex Red assay (as described in Materials and Methods) we detected an increased amount of H2O2 released by hypertrophied hearts. Strikingly, mitoKATP opening by diazoxide treatment suppressed H2O2 pro-duction by ventricular blocks. This effect was reversed by concomitant treatment with 5-hydroxydecanoate treatment (Fig. 2b). H2O2is generated by dismutation of superoxide radical anions and removed by the enzymatic activity of catalase and glutathione peroxidase. Previously, we found that mitoKATP opening had no effect on catalase activity[16]. To further address the impact of isoproterenol-induced cardiac hypertrophy on other H2O2 removal systems we measured the activity of glutathione peroxidase. Glutathione peroxidase activity was suppressed during cardiac hypertrophy (Fig. 2c), a result which at least partially ex-plains the higher H2O2release by hypertrophic hearts. Strikingly, diazoxide treatment enhanced cardiac glutathione peroxidase ac-tivity. As expected, 5-hydroxydecanoate downregulated it (Fig. 2c).
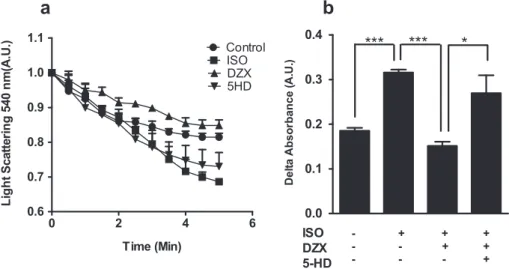
3.3. MitoKATP protects mitochondria against calcium-induced MPTP opening during hypertrophy
In order to examine if cardiac mitochondria from treated mice had increased susceptibility to Ca2þ
-induced mPTP, we treated isolated mitochondria from mice treated as in Materials and
Fig. 1. MitoKATP opening attenuates cardiac hypertrophy in vivo. a, Bar graph showing quantitative analysis of cross-sectional area of myocytes stained with hematoxylin and eosin (400) 8 days after isoproterenol (ISO), diazoxide (DZX), or 5-hydroxydecanoate (5-HD) treatments. b, Heart weight/tibia length ratio 8 days after isoproterenol (ISO),
Methods with a Ca2þ
bolus (2
m
M/0.2 mg mitochondrial protein). Cardiac isolated mitochondria from mice treated with isoproter-enol presented increased susceptibility to mPTP opening, seen as decreased light scattering in 540 nm (Fig. 3). This effect was reversed by mitoKATP opening using diazoxide, in a 5-hydroxydecanoate-inhibited manner.Taken together, these results indicate that mitoKATP opening exerts counter-regulatory effects on cardiac hypertrophy, in part by avoiding mitochondrial oxidant generation and damage.
4. Discussion
The present study shows that cardiac hypertrophy induces oxidative imbalance with decreased glutathione peroxidase activity and mitochondrial swelling in response to high Ca2þ
loads. Treating mice with diazoxide (a mitoKATP opener) prevented
isoproterenol-induced hypertrophy[16]in a manner accompanied by the pres-ervation of glutathione peroxidase activity, low H2O2release and inhibition of calcium-induced mitochondrial swelling. Our data, taken together with other studies[16,24,25]suggest that protecting mitochondria by mitoKATP opening can be a valuable tool to avoid cardiac hypertrophy and associated functional loss. Evidence for this hypothesis emerged in 2004 when Xia and colleagues [24] demonstrated that diazoxide inhibited phenylefrine-induced hy-pertrophy in isolated rat cardiomyocytes by modulation of Na-H exchanger isoform 1. Moreover, mitoKATP opening has been asso-ciated with the antihypertrophic effect of K-opioid receptor acti-vation[25]. Our study extends these observations by showing that mitoKATP exert anti-hypertrophic effects in a manner associated to the maintenance of intracellular redox balance and mitochondrial function.
