ANÁLISE DOS FATORES DETERMINANTES PARA A
QUALIDADE DA ANOTAÇÃO GENÔMICA AUTOMÁTICA
Dissertação apresentada ao Programa de Pós-Graduação “Strictu Sensu” em Biotecnologia e Ciências Genômicas da Universidade Católica de Brasília como requisito para a obtenção do Título de Mestre em Biotecnologia e Ciências Genômicas
Orientador: Prof. Dr. Georgios J. Pappas Jr.
TERMO DE APROVAÇÃO
Dissertação defendida e aprovada como requisito parcial para obtenção do Título de
Mestre em Ciências Genômicas e Biotecnologia, defendida e aprovada em 03 de setembro de
2004 pela banca examinadora constituída por:
________________________________________ Prof. Dr. Georgios J. Pappas Jr. (Orientador)
_________________________________________ Prof. Dr. Marcos Mota do Carmo Costa
A meus pais,
Pelo apoio e incentivo em todas as horas e por terem despertado em mim desde cedo a curiosidade e vontade de aprender.
À Keila,
Simplesmente pela pessoa maravilhosa que é.
Ao meu orientador Georgios,
Pela motivação e pelas importantes e decisivas sugestões.
A todos professores,
Pelo exemplo de persistência e coragem ao fazer pesquisa no Brasil.
Aos amigos e colegas,
Por entenderem esses tempos de reclusão e continuarem a ser amigos.
Aos funcionários pela competência e dedicação.
“Porque ele está lá”
Resumo
A genômica é uma disciplina recente da Biologia que diz respeito à caracterização
molecular de genomas na sua totalidade. A bioinformática é uma área interdisciplinar
envolvendo biologia, informática, matemática e estatística, que utiliza métodos
computacionais para analisar os dados de seqüências biológicas gerados pela genômica e
predizer a função e estrutura de macromoléculas.
A anotação genômica é um processo que consiste em adicionar análises de
bioinformática para gerar interpretações biológicas sobre as seqüências brutas de DNA. A
determinação ou predição da função de seqüências de aminoácidos, chamada de anotação
funcional, é uma das sub divisões da anotação genômica. A predição da função de uma
proteína é, via de regra, feita com base na comparação com seqüências de proteínas
previamente caracterizadas e armazenadas em bancos de dados biológicos. Essa comparação
é, quase sempre, feita com programas computacionais de alinhamento de seqüências, entre os
quais, o BLAST é um dos mais conhecidos. A anotação funcional é uma tarefa bastante
complexa e sujeita a erros por vários motivos. Um deles, diz respeito à falta de padronização
na interpretação dos resultados do BLAST. Outro problema é a propagação de erros, que é a
predição da função de proteínas baseada em proteínas que também foram preditas mas que
podem estar com a anotação imprecisa ou até incorreta.
A genômica gera uma grande quantidade de dados os quais são armazenados em
bancos de dados biológicos. Para a anotação funcional de proteínas, os bancos de dados de
seqüências de aminoácidos são obviamente fundamentais, pois conforme mencionado, são
usados na busca por seqüências similares. Atualmente, a taxa de crescimento desses bancos
de dados é exponencial, tornando a atividade de anotação funcional dinâmica. Por esse
motivo, a reanotação genômica - o processo no qual anota-se novamente uma proteína usando
um banco de dados mais recente - é uma atividade cada vez mais necessária e explorada em
A maioria dos trabalhos que abordam a reanotação genômica preocupa-se apenas em
reanotar as proteínas de um organismo e comparar os resultados obtidos com os resultados
originais, apontando as evoluções e os erros encontrados. Na primeira parte desse trabalho,
abordou-se a anotação genômica através de um enfoque temporal. Buscou-se entender de que
forma o crescimento constante dos bancos de dados biológicos afeta a anotação genômica
através de análises quantitativas e qualitativas a respeito dessa relação direta entre os dois. A
principal conclusão dessa primeira parte é a confirmação de que a medida que os bancos de
dados aumentam de tamanho (por isso, o enfoque temporal), os alinhamentos entre seqüências
alvo (seqüências de entrada do BLAST) com as seqüências depositadas nos bancos aumentam
em similaridade. Isso foi medido através do bit-escore dos alinhamentos. Porém, duas
constatações foram encontradas. Primeiro, a taxa de aumento na similaridade é muito inferior
à taxa de aumento no tamanho dos bancos de dados, tendendo a uma saturação. Segundo, o
aumento da quantidade de seqüências e conseqüentemente o aumento na similaridade entre
seqüências não garantem automaticamente uma melhora na qualidade da anotação funcional.
Na segunda parte do trabalho, foram feitos estudos acerca de fontes de erro na
anotação genômica. São sugeridos procedimentos para melhor interpretar os resultados do
BLAST, incluindo um estudo do comportamento do parâmetro do BLAST conhecido como
bit-escore, o qual pode ser usado preferencialmente ao invés do E-Value.
Abstract
Genomics is a recent discipline that refers to the molecular characterization of entire
genomes. Bioinformatics is an interdisciplinary field that uses computational methods to
analyze biological sequence data generated from genomics with the aim to predict their
function and structure.
Genomic annotation is a process that consists in the addition of bioinformatics
analyses to generate biological interpretations concerning raw DNA sequences. The
determination or prediction of an amino acid sequence function, called functional annotation,
is one of the fields of genomic annotation. The prediction of protein function is generally
done based on confrontation with protein sequences already characterized and stored in
biological databases, which is often done with the aid of similarity search programs, such as
BLAST
Genomics generates a huge amount of data that is stored in biological databases,
which provide the comparative pillar for functional annotation. Nowadays, the growth rate of
these databases is exponential, making the functional annotation activity very dynamic. For
this reason, the genomic re-annotation (the task that consists to annotate again a protein using
a more recent database) is an increasingly important procedure that has been exploited in
many studies.
In this study we try to identify and evaluate factors affecting the quality of genomic
annotation. In the first half of this work, we made a temporal approach to analyze how the
constant growth of biological databases affects the genomic annotation. The main conclusion
of this part is the observation that the database growth seemed to reach a saturation level in
terms of providing sequences suitable for annotation. Therefore the increase in database size
Finally, we evaluated the quality of an automatic re-annotation of Chlamydia
trachomatis genome, assessing its protocols, performance, sources of errors, and studying
BLAST parameters in order to optimize the procedure.
Sumário
RESUMO ... v
ABSTRACT ... vii
1. INTRODUÇÃO... 14
1.1. GENÔMICA... 14
1.2. BIOINFORMÁTICA... 15
1.2.1. O Caráter multidisciplinar da bioinformática... 18
1.2.2. Genômica e bioinformática no Brasil... 19
1.2.3. Bioinformática na industria farmacêutica e agricultura ... 20
1.3. BANCOS DE DADOS BIOLÓGICOS... 21
1.3.1. Bancos de dados de nucleotídeos ... 21
1.3.2. Bancos de dados de proteínas ... 22
1.4. COMPARAÇÃO DE PROTEÍNAS POR SIMILARIDADE DE SEQÜÊNCIA X ESTRUTURA... 28
1.4.1. Termos usados para comparar seqüências e estruturas de proteínas ... 29
1.5. EVOLUÇÃO DIVERGENTE E CONVERGENTE... 33
1.5.1. A evolução produziu um número relativamente limitado de dobramentos de proteínas e mecanismos catalíticos ... 33
1.5.2. Proteínas que diferem na seqüência e estrutura podem ter convergido para sítios ativos, mecanismos catalíticos e funções bioquímicas similares ... 34
1.5.3. Proteínas com baixa similaridade de seqüência mas estrutura e sítios ativos similares são provavelmente homólogos ... 34
1.5.4. Casos de evolução convergente e divergente são, às vezes, difíceis de distinguir ... 35
1.5.5. Evolução divergente pode produzir proteínas com similaridade de seqüência e estrutura mas com diferenças nas funções ... 36
1.6. ALINHAMENTO E COMPARAÇÃO DE SEQÜÊNCIAS... 37
1.6.1. Alinhamento global e local... 39
1.7. FERRAMENTAS DE BIOINFORMÁTICA PARA BUSCA EM BANCO DE DADOS... 40
1.7.1. FASTA e BLAST ... 40
1.7.2. PSI-BLAST e RPS-BLAST ... 44
1.7.3. Arquivos no formato FASTA... 45
1.7.4. Problemas e armadilhas do BLAST... 45
1.7.5. HMMER... 49
1.8. ANOTAÇÃO GENÔMICA... 49
1.8.1. Anotação em nível de nucleotídeos... 49
1.8.2. Anotação em nível de proteínas... 52
1.8.3. Anotação em nível de processo... 56
1.8.4. Reanotação genômica... 57
1.9. ERROS E LIMITAÇÕES EM ANOTAÇÃO FUNCIONAL DE PROTEÍNAS... 58
1.10. ONTOLOGIA... 60
1.10.2. Nomenclatura de enzimas ... 63
1.11. A FAMÍLIA CHLAMYDIAE... 64
1.11.1. Chlamydia trachomatis... 65
1.11.2. Anotação manual da Chlamydia trachomatis... 66
2. OBJETIVOS DO TRABALHO... 68
3. MÉTODOS... 69
3.1. OBTENÇÃO DOS DADOS DA CHLAMYDIA TRACHOMATIS... 69
3.2. MÉTODOS PARA OBTENÇÃO DOS DADOS PARA A ANÁLISE TEMPORAL DA ANOTAÇÃO GENÔMICA... 69
3.2.1. Obtenção das versões antigas do Swiss-Prot ... 70
3.2.2. Seleção das 8 versões do Swiss-Prot e procedimentos para preparar os arquivos para serem usados pelo BLAST ... 71
3.2.3. Execução do BLAST ... 72
3.3. COMPARAÇÃO ENTRE ANOTAÇÃO FINAL E “MELHORES HITS” DO BLAST ... 73
3.4. PESQUISA POR DOMÍNIOS DE PROTEÍNAS... 73
4. RESULTADOS ... 75
4.1. ANÁLISE DA EVOLUÇÃO DA ANOTAÇÃO GENÔMICA USANDO BANCOS DE DADOS ANTIGOS... 75
4.1.1. Análise quantitativa da evolução da anotação genômica ... 76
4.1.2. Análise qualitativa da evolução da anotação genômica ... 84
4.1.3. Contribuição de organismos para os “hits” do BLAST ... 87
4.2. ANÁLISE ESTÁTICA DE FONTES DE ERRO NA ANOTAÇÃO GENÔMICA... 91
4.2.1. Hits com E-Value não conclusivo mas com anotação correta ... 94
4.2.2. Hits com E-Value significativo mas com anotação incorreta... 99
4.2.3. Funcionalidade x função de proteínas... 104
4.2.4. Estimativa de limiar para uso do bit-escore... 105
4.2.5. Reanotação das ORFs da categoria “sem anotação final” ... 107
4.2.6. Valores de bit-escore na zona de incerteza ... 111
4.2.7. Outros problemas de ontologia ... 116
4.3. OUTROS RESULTADOS... 117
4.3.1. O uso de banco de dados não curados ... 117
4.3.2. Erros de atualização de anotação entre versões do Swiss-Prot ... 118
5. CONCLUSÃO E DIRECIONAMENTOS FUTUROS... 120
5.1. EVOLUÇÃO DA ANOTAÇÃO GENÔMICA... 120
5.2. BLAST COMO FERRAMENTA AUXILIAR NA ANOTAÇÃO... 122
5.3. PROBLEMAS E ERROS NA ANOTAÇÃO FUNCIONAL COMPUTACIONAL... 123
5.3.1. Ontologia... 123
5.3.2. Pequena quantidade de proteínas anotadas experimentalmente... 123
5.3.3. Alternativas para uma anotação genômica mais precisa ... 124
Figuras
Figura 1: Crescimento exponencial do GenBank... 18
Figura 2: Exemplo de formato de um registro do Swiss-Prot ... 25
Figura 3: Representação de domínios no Pfam ... 26
Figura 4: Homologia ... 32
Figura 5: Diagrama de fita da estrutura de um monômero de benzoilformate descarboxilase (BFD) e piruvato descarboxilase (PDC)... 35
Figura 6: Superposição das estruturas tridimensionais das moléculas: steroid-delta-isomerase, fator de transporte nuclear-2 e scytalone desidratase... 37
Figura 7: Exemplo de alinhamento global entre duas seqüências ... 39
Figura 8: Exemplo de alinhamento local entre duas seqüências ... 40
Figura 9: Algoritmo do BLAST ... 43
Figura 10: Exemplo de arquivo no formato FASTA com 2 registros ... 45
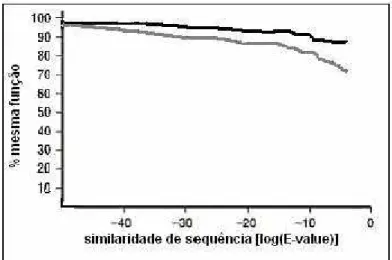
Figura 11: Percentual de similaridade de função de acordo com o E-Value... 48
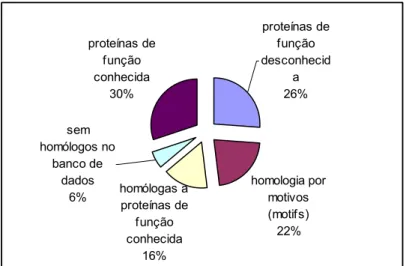
Figura 12: Relação entre similaridade de seqüência e similaridade de função. ... 53
Figura 13: Análise das funções de seqüências codantes do genoma da levedura.... 54
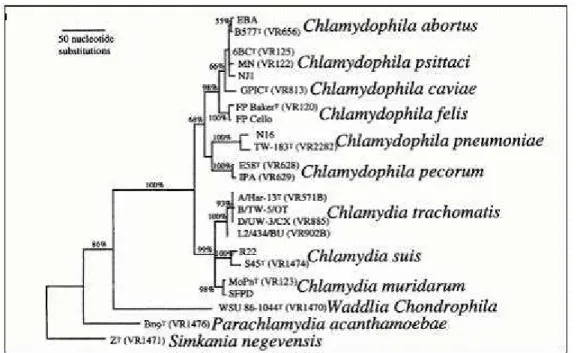
Figura 14: Relacionamentos evolucionários nas Chlamydiales. ... 65
Figura 15: Filogenia baseada no TTSS ... 66
Figura 16: Método principal utilizado para a análise temporal da anotação genômica ... 70
Figura 17: Histograma dos bit-escores dos bancos de dados representativos ... 78
Figura 18: Enquadramento das proteínas geradoras de alinhamento com bit-escore em torno de 50 e acima de 150 nos bancos de dados DB1 e DB4. ... 80
Figura 19: Análise de componentes principais das séries compostas pelos 893 bit-escores do BLAST contra os bancos DB1 a DB8... 84
Figura 20: Comparação qualitativa da evolução da anotação ao longo do tempo ... 86
Figura 21: Evolução dos 5 organismos que mais contribuíram para os “melhores hits” do BLAST.... 89
Figura 22 a) Comparação entre os “hits” de BLAST de seqüências dos genomas de B. subtilis e E. coli contra seqüências de C. trachomatis. b) Comparação de seqüências dos genomas de C. muridarum e C. pneumoniae contra seqüências de C. trachomatis. ... 90
Figura 23: Distribuição dos resultados em 4 categorias... 93
Figura 24: Resultados do programa CD-Search mostrando a similaridade da ORF 3328739 de C. trachomatis com a família SecE (Preprotein translocase subunit SecE) no banco COG... 98
Figura 25: Resultados do programa CD-Search mostrando a similaridade da ORF 3329239de C. trachomatis com o domínio Acyltransferase no banco PFAM. ... 98
Figura 26: Representação dos domínios encontrados pelo CD-Search para a cadeia A da proteína em análise. ... 104
Figura 28: Histograma dos bit-escores das categorias “conferem” e “não conferem”... 107
Figura 29: Relação entre E-Value e quantidade de proteínas hipotéticas. Verifica-se que proteínas com E-Value >10e-4 possuem alto percentual de proteínas não hipotéticas... 111
Figura 30 : Pesquisa por uma proteína no sítio do NCBI... 113
Figura 31: Resultado do BLAST para a seqüência gi=3328865 ... 113
Figura 32: Resultado do alinhamento local usando o programa “blast2seq” ... 114
Figura 33: Resultado da busca por domínios conservados para a ORF 3328865 ... 115
Tabelas
Tabela 1: Dados e tópicos de bioinformática (2004) ... 16
Tabela 2: Família de programas BLAST-NCBI... 44