hypertrophy [4,5,12e16]. Enhanced oxidant production exacer-bates hypertrophy[5,13e15]while antioxidants blocked it[4,12]. More specifically, treatment with N-2-mercaptopropionylglycine, an antioxidant, alleviates cardiac hypertrophyin vivo[4]. In addi-tion, the inhibition of SOD[26]or its cardiac specific knockout[27] causes cardiac hypertrophy. Moreover, direct administration of H2O2 induces hypertrophy in isolated cardiomyocytes [14]. Oxi-dants are also implicated in the expression of the hypertrophic marker beta-myosin heavy chain via MAPK pathway [15]. Our studies suggested a simultaneous attenuation of antioxidant en-zymes catalase and superoxide dismutase (SOD) during hypertro-phy, while mitoKATP opening leads to an upregulation of superoxide dismutase [16]. Here we add to this evidence by showing that mitoKATP activation promotes the upregulation of another antioxidant enzyme (glutathione peroxidase). This enzyme transforms H2O2to H2O, which partially explains the lower H2O2 release seen with mitoKATP opening. Furthermore, together with prior results showing that mitoKATP activation increases SOD ac-tivity[16]our results suggest that opening of this channel enhances the full system necessary for the removal of both superoxide rad-icals and H2O2produced by their dismutation. Even though the mechanism by which mitoKATP channel opening leads to the maintenance of glutathione peroxidase has not been clarified in the present study we believe that the mitochondrial protection induced by diazoxide will, at least in part, be responsible for the maintenance of Glutathione Peroxidase activity on hypertrophic hearts. Glutathione peroxidase exists in cytosolic and mitochon-drial forms [28,29]and plays a critical role in H2O2and organic peroxide metabolism in the heart. On the other hand, the exact mechanism(s) by which diazoxide enhance glutathione peroxidase in our model of hypertrophy in unknown and remains to be elucidated.
Upregulation of cardiac contractility with isoproterenol leads to mitochondrial Ca2þ
overload and oxidative imbalance with conse-quent mPTP opening[22]. Under high intracellular Ca2þconditions,
mitochondria may function as a buffer to control cytosolic Ca2þ
concentrations[30]. It is therefore possible that mitoKATP opening during hypertrophy may result in mitochondria more resistant against Ca2þ
overload[31]and ROS generation[32]. Indeed, mito-chondria from hypertrophied hearts had greater vulnerability to stress-induced opening of the mPTP[33]in a manner reversed by mitoKATP opening promoted by diazoxide treatment (this
manuscript). Diazoxide treatmentin vivoalso attenuatesin vitro mPTP formation, while its inhibition sensitizes mitochondria to mPTP opening.
Perhaps a more important consideration is related to the multifactorial nature of the cardiac hypertrophic response itself. Clinically used drugs such as blockers of beta receptors, calcium channels and angiotensin converting enzymes have other effects that might be beneficial for hypertrophy treatment. For example, carvedilol (a beta receptor blocker) protects heart mitochondria from oxidative stress-induced injury [34] and mitochondrial swelling[35]and reverses cardiac hypertrophy[36]. Mitochondrial swelling was also inhibited by the angiotensin receptor type I blocker losartan[37]. Here we found that mitoKATP opening (using diazoxide) avoids isoproterenol-induced cardiac hypertrophy by decreasing oxidant formation and mitochondrial swelling. This drug has no documented actions over
b
receptors and angiotensin receptors. Even thoughb
receptors and angiotensin receptor blockers do not target mitochondria alone, our results suggest that mitochondrial protection represents an efficient anti-hypertrophic therapeutic approach. Therefore, we conclude that mitoKATP opening negatively regulates cardiac hypertrophy by avoiding oxidative impairment and mitochondrial damage.Conflict of interest statement
The authors declare that they do not have any conflict of interest.
Acknowledgements
This research was supported by Conselho Nacional de Desen-volvimento Científico e Tecnologico e CNPq to Heberty Tarso Facundo (Grant number 443773/2014-9) and to Claudio G da Silva (Grant number 441965/2014-8). Alicia J. Kowaltowski is supported by The Centro de Pesquisa, Inovaç~ao e Difus~ao de Processos Redox em Biomedicina (FAPESP 13/07937-8) and CNPq. We acknowledge the technical assistance of Ant^onio F. R. Santos. Iago M. R. Leite and Ana B. T. Filgueiras are recipients of research scholarship from CNPq. Aline M. Lucas is a scholarship holder from Fundaç~ao Cear-ense de Apoio ao Desenvolvimento Científico e Tecnologico (Fun-cap). Camille C. da Silva is gratefully acknowledged for providing access to research consumables.