Tabela 3: Lista dos banco de dados selecionados ... 72
Tabela 4 : Contribuição, por espécie, para o “melhor hit” do BLAST em cada banco de dados (em quantidade de “melhores hits”)... 87
Tabela 5: “Melhores hits” com E-Value acima do limiar mas com anotação correta... 95
Tabela 6: “Melhores hits” com E-Value significativo mas com anotação incorreta... 99
Tabela 7: Estatística das 4 categorias (valores de bit-escore) ... 106
Tabela 8: Resultado parcial do BLAST contra o TrEmbl. Os registros marcados com ► são os registros nos quais a anotação pode ser aproveitada para as ORFs que não tinham anotação final. ... 108
Tabela 9: Resultado BLAST contra o TrEmbl (seqüências com bit-escore entre 70 e 100). Os candidatos para investigação detalhada tem a marca “►“ ... 111
Tabela 10: Exemplos de problema de ontologia ... 116
1. Introdução
1.1. Genômica
A genômica é uma disciplina recente da Biologia que diz respeito à caracterização
molecular de genomas na sua totalidade. Para isso, técnicas experimentais especiais foram
desenvolvidas para tratar a difícil tarefa de manipular e caracterizar a grande quantidade de
dados. A genômica pode dividida em duas vertentes principais: genômica estrutural,
caracterizando a natureza física de genomas completos e genômica funcional, caracterizando
as regiões codificadoras e padrões globais de expressão de genes (Griffiths et al., 1999). A
caracterização de genomas completos é importante por duas razões. Primeiro, proporciona
uma maneira de se obter uma visão global da arquitetura genética de um organismo e,
segundo, provê todos os dados para a descoberta de novos genes, como por exemplo, os que
estão envolvidos em doenças. Por outro lado, a caracterização somente de seqüências
expressas fornece uma representação dos componentes funcionais que são determinantes para
a fisiologia celular em um determinado instante (Brown, 2002).
Durante a década passada, mais de 800 organismos foram objeto de projetos de
seqüenciamento de genoma. Hoje, tem-se disponível as seqüências completas de DNA do
genoma de mais de 100 espécies de bactérias e árqueo bactérias, incluindo alguns patógenos
importantes e três leveduras, entre elas a Saccharomyces cerevisiae. Também existem seqüências de genomas parciais ou completos de parasitas protozoários como o Plasmodium
falciparum, causador da malária. Entre os organismos multicelulares, os genomas da
Caenorhabditis elegans (nematóide), da Drosophila melanogaster (mosca de fruta) e as
plantas Arabidopsis thaliana e o arroz também já foram totalmente sequenciados. O genoma
Humano está atualmente completamente terminado, bem como o de Mus musculus
A genômica também está gerando uma crescente contribuição ao estudo da estrutura e
função de proteínas. Os programas de seqüenciamento de genoma estão gerando grande
quantidade de seqüências inferidas de aminoácidos de função desconhecida. Ao mesmo
tempo, muitas ferramentas experimentais e computacionais estão disponíveis para comparar
essas seqüências com outras de função conhecida. Também existem esforços para predizer a
estrutura tridimensional dessas estruturas, suas localizações sub celulares, seus parceiros de
interação. Entretanto, a comparação de seqüência e estrutura, geralmente fornece informações
limitadas. A caracterização completa de uma proteína na célula ou organismo sempre
requererá investigações experimentais adicionais nas proteínas purificadas in vitro assim
como estudos in vivo (Petsko e Ringe, 2003).
1.2. Bioinformática
A bioinformática tem várias definições. De forma geral mas não ampla, a
bioinformática é uma área interdisciplinar envolvendo biologia, informática, matemática e
estatística com o objetivo de analisar dados de seqüências biológicas e predizer a função e
estrutura de macromoléculas biológicas. Uma das formas de definir bioinformática é explicar
os seus objetivos. Primeiramente visa organizar dados de uma maneira que permita aos
pesquisadores acessar as informações existentes e submeter novos dados assim que são
produzidos. Essa quantidade de dados é cada vez maior, tanto de proteínas quanto de ácidos
nucléicos (vide figura 1 na página 18). Entretanto, as informações armazenadas em banco de
dados por si só não são muito úteis se não forem analisadas. Portanto, um segundo objetivo da
bioinformática é desenvolver ferramentas e recursos que ajudem na análise dos dados. Desse
modo, os biólogos moleculares podem passar a utilizar ferramentas computacionais capazes
de analisar grandes quantidades de dados biológicos, de predizer funções dos genes e de
demonstrar relações entre genes e proteínas. O terceiro objetivo é usar essas ferramentas para
Tradicionalmente, estudos biológicos examinavam sistemas individuais em detalhes e
comparavam os estudos com poucos outros relacionados. Com a bioinformática, podemos
conduzir análises globais de todos os dados disponíveis com o objetivo de desvendar
princípios comuns que se aplicam em vários sistemas e destacar novas características. A tabela
1 lista os tipos de dados que são analisados em bioinformática e o espectro de tópicos que
fazem parte do campo (Luscombe et al., 2001).
Tabela 1: Dados e tópicos de bioinformática (2004)
Fonte de dados Tamanho dos dados Tópicos da bioinformática
Seqüências brutas de DNA ~30 milhões de seqüências, 36 bilhões de bases
• Separação de regiões codantes e não codantes
• Identificação de íntrons e éxons
• Predição de produtos gênicos
• Análises forenses
Seqüência de proteínas ~900000 seqüências (~300
aminoácidos cada)
• Algoritmos de comparação de
seqüências
• Anotação funcional de proteínas
• Alinhamento múltiplo de
seqüências
• Identificação de seqüências de
motivos conservadas
Estrutura de macromoléculas ~26000 estruturas (~1000
coordenadas atômicas cada)
• Predição de estrutura secundaria, terciária
• Algoritmos de alinhamento de
estrutura 3D
• Medições de geometria de
proteínas
• Cálculo de volume e forma da
superfície
• Interações inter moleculares
• Simulações moleculares (cálculo
de campos de força, movimentos moleculares e predição de “docking”)
Genomas 100 genomas completos (1,6
milhão a 3 bilhões de bases cada)
• Caracterização de repetições
• Designação estrutural aos genes
• Análises filogenéticas
• Caracterização de vias metabólicas
• Análise de relação de genes
específicos com doenças
Expressão gênica • Correlação de padrões de
expressão
• Mapeamento de dados de
expressão para dados bioquímicos, de seqüência, e de estrutura. Literatura
Vias metabólicas
11 milhões de citações • Bibliotecas digitais para busca
automatizada de bibliografia
O auxílio no seqüenciamento de genomas foi uma das primeiras áreas em que a
bioinformática foi utilizada. Os seqüenciadores automáticos de DNA têm limitações técnicas
que os impedem de seqüenciar regiões maiores que 1000 bases por vez. Desta forma, para
viabilizar o seqüenciamento completo do genoma, deve-se, primeiramente, tratar o DNA das
células de forma a criar inúmeros fragmentos. Esses fragmentos devem ser individualmente
seqüenciados e, posteriormente, montados com o auxílio da bioinformática, como verdadeiras
peças de um quebra-cabeça (Luscombe et al., 2001).
O papel da bioinformática no auxílio para a decodificação de um gene é de suma
importância devido à velocidade com que a tarefa pode ser realizada. O Genoma Humano,
inicialmente previsto para ser desenvolvido e concluído em 15 anos, acabou sendo antecipado
em cerca de 5 anos, tendo sido dado por decifrado, em seus aspectos essenciais, em 2000. Um
novo gene, com cerca de 12 mil pares de bases, pode ter sua seqüência decifrada em 1 minuto,
quando há 4 anos atrás a mesma tarefa levaria 20 minutos e há 20 anos, em torno de 1 ano
(Vogt, 2003).
O resultado final foi o imenso acúmulo de seqüências sem a contrapartida funcional. A
disponibilidade das seqüências é um avanço significativo, mas por si só não é garantia que se
possa entender o funcionamento do organismo, mesmo no caso dos genomas completos. Isto
se deve, primariamente, ao fato dos sistemas biológicos serem extremamente complexos,
onde cada gene participa de uma rede intrincada de interações. Atributos como
não-linearidade, contextualidade e plasticidade caracterizam o papel dos genes na célula. A
conseqüência mais importante é o fato que para se determinar a função de um gene se fazem
necessários uma série de procedimentos experimentais, que, em sua maioria, requerem uma
grande parcela de tempo. Por isso, e também pelo fato de que seqüências acumuladas sem
interpretação não terem muito valor, a anotação genômica ganhou importância nos últimos
A figura 1 fornece uma amostra do ritmo de crescimento da deposição de dados no
banco de dados GenBank, um dos principais bancos de dados de seqüências de nucleotídeos
(Benson et al., 2004).
Figura 1: Crescimento exponencial do GenBank
(fonte: <http://www.ncbi.nlm.nih.gov/Genbank/genebankstats.html>)
1.2.1. O Caráter multidisciplinar da bioinformática
Assim, como outras áreas emergentes de trabalho, a bioinformática se enquadra em
um contexto onde o problema não é hardware, nem software, mas peopleware, ou seja,
formação de pessoal qualificado na área. Um profissional de bioinformática bem colocado
precisa ter um bom conhecimento em ciência da computação e conhecer os princípios da
biologia molecular e as diferentes técnicas de bancada nos diferentes domínios onde a
bioinformática se faz necessária. Algumas das áreas da computação mais utilizadas pela
bioinformática são: banco de dados, inteligência artificial e computação paralela. Da biologia,
são usados principalmente conhecimentos de biologia molecular, biofísica, bioquímica e
1.2.2. Genômica e bioinformática no Brasil
No Brasil, a área iniciou a partir dos diversos projetos do Programa Genoma
patrocinados pela Fapesp (Fundação de Amparo à Pesquisa de São Paulo), reunidos desde
maio de 1997 na Rede Onsa (sigla, em inglês, para “Organization for Nucleotide Sequencing
and Analysis”). Com a Rede Onsa, foram estabelecidas as bases para o funcionamento de um
instituto virtual e dinâmico que congregou, inicialmente, cerca de 30 laboratórios de
diferentes instituições do estado de São Paulo, e que hoje, com uma participação cada vez
maior de novos grupos de pesquisa em novos projetos, acabou por consolidar uma nova
concepção do desenho institucional da pesquisa no país.
A bioinformática esteve presente como instrumento indispensável nos projetos do
genoma da Xylella fastidiosa, da Cana, do Genoma Humano do Câncer, do genoma Clínico
do Câncer, do genoma Xanthomonas, do Eucalipto, do Schistomona Mansoni, da bactéria
Leifsonia Xyli. Esses projetos fazem parte de um projeto abrangente dos Genomas
Agronômicos e Ambientais (AEG, sigla para o inglês Agronomical and Environmental
Genomics), criado em 2000, a partir do projeto feito em conjunto com o Departamento de
Agricultura dos EUA para o seqüenciamento de uma variante da Xylella fastidiosa. O
Genoma Bovino, anunciado mais recentemente, também marca as etapas dessas progressivas
conquistas científicas e tecnológicas da genômica no Brasil.
Desde o lançamento da Rede Onsa, vários centros de pesquisa e desenvolvimento em
bioinformática foram se constituindo e se consolidando em São Paulo, acompanhando a
espiral de crescimento da cultura genômica no país. Assim foi na Unicamp, na USP, na
Unesp, no Instituto Ludwig, no Laboratório Nacional de Computação Científica, no Rio de
Janeiro, na Universidade Federal de Pernambuco, na Federal de Minas Gerais, na Federal do
Rio Grande do Sul, na Universidade de Brasília e na Universidade Católica de Brasília. (Vogt,
Em particular, tem se verificado um grande crescimento científico na área genômica
no centro-oeste do Brasil. Instituições da região coordenam e participam de diversos projetos
genoma de alcance nacional ou regional, tais como os genomas do eucalipto (genolyptus), café, banana (promusa) e do fungo Paracoccidioidesbrasiliensis.
Quem aparece em primeiro lugar, em pesquisa, tecnologia e ensino em biotecnologia
são os Estados Unidos, representados pelo Centro Nacional de Informação Biotecnológica
(NCBI), seguido pela Inglaterra, através do Instituto Sanger, que faz parte do Instituto
Europeu de Bioinformática. Em seguida, no mesmo nível, vem a França (Instituto Pasteur), a
Alemanha (Heilderberg), a Suíça com seu Instituto Suíço de Bioinformática e o Japão com o
Banco de Dados de DNA (DDBJ). O Brasil, Cingapura e Índia, são os representantes mais
importantes de países em desenvolvimento que desenvolvem e investem em bioinformática.
1.2.3. Bioinformática na industria farmacêutica e agricultura
O setor farmacêutico foi o grande motor do desenvolvimento da bioinformática e
ainda hoje é o maior consumidor de bioinformática no mundo, respondendo por mais de 95%
dos recursos aí investidos. O processo de desenvolvimento de um medicamento inicia-se com
uma grande quantidade de moléculas potenciais. A cada fase, muitas delas são descartadas por
não atenderem às especificações necessárias. Ao final, apenas algumas terão restado, das
quais a mais adequada servirá de base para o medicamento. Às vezes não se identifica
nenhum candidato. Em qualquer caso, fica claro que grande parte dos gastos é empregada em
avaliar e testar substâncias que no final das contas serão descartadas. Estima-se que 75% do
custo de desenvolver um novo remédio é usado para pagar por todas as potenciais moléculas
descartadas. Se as empresas conseguirem eliminar precocemente uma molécula inadequada,
poderão então melhorar significativamente seu retorno comercial. É aí que a bioinformática se
faz presente. Permitindo a detecção de moléculas fadadas ao descarte, a bioinformática pode
guiar os pesquisadores na direção das mais promissoras. No Brasil, esse cenário ainda não
Embora a indústria farmacêutica seja a maior demandante de bioinformática, o setor
da agricultura e pecuária está ganhando forte impulso como utilizador de técnicas
moleculares, especialmente para melhoramento genético. Nesta arena, o Brasil saiu na frente,
pois enquanto os países mais ricos empregavam grande parte dos seus investimentos no setor
de saúde humana, deixando à margem os desenvolvimentos na área agrícola, aqui foram
lançados programas com o objetivo inicial de desvendar a seqüência genética de organismos
de interesse para as culturas nas quais o Brasil é líder mundial (Meidanis, 2003).