Fig. 3. MitoKATP protects mitochondria against calcium-induced mPTP opening during hypertrophy. Mouse heart mitochondria were isolated after 8 days following vehicle (Control), isoproterenol (ISO), diazoxide (DZX) or 5-hydroxydecanoate (5-HD) treatments. Mitochondria (0.2 mg protein/mL) were incubated in 100 mM KCl, 10 mM Hepes, 2 mM succinate, 2 mM MgCl2, 2 mM KH2PO4, 1mg/mL oligomycin, pH 7.2. mPTP opening was induced by the addition of 2mM Ca2þ
Transparency document
Transparency document related to this article can be found online athttp://dx.doi.org/10.1016/j.cbi.2016.11.012.
Appendix A. Supplementary data
Supplementary data related to this article can be found athttp:// dx.doi.org/10.1016/j.cbi.2016.11.012.
References
[1] N. Frey, E.N. Olson, Cardiac hypertrophy: the good, the bad, and the ugly, Annu. Rev. Physiol. 65 (2003) 45e79, http://dx.doi.org/10.1146/ annurev.physiol.65.092101.142243.
[2] B.A. Vakili, P.M. Okin, R.B. Devereux, Prognostic implications of left ventricular hypertrophy, Am. Heart J. 141 (2001) 334e341,http://dx.doi.org/10.1067/ mhj.2001.113218.
[3] A.S. Go, D. Mozaffarian, V.L. Roger, E.J. Benjamin, J.D. Berry, W.B. Borden, D.M. Bravata, S. Dai, E.S. Ford, C.S. Fox, S. Franco, H.J. Fullerton, C. Gillespie, S.M. Hailpern, J.A. Heit, V.J. Howard, M.D. Huffman, B.M. Kissela, S.J. Kittner, D.T. Lackland, J.H. Lichtman, L.D. Lisabeth, D. Magid, G.M. Marcus, A. Marelli, D.B. Matchar, D.K. McGuire, E.R. Mohler, C.S. Moy, M.E. Mussolino, G. Nichol, N.P. Paynter, P.J. Schreiner, P.D. Sorlie, J. Stein, T.N. Turan, S.S. Virani, N.D. Wong, D. Woo, M.B. Turner, Heart disease and stroke statisticse2013 update: a report from the American Heart Association, Circulation 127 (2013) e6ee245,http://dx.doi.org/10.1161/cir.0b013e31828124ad.
[4] M. Date, T. Morita, N. Yamashita, K. Nishida, O. Yamaguchi, Y. Higuchi, S. Hirotani, Y. Matsumura, M. Hori, M. Tada, K. Otsu, The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model, J. Am. Coll. Cardiol. 39 (2002) 907e912. [5] A.K. Dhalla, M.F. Hill, P.K. Singal, Role of oxidative stress in transition of
hy-pertrophy to heart failure, J. Am. Coll. Cardiol. 28 (1996) 506e514,http:// dx.doi.org/10.1016/0735-1097(96)00140-4.
[6] A.K. Dhalla, P.K. Singal, Antioxidant changes in hypertrophied and failing Guinea pig hearts, Am. J. Physiol. 266 (1994) H1280eH1285.
[7] S. Hirotani, K. Otsu, K. Nishida, Y. Higuchi, T. Morita, H. Nakayama, O. Yamaguchi, T. Mano, Y. Matsumura, H. Ueno, M. Tada, M. Hori, Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-pro-tein-coupled receptor agonist-induced cardiomyocyte hypertrophy, Circula-tion 105 (2002) 509e515.
[8] D.R. Pimentel, J.K. Amin, L. Xiao, T. Miller, J. Viereck, J. Oliver-Krasinski, R. Baliga, J. Wang, D.A. Siwik, K. Singh, P. Pagano, W.S. Colucci, D.B. Sawyer, Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes, Circ. Res. 89 (2001) 453e460.
[9] K. Nakamura, K. Fushimi, H. Kouchi, K. Mihara, M. Miyazaki, T. Ohe, M. Namba, Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertro-phy induced by tumor necrosis factor-alpha and angiotensin II, Circulation 98 (1998) 794e799.
[10] N.S. Rajasekaran, P. Connell, E.S. Christians, L.-J. Yan, R.P. Taylor, A. Orosz, X.Q. Zhang, T.J. Stevenson, R.M. Peshock, J.A. Leopold, W.H. Barry, J. Loscalzo, S.J. Odelberg, I.J. Benjamin, Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice, Cell 130 (2007) 427e439,http://dx.doi.org/10.1016/j.cell.2007.06.044.