1.3. Bancos de dados biológicos
Bancos de dados biológicos podem conter, entre outras, informações sobre a
seqüência, estrutura, e função de biomoléculas. Além dos organismos modelos citados
anteriormente, milhares de outros tiveram regiões seqüenciadas e depositadas em bancos de
dados públicos. Os bancos de dados estão crescendo continuamente (vide figura 1) e
armazenam informações muito úteis para os pesquisadores.
1.3.1. Bancos de dados de nucleotídeos
O banco de dados GenBank (Benson, et al., 2004) contém seqüências de DNA
disponíveis publicamente de mais de 140.000 organismos diferentes. Essas seqüências são
obtidas principalmente através de submissões de dados de seqüência de laboratórios
individuais e submissões em lotes de projetos de seqüenciamento de grande escala. O
GenBank é mantido pelo “National Center for Biotechnology Information” (NCBI), uma
divisão do “National Library of Medicine” (NLM), localizado no campus do “US National
Institutes of Health” (NIH) em Bethesda (EUA). Este banco mantém uma colaboração
internacional com outros dois bancos de dados: EMBL na Inglaterra (Stoesser et al., 2002) e
DDBJ (DNA Data Bank of Japan), a qual inclui uma troca diária de dados entre os três bancos
para que se mantenham sincronizados. Em agosto de 2003, o GenBank continha mais de 33.9
espécies estão representadas e novas são adicionadas a uma taxa de mais 1.700 por mês.
Aproximadamente 26% das seqüências são de origem humana. Depois do Homosapiens, as
espécies que mais aparecem são: Mus musculus, Rattus norvegicus, Danio rerio, Oryza sativa, Drosophilamelanogaster, Zeamays, Arabidopsisthaliana e Gallusgallus.
O banco de dados não-redundante “nr” de nucleotídeos contém todos os registros do
próprio GenBank adicionados aos registros dos bancos de dados: EMBL, DDBJ e PDB
(Protein Data Bank) (Berman et al., 2000). Não contém, porém, seqüências de EST
(“Expressed Sequence Tags”), STS (“Sequence Tagged Site”), GSS (“Genome Survey
Sequence”) e HTGS (“High Throughput Genomic Sequences”). O banco EST (Boguski et al.,
1993) contém “expressed sequence tags”. O GSS contém leituras aleatórias de fragmentos
genômicos e seqüências de cosmídeos, BAC e YAC.
1.3.2. Bancos de dados de proteínas
Projetos de seqüenciamento de genoma levaram a um rápido aumento na quantidade
de informação acerca de seqüência de proteínas. Dentre os vários bancos de dados de
proteínas, são listados, a seguir, os bancos de dados que foram usados nesse trabalho.
1.3.2.1. Swiss-Prot e TrEmbl
O banco de dados Swiss-Prot (Boeckmann et al., 2003) contém seqüências de
aminoácidos em conexão com o conhecimento atual das ciências naturais. O SwissProt é
curado por um grande número de especialistas e, por isso, torna-se um banco com
informações mais confiáveis. Cada registro de proteína provê uma visão geral interdisciplinar
de importantes informações trazendo, junto com o registro, resultados experimentais,
características computadas e, algumas vezes, até conclusões contraditórias. Outras
informações detalhadas que estão além do escopo do Swiss-Prot ficam disponíveis através de
ligações para banco de dados especializados. O Swiss-Prot provê registros anotados para
outros organismos modelos, a fim de assegurar a presença de anotações de alta qualidade de
membros representativos de todas as famílias de proteínas. Famílias e grupos de proteínas são
regularmente revisados para que se mantenham atualizados com os atuais descobrimentos
científicos. Complementarmente, o TrEMBL esforça-se em abranger todas seqüências de
proteínas que não estão ainda representadas no Swiss-Prot. Ao mesmo tempo em que é
necessário manter uma alta qualidade de anotação no Swiss-Prot, é também vital tornar as
seqüências disponíveis o mais rápido possível. Essa é a função do TrEMBL, que é formado
por registros anotados computacionalmente derivados da tradução de todas as seqüências
codantes (CDS) no banco de dados EMBL.
A figura 2 contém um exemplo do formato de um registro do Swiss-Prot. Cada linha
do registro tem um significado próprio. Dentre essas informações, as linhas que foram
extraídas e usadas nesse trabalho são:
• Linha ”ID” (Identificação): A linha “ID” é sempre a primeira linha no registro. Essa
linha contém três informações separadas por ponto-e-vírgula: O nome do registro, o tipo
de molécula (nesse caso o valor é sempre “PRT”; proteína) e o tamanho da proteína
(quantidade de aminoácidos).
• Linha “AC” (Número de acesso): O objetivo do número de acesso é prever uma maneira
estável de identificar os registros de versão para versão. A linha AC pode ter mais de
um número de acesso se os registros foram unidos ou separados. Por exemplo, quando
dois registros são unidos em um único, os números de acesso de ambos são mantidos
nessa linha. Se um registro é dividido em dois (uma ocorrência rara), os números de
acesso originais são mantidos nos novos registros criados e um novo número de acesso
é criado para ambos novos registros. Um número de acesso é eliminado somente
quando os dados aos quais ele designa forem completamente removidos do banco de
• Linha “DE” (descrição): Contém informações descritivas gerais sobre a seqüência
armazenada. Essa informação é geralmente suficiente para identificar a proteína com
precisão. A descrição tem formato livre e sempre começa com o nome proposto oficial.
Sinônimos são colocados entre parênteses (vide figura 2). Quando é sabido que uma
proteína é clivada em componentes multifuncionais, a descrição começa com o nome da
proteína precursora, seguida por uma seção delimitada por “[Contains:...]”. Todos os
componentes individuais são listados nessa seção. Quando é sabido que uma proteína
inclui domínios com múltiplas funções, com cada um deles descrito por um nome
diferente, a descrição começa com o nome da proteína total, seguido por uma seção
delimitada por “[Includes:]”. Todos os domínios são listados nessa seção. Em casos
raros, os domínios funcionais de uma proteína são clivados, mas a atividade catalítica só
pode ser observada quando as cadeias individuais reorganizam-se em um complexo.
Essas proteínas são descritas na linha “DE” por uma combinação de “[Includes:...]” e
“[Contains:...]”.
• Linha “OS” (espécie do organismo): especifica o(s) organismo(s) que foi (foram) a
fonte da seqüência armazenada. A designação da espécie consiste, na maioria dos casos,
pela designação em latim da espécie seguida do nome em Inglês entre parênteses. No
caso de vírus, somente o nome em Inglês é fornecido.
O registro é sempre terminado por uma linha com “//”.
ID KPYK_CHLMU STANDARD; PRT; 481 AA. AC Q9PK61;
DT 16-OCT-2001 (Rel. 40, Created)
DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 15-MAR-2004 (Rel. 43, Last annotation update) DE Pyruvate kinase (EC 2.7.1.40) (PK).
GN PYK OR TC0609. OS Chlamydia muridarum.
OC Bacteria; Chlamydiae; Chlamydiales; Chlamydiaceae; Chlamydia. OX NCBI_TaxID=83560;
RN [1]
RP SEQUENCE FROM N.A. RC STRAIN=MoPn / Nigg;
RX MEDLINE=20150255; PubMed=10684935;
RA Read T.D., Brunham R.C., Shen C., Gill S.R., Heidelberg J.F.,
RA White O., Hickey E.K., Peterson J., Utterback T., Berry K., Bass S., RA Linher K., Weidman J., Khouri H., Craven B., Bowman C., Dodson R., RA Gwinn M., Nelson W., DeBoy R., Kolonay J., McClarty G., Salzberg S.L., RA Eisen J.A., Fraser C.M.;
RT pneumoniae AR39.";
RL Nucleic Acids Res. 28:1397-1406(2000).
CC -!- CATALYTIC ACTIVITY: ATP + pyruvate = ADP + phosphoenolpyruvate. CC -!- COFACTOR: Requires magnesium and potassium.
CC -!- PATHWAY: Glycolysis; final step. CC -!- SUBUNIT: Homotetramer (By similarity).
CC --- CC This SWISS-PROT entry is copyright. It is produced through a collaboration CC between the Swiss Institute of Bioinformatics and the EMBL outstation - CC the European Bioinformatics Institute. There are no restrictions on its CC use by non-profit institutions as long as its content is in no way CC modified and this statement is not removed. Usage by and for commercial CC entities requires a license agreement (See http://www.isb-sib.ch/announce/ CC or send an email to license@isb-sib.ch).
CC --- DR EMBL; AE002329; AAF39440.1; -.
DR PIR; F81684; F81684. DR HSSP; P14178; 1E0T. DR TIGR; TC0609; -.
DR InterPro; IPR001697; Pyruvate_kinase. DR Pfam; PF00224; PK; 1.
DR PRINTS; PR01050; PYRUVTKNASE.
DR ProDom; PD001009; Pyruvate_kinase; 1. DR TIGRFAMs; TIGR01064; pyruv_kin; 1.
DR PROSITE; PS00110; PYRUVATE_KINASE; FALSE_NEG.
KW Transferase; Kinase; Glycolysis; Magnesium; Complete proteome. FT ACT_SITE 219 219 By similarity.
FT METAL 221 221 Magnesium (By similarity). FT METAL 242 242 Magnesium (By similarity). FT METAL 243 243 Magnesium (By similarity). SQ SEQUENCE 481 AA; 53175 MW; 9222E5DAED557B51 CRC64; MIARTKIICT IGPATNTPEM LEKLLDAGMN VARLNFSHGT HESHGRTIAI LKELREKRQV PLAIMLDTKG PEIRLGQVES PIKVKPGDRL TLTSKEILGS KEAGVTLYPS CVFPFVRERA PVLIDDGYIQ AVVVNAQEHL IEIEFQNSGE IKSNKSLSIK DIDVALPFMT EKDITDLKFG VEQELDLIAA SFVRCNEDID SMRKVLENFG RPNMPIIAKI ENHLGVQNFQ EIAKASDGIM IARGDLGIEL SIVEVPALQK FMARVSRETG RFCITATQML ESMIRNPLPT RAEVSDVANA IHDGTSAVML SGETASGTYP IEAVKTMRSI IQETEKSFDY QAFFQLNDKN SALKVSPYLE AIGASGIQIA EKASAKAIIV YTQTGGSPMF LSKYRPYLPI IAVTPNRNVY YRLAVEWGVY PMLTSESNRT VWRHQACVYG VEKGILSNYD KILVFSRGAG MQDTNNLTLT TVNDVLSPSL E
//
Figura 2: Exemplo de formato de um registro do Swiss-Prot
O Swiss-Prot e o TrEMBL são complementares e, juntos, formam um banco de dados
não redundante. O Swiss-Prot aumenta de tamanho continuamente quando novas seqüências
anotadas são adicionadas. O banco de dados TrEMBL diminui de tamanho assim que algumas
de suas seqüências são anotadas e movidas para o Swiss-Prot. Entretanto, uma característica
marcante do Swiss-Prot é que a sua taxa de crescimento é a menor entre os demais bancos
biológicos em função da necessidade de intervenção de diversos especialistas humanos para
construir uma entrada. Quatro vezes por ano, uma nova versão do TrEMBL é construída no
EBI. Nesse momento, o TrEMBL aumenta de tamanho pois ele passa a incluir todos os novos
registros que estavam acumulados desde a última versão. Swiss-Prot e TrEMBL
compartilham o mesmo sistema de números de acesso. Dessa forma, não existirá duplicação
mantém disponível na Internet, além da versão atual, as suas versões antigas. Essas versões
antigas foram muito úteis e foram usadas intensamente nesse trabalho.
1.3.2.2. PFAM
O Pfam (Protein families database of alignments and HMMs) é uma grande coleção de
alinhamentos múltiplos de seqüências e “Hidden Markov Models” cobrindo as mais comuns
famílias e domínios de proteínas (Beteman et al.,2002). HMM são modelos estatísticos que
capturam informações específicas da posição de quanto cada coluna do alinhamento é
conservada e quais os resíduos contidos. Para cada família em Pfam, pode-se consultar
alinhamentos múltiplos, ver a arquitetura de domínios de proteínas, examinar a distribuição de
espécies, olhar estruturas de proteínas conhecidas. Pfam pode ser usado para ver a
organização de domínios de proteínas. Um exemplo típico é mostrado na figura 3. Note que
uma única proteína pode pertencer a várias famílias do Pfam.
Figura 3: Representação de domínios no Pfam
Aproximadamente 74% das seqüências de proteínas conhecidas têm ao menos uma
entrada no Pfam. O Pfam tem duas partes: a primeira (Pfam-A) é a parte curada do banco de
dados contendo aproximadamente 7316 famílias de proteínas. A segunda parte (Pfam-B) é um
suplemento que contém um grande número de pequenas famílias adquiridas do banco de
dados PRODOM (Servant et al., 2002) as quais não têm sobreposição com Pfam-A. Embora
tenha menos qualidade, B pode ser útil quando nenhuma família é encontrada em
Pfam-A.
1.3.2.3. NR (Banco de dados não-redundante de proteínas) do NCBI
O banco de dados “nr” (non-redundant database) do NCBI reúne todas as traduções
(Berman et al., 2000), Swiss-Prot, PIR (Protein Information Resource, Wu et al., 2003) e PRF (Protein Research Foundation).
Cada seqüência protéica é identificada por um “protein id” no formato
“accession.version”, sistema esse que foi implementado pelo GenBank, EMBL, e DDBJ em
fevereiro de 1999. O “protein id” consiste de três letras seguidas por cinco dígitos, um ponto,
e o número da versão. Se ocorrer qualquer mudança na seqüência (mesmo em um único
aminoácido), o número da versão é incrementado, mas a porção de acesso (“accession”)
permanece estável (por exemplo, AAA98665.1 mudará para AAA98665.2).
Paralelamente, toda seqüência também é identificada por um “gi” (GenInfo Identifier).
O sistema GI de identificação de seqüências corre em paralelo com o sistema no formato
“protein id”. Portanto, se a seqüência protéica for modificada de qualquer modo, ela receberá
um novo numero GI e o sufixo do “protein id” será incrementado de um.
1.3.2.4. COG
O banco de dados COG (“Clusters of Orthologous Groups”) apresenta uma
classificação filogenética de proteínas de procariontes e eucariontes unicelulares. Os "COGs"
são derivados de comparação do tipo "todos contra todos" entre as seqüências das proteínas.