[11] S. Matsushima, J. Kuroda, T. Ago, P. Zhai, J.Y. Park, L.-H. Xie, B. Tian, J. Sadoshima, Increased oxidative stress in the nucleus caused by Nox4 me-diates oxidation of HDAC4 and cardiac hypertrophy, Circ. Res. 112 (2013) 651e663,http://dx.doi.org/10.1161/circresaha.112.279760.
[12] D.-F. Dai, L.F. Santana, M. Vermulst, D.M. Tomazela, M.J. Emond, M.J. MacCoss, K. Gollahon, G.M. Martin, L.A. Loeb, W.C. Ladiges, P.S. Rabinovitch, Over-expression of catalase targeted to mitochondria attenuates murine cardiac aging, Circulation 119 (2009) 2789e2797, http://dx.doi.org/10.1161/ circulationaha.108.822403.
[13] Z. Xie, P. Kometiani, J. Liu, J. Li, J.I. Shapiro, A. Askari, Intracellular reactive oxygen species mediate the linkage of Naþ
/Kþ
-ATPase to hypertrophy and its marker genes in cardiac myocytes, J. Biol. Chem. 274 (1999) 19323e19328. [14] S.H. Kwon, D.R. Pimentel, A. Remondino, D.B. Sawyer, W.S. Colucci, H(2)O(2)
regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways, J. Mol. Cell Cardiol. 35 (2003) 615e621. [15] N.L. Shih, T.H. Cheng, S.H. Loh, P.Y. Cheng, D.L. Wang, Y.S. Chen, S.H. Liu,
C.C. Liew, J.J. Chen, Reactive oxygen species modulate angiotensin II-induced beta-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes, Biochem. Bio-phys. Res. Commun. 283 (2001) 143e148, http://dx.doi.org/10.1006/ bbrc.2001.4744.
[16] F.R. Lemos Caldas, I.M. Rocha Leite, A.B. Tavarez Filgueiras, I.L. de Figueiredo
Júnior, T.A. Gomes Marques de Sousa, P.R. Martins, A.J. Kowaltowski, H. di, T. Fernandes Facundo, Mitochondrial ATP-sensitive potassium channel opening inhibits isoproterenol-induced cardiac hypertrophy by preventing oxidative damage, J. Cardiovasc. Pharmacol. 65 (2015) 393e397, http:// dx.doi.org/10.1097/fjc.0000000000000210.
[17] M. Osterholt, T.D. Nguyen, M. Schwarzer, T. Doenst, Alterations in mito-chondrial function in cardiac hypertrophy and heart failure, Heart Fail. Rev. 18 (2013) 645e656,http://dx.doi.org/10.1007/s10741-012-9346-7.
[18] R. Ventura-Clapier, A. Garnier, V. Veksler, Energy metabolism in heart failure, J. Physiol. 555 (2004) 1e13,http://dx.doi.org/10.1113/jphysiol.2003.055095. [19] N. Frey, T.A. McKinsey, E.N. Olson, Decoding calcium signals involved in
car-diac growth and function, Nat. Med. 6 (2000) 1221e1227,http://dx.doi.org/ 10.1038/81321.
[20] H.T.F. Facundo, J.G. de Paula, A.J. Kowaltowski, Mitochondrial ATP-sensitive Kþ channels prevent oxidative stress, permeability transition and cell death, J. Bioenerg. Biomembr. 37 (2005) 75e82, http://dx.doi.org/10.1007/s10863-005-4130-1.
[21] G. Gonzalez, D. Zaldívar, E. Carrillo, A. Hernandez, M. García, J. Sanchez, Pharmacological preconditioning by diazoxide downregulates cardiac L-type Ca(2þ)channels, Br. J. Pharmacol. 161 (2010) 1172e1185,http://dx.doi.org/ 10.1111/j.1476-5381.2010.00960.x.
[22] M. Izem-Meziane, B. Djerdjouri, S. Rimbaud, F. Caffin, D. Fortin, A. Garnier, V. Veksler, F. Joubert, R. Ventura-Clapier, Catecholamine-induced cardiac mitochondrial dysfunction and mPTP opening: protective effect of curcumin, Am. J. Physiol. Heart Circ. Physiol. 302 (2012) H665eH674,http://dx.doi.org/ 10.1152/ajpheart.00467.2011.