Cada COG consiste de proteínas individuais ou grupos de ortólogas de no mínimo três
linhagens e, portanto, é considerado como correspondente a um domínio ancestral
conservado. O banco de dados é projetado para dar suporte a pesquisa em evolução de
genomas assim como em anotação funcional. Também existe um banco chamado KOG que
consiste de “clusters” de ortólogos preditos de 7 genomas eucariontes.
A coleção COG consiste, atualmente, de 138.458 proteínas, as quais formam 4.873
COGs e compreendem 75% das 185.505 proteínas preditas codificadas em 66 genomas de
organismos unicelulares. Os grupos ortólogos de eucariontes (KOGs) incluem proteínas de 7
genomas eucariontes: Caenorhabditis elegans, Drosophila melanogaster e Homo sapiens,
Schizosaccharomyces pombe) e o parasita intracelular Encephalitozoon cuniculi. O KOG
atual consiste de 4.852 agrupamentos de ortólogos, os quais incluem 59.838 proteínas, ou
aproximadamente 54% dos 110.655 produtos gênicos de eucariontes analisados.
Exames nos padrões filéticos dos KOGs revelam um núcleo conservado representado
em todas as espécies analisadas e que consiste em ~20% do conjunto do KOG. Essa porção
conservada do KOG é muito maior do que a porção ubíqua do conjunto COG (~1% do COG).
Em parte, essa diferença é provavelmente devida a menor quantidade de genomas eucariontes
incluídos, mas também pode refletir a relativa compacidade e maior estabilidade
evolucionária dos genomas eucariontes (Tatusov et al., 2003).
1.3.2.5. Uniprot
O consórcio UniProt (Apweiler, 2004) é formado pelo EBI (“European Bioinformatics
Institute”), SIB (“Swiss Institute of Bioinformatics”) e PIR (“Protein Information Resource”).
Foi criado para unir as atividades dos bancos de dados Swiss-Prot, TrEMBL e PIR em um
novo recurso capaz de prover uma visão estável e compreensiva de seqüências e funções de
proteínas. Esse recurso é composto de três camadas: um arquivo de seqüências de proteínas,
uma base de conhecimentos e um banco de dados de referencias não redundante.
1.4. Comparação de proteínas por similaridade de seqüência x
estrutura
As proteínas podem ser comparadas com outras usando similaridade de seqüência ou
similaridade de estrutura. A primeira abordagem foi, historicamente, a primeira a ser usada. É
baseada em alinhamento de seqüências protéicas. Inicialmente, a busca por similaridade era
feita sobre toda a seqüência. Mais tarde, as proteínas começaram a ser analisadas na base de
ocorrências de padrões conservados de aminoácidos. Na comparação por similaridade de
conhecidas (Mount, 2001). Nesse trabalho, o enfoque é predominantemente baseado em
comparação por similaridade de seqüência.
1.4.1. Termos usados para comparar seqüências e estruturas de proteínas
Durante a comparação de seqüências, vários termos são usados para descrever relações
evolucionárias e estruturais entre proteínas. Nesse item, descrevemos os mais importantes.
Outros podem ser encontrados no sítio CATH (http://www.biochem.ucl.ac.uk
/bsm/cath/lex/glossary.html), no sítio SCOP (“Structural Classification of Proteins”)
encontrado em (http://pdb.wehi.edu.au/scop/gloss.html) e no tutorial situado no sítio do Swiss
Bioinformatics Institute (http://www.expasy.ch/swissmod/course/course-index.htm).
Principais termos:
• Sitio ativo: é uma combinação localizada de cadeias laterais de aminoácidos dentro
da estrutura terciária ou quaternária a qual pode interagir com um substrato químico
específico e que provê atividade biológica à proteína. Proteínas de diferentes
seqüências de aminoácidos podem formar estruturas que produzem sítios ativos
semelhantes.
• Blocos: (“blocks”) é um termo usado para descrever um padrão de seqüência de
aminoácidos conservado em uma família de proteínas. O padrão inclui uma série de
possíveis coincidências em cada posição nas seqüências representadas, mas não há
nenhuma posição inserida ou removida no padrão ou nas seqüências. Em contraste,
perfis de seqüência são um tipo de matriz de escores que representam um conjunto
similar de padrões que incluem inserções e remoções. Perfis HMMs são “Hidden
Markov Models” desse segundo tipo de padrão.
• Domínio (contexto de seqüência): Nesse contexto, o termo está associado à
homologia. Domínios homólogos referem-se a um padrão estendido de seqüência,
homólogo indica uma origem evolucionária comum dentre as seqüências alinhadas.
Um domínio é geralmente mais longo do que um motivo. O domínio pode incluir
uma porção ou a seqüência inteira da proteína. Alguns domínios são complexos e
feitos de diversos domínios homólogos menores que, durante a evolução, se ligam
formando domínios maiores.
• Domínio (contexto de estrutura): Refere-se a um segmento de uma cadeia
polipeptídica que pode formar uma estrutura tridimensional independente da
presença de outros segmentos da cadeia. Os domínios separados de uma proteína
podem interagir extensivamente ou podem estar conectados somente por uma
cadeia polipeptídica. Uma proteína com diversos domínios pode usá-los para
interações funcionais com diferentes moléculas.
• Família (contexto de seqüência): é um grupo de proteínas de função bioquímica
similar e que têm alta similaridade de seqüência. Uma família de proteínas engloba
proteínas com a mesma função em organismos diferentes (seqüências ortólogas)
mas também pode incluir proteínas do mesmo organismo derivadas de duplicação
de genes e rearranjos (seqüências parálogas). Famílias podem ser subdivididas em
subfamílias ou agrupadas em superfamílias em função do nível de alinhamento
obtido entre as seqüências. Quando seqüências de proteínas com a mesma função
são examinadas em detalhes, algumas compartilham alta similaridade de seqüência.
Elas são, obviamente, membros da mesma família de acordo com o critério acima.
Entretanto, outras têm pouca ou quase insignificante similaridade de seqüência com
os outros membros da família. Nesses casos, o relacionamento entre dois membros
distantes da família A e C pode ser demonstrado por um membro da família B que
compartilha similaridade significativa com ambos A e C. Assim, B provê uma
• Família (contexto de estrutura): refere-se a duas estruturas que em um nível
relevante de similaridade estrutural mas não necessariamente similaridade
significativa de seqüência.
• Motivo (“motif”; contexto de seqüência): refere-se a um padrão conservado de
uma seqüência pequena de aminoácidos que é encontrado em duas ou mais
proteínas. No catálogo do PROSITE (Hulo et al., 2004), um motivo é um padrão de
aminoácidos que é encontrado em um grupo de proteínas que têm uma atividade
bioquímica similar.
• Motivo (“motif”; contexto de estrutura): refere-se a uma combinação de diversos
elementos de estrutura secundária produzidos pelo dobramento de seções
adjacentes da cadeia do polipeptídio em uma configuração tridimensional
específica. Um exemplo é o motivo “helix-turn-helix”, encontrados em proteínas
que se ligam a DNA. Motivos de estrutura são também conhecidos como estruturas
supersecundárias.
• Perfil (“profile”; contexto de seqüência): É uma matriz de escores que representa
um alinhamento múltiplo de seqüências de uma família de proteínas. O perfil é
geralmente obtido de uma região bem conservada em um alinhamento múltiplo. O
perfil tem a forma de uma matriz em que cada coluna representa uma posição no
alinhamento e cada linha representa um aminoácido. Os valores da matriz dão a
probabilidade de cada aminoácido estar na posição correspondente no alinhamento.