[23] H.T.F. Facundo, J.G. de Paula, A.J. Kowaltowski, Mitochondrial ATP-sensitive Kþ channels are redox-sensitive pathways that control reactive oxygen species production, Free Radic. Biol. Med. 42 (2007) 1039e1048,http://dx.doi.org/ 10.1016/j.freeradbiomed.2007.01.001.
[24] Y. Xia, V. Rajapurohitam, M.A. Cook, M. Karmazyn, Inhibition of phenylephrine induced hypertrophy in rat neonatal cardiomyocytes by the mitochondrial KATP channel opener diazoxide, J. Mol. Cell Cardiol. 37 (2004) 1063e1067, http://dx.doi.org/10.1016/j.yjmcc.2004.07.002.
[25] L. Zhang, H. Wang, M. Lu, G. Wu, Y. Yang, C. Liu, L.N. Maslov, K(ATP) channels mediate the antihypertrophic effects afforded byk-opioid receptor stimula-tion in neonatal rat ventricular myocytes, Exp. Ther. Med. 4 (2012) 261e266, http://dx.doi.org/10.3892/etm.2012.578.
[26] D.A. Siwik, J.D. Tzortzis, D.R. Pimental, D.L. Chang, P.J. Pagano, K. Singh, D.B. Sawyer, W.S. Colucci, Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro, Circ. Res. 85 (1999) 147e153.
[27] H. Koyama, H. Nojiri, S. Kawakami, T. Sunagawa, T. Shirasawa, T. Shimizu, Antioxidants improve the phenotypes of dilated cardiomyopathy and muscle fatigue in mitochondrial superoxide dismutase-deficient mice, Molecules 18 (2013) 1383e1393,http://dx.doi.org/10.3390/molecules18021383. [28] R.S. Esworthy, Y.S. Ho, F.F. Chu, The Gpx1 gene encodes mitochondrial
glutathione peroxidase in the mouse liver, Arch Biochem. Biophys 340 (1997) 59e63.
[29] M. Arai, H. Imai, D. Sumi, T. Imanaka, T. Takano, N. Chiba, Y. Nakagawa, Import into mitochondria of phospholipid hydroperoxide glutathione peroxidase requires a leader sequence, Biochem. Biophys. Res. Commun. 227 (1996) 433e439,http://dx.doi.org/10.1006/bbrc.1996.1525.
[30] G.S.B. Williams, L. Boyman, A.C. Chikando, R.J. Khairallah, W.J. Lederer, Mito-chondrial calcium uptake, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 10479e10486,http://dx.doi.org/10.1073/pnas.1300410110.
[31] S. Javadov, M. Karmazyn, Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for car-dioprotection, Cell Physiol. Biochem. 20 (2007) 1e22, http://dx.doi.org/ 10.1159/000103747.
[32] T.-I. Peng, M.-J. Jou, Oxidative stress caused by mitochondrial calcium over-load, Ann. N. Y. Acad. Sci. 1201 (2010) 183e188,http://dx.doi.org/10.1111/ j.1749-6632.2010.05634.x.
[33] M. Marcil, A. Ascah, J. Matas, S. Belanger, C. Deschepper, Y. Burelle, Compensated volume overload increases the vulnerability of heart mito-chondria without affecting their functions in the absence of stress, J. Mol. Cell Cardiol. 41 (2006) 998e1009,http://dx.doi.org/10.1016/j.yjmcc.2006.08.117. [34] P.J. Oliveira, A.P. Rolo, C.M. Palmeira, A.J. Moreno, Carvedilol reduces
mito-chondrial damage induced by hypoxanthine/xanthine oxidase: relevance to hypoxia/reoxygenation injury, Cardiovasc. Toxicol. 1 (2001) 205e213. [35] P.J. Oliveira, T. Esteves, A.P. Rolo, C.M. Palmeira, A.J.M. Moreno, Carvedilol
inhibits the mitochondrial permeability transition by an antioxidant mecha-nism, Cardiovasc. Toxicol. 4 (2004) 11e20.
[36] F.C. Barone, R.N. Willette, A.H. Nelson, E.H. Ohlstein, D.P. Brooks, R.W. Coatney, Carvedilol prevents and reverses hypertrophy-induced cardiac dysfunction, Pharmacology 80 (2007) 166e176,http://dx.doi.org/10.1159/000103384. [37] N. Mariappan, C.M. Elks, M. Haque, J. Francis, Interaction of TNF with