Espaços (“gaps”) são permitidos durante o alinhamento e uma penalidade ao espaço
é incluída nesse caso como um escore negativo quando nenhum aminoácido
coincidente é encontrado. Um perfil de seqüência pode também ser representado
• Superfamília: é um grupo de famílias de proteínas que são relacionados por distante
mas ainda detectável similaridade de seqüência. Proteínas com poucas identidades
em um alinhamento de seqüências, mas com um convincente número de
características funcionais e estruturais em comum são colocadas na mesma
superfamília.
• Seqüências homólogas: São seqüências com similaridade atribuída devido à
descendência de um ancestral comum.
• Seqüências ortólogas: São seqüências homólogas em espécies diferentes que
advém de um gene ancestral comum durante a especiação. Podem ou não ser
responsável por uma função similar.
• Seqüências parálogas: São seqüências homólogas dentro da uma mesma espécie
que advém da duplicação de genes.
A figura 4 mostra um exemplo dessas últimas definições.
Gene ancestral da hemoglobina
Gene cadeia ß
Gene cadeia a
sapo a Chick a camun-dongo a camun-dongo ß Chick ß sapo ß
Duplicação gênica homólogos
parólogos
ortólogos ortólogos
Figura 4: Homologia
1.5. Evolução divergente e convergente
Evolução convergente é a evolução de estruturas, não relacionadas por antepassados,
para uma função em comum e que é refletida em uma estrutura semelhante. Evolução
divergente é evolução a partir de um ancestral comum.
1.5.1. A evolução produziu um número relativamente limitado de dobramentos
de proteínas e mecanismos catalíticos
Embora a quantidade total de atividades enzimáticas diferentes em qualquer célula
viva seja grande, elas envolvem um pequeno número de classes de transformações químicas.
Para cada uma dessas transformações, existe um número ainda menor de diferentes
mecanismos catalíticos através dos quais eles podem ser obtidos. Isso tudo sugere que a
maioria das enzimas deve estar relacionada na seqüência e estrutura com muitas outras de
mecanismos similares, mesmo que seus substratos sejam diferentes.
Duas proteínas com alta identidade de seqüência podem ser assumidas como terem
surgido através de evolução divergente de um ancestral em comum. Nesse caso, pode-se
predizer que terão estruturas muito parecidas ou até idênticas. Em geral, se a identidade entre
duas seqüências for maior do que aproximadamente 40% sem a necessidade de introduzir
muitos espaços no alinhamento e se essa identidade é espalhada ao longo das seqüências,
então a expectativa é de que elas codifiquem para proteínas com dobramento similar.
Entretanto, problemas em deduzir relações evolucionárias e em predizer função a partir da
seqüência e estrutura surgem quando a identidade é inferior a 40%. Além disso, mesmo
proteínas com identidade de seqüência maior que 90%, as quais devem ter estrutura e sítios
1.5.2. Proteínas que diferem na seqüência e estrutura podem ter convergido
para sítios ativos, mecanismos catalíticos e funções bioquímicas similares
A estrutura do sítio ativo determina a função bioquímica de uma enzima e, em muitas
proteínas homólogas, os resíduos do sítio ativo e a estrutura são conservados mesmo quando o
resto da seqüência divergiu a ponto de não ser mais reconhecida. Portanto, poderíamos supor
que todas proteínas com sítios ativos e mecanismos catalíticos similares fossem homólogas .
Entretanto, esse não é o caso. Se duas proteínas têm dobramentos muito diferentes e baixa
similaridade de seqüência, é provável que sejam exemplos de evolução convergente. Ou seja,
elas não divergiram de um ancestral comum mas apareceram independentemente e
convergiram na mesma configuração de sítio ativo como resultado de seleção natural de uma
função bioquímica particular. Exemplos claros de evolução convergente são encontrados entre
serino-proteases e aminotransferases.
1.5.3. Proteínas com baixa similaridade de seqüência mas estrutura e sítios
ativos similares são provavelmente homólogos
Pode ser difícil perceber homologia a partir da seqüência somente, porque seqüências
mudam mais rapidamente com a evolução do que a estrutura 3D (vide figura 5). De fato,
proteínas com nenhuma detecção de similaridade de seqüência, mas com as mesmas
estruturas e funções bioquímicas, já foram encontradas. Entre os numerosos exemplos estão
as glicosiltransferases, as quais transferem um monosacarídeo de um açúcar ativado doador
para um sacarídeo, proteína, lipídeo, DNA ou pequena molécula aceptora. Algumas
glicosiltransferases que operam em diferentes substratos e não têm similaridade de seqüência,
mas têm estruturas similares são tidos como terem um ancestral comum. Em alguns,
provavelmente a maioria, dos casos, baixa homologia de seqüência combinada com alta
similaridade de estrutura reflete conservação seletiva de resíduos funcionais importantes em
seqüências homólogas genuínos, mas altamente divergentes. Exemplos: mandelato racemase
similares. As reações que eles catalisam compartilham uma fase central. Essa fase é catalisada
da mesma forma pelas enzimas, implicando que elas provavelmente divergiram de um
ancestral comum.
1.5.4. Casos de evolução convergente e divergente são, às vezes, difíceis de
distinguir
Em alguns casos, existe equivalência espacial no sítio funcional, mas pouca ou
nenhuma conservação de seqüência dos resíduos de importância funcional. Nesses casos,
distinguir entre evolução convergente e divergente pode ser difícil. Por exemplo, as enzimas
benzoilformato descarboxilase (BFD) e piruvato descarboxilase (PDC) têm somente 21% de
identidade de seqüência mas têm, essencialmente, dobramentos idênticos (vide figura 5). As
cadeias laterais de aminoácidos catalíticos são conservadas na mesma posição espacial na
estrutura 3D, mas não na seqüência.
Figura 5: Diagrama de fita da estrutura de um monômero de benzoilformate descarboxilase (BFD) e piruvato descarboxilase (PDC).
É possível que as duas proteínas tenham evoluído independentemente e tenham
convergido para a mesma solução química para o problema de descarboxilar um
alfa-cetoácido. Mas, sua grande similaridade na estrutura seria vista como indicativo de terem
divergido de um ancestral comum. O nível de identidade de seqüência entre eles é, no entanto,
muito baixo para distinguir entre essas duas possibilidades com confiança (Petsko e Ringe,
2003).
1.5.5.Evolução divergente pode produzir proteínas com similaridade de seqüência
e estrutura mas com diferenças nas funções
Existem proteínas com funções bioquímicas muito diferentes mas que, apesar disso,
possuem muita similaridade tridimensional e suficiente identidade de seqüência para
pressupor homologia. Tais casos sugerem que a estrutura diverge mais lentamente do que a
função durante a evolução. Por exemplo, esteroid-delta-isomerase, fator de transporte
nuclear-2 e scitalone desidratase compartilham muitos detalhes estruturais (vide figura 6) e são
considerados homólogos, ainda que as duas enzimas, a isomerase e a desidratase, não tenham
resíduos catalíticos essenciais em comum. Isto sugere que são as características gerais da
cavidade do sítio ativo dessa proteína que têm a habilidade potencial de catalisar reações
químicas diferentes dados resíduos de sítios ativos diferentes. A terceira proteína desse
conjunto de homólogos, um fator de transporte nuclear-2, não é propriamente uma enzima,
mas sua cavidade contém resíduos que estão presentes nos sítios catalíticos de ambas
enzimas. Portanto, determinação de função, a partir da seqüência e estrutura, é complicado
pelo fato de proteínas com estrutura similar poderem não ter a mesma função mesmo quando
Figura 6: Superposição das estruturas tridimensionais das moléculas: steroid-delta-isomerase, fator de transporte nuclear-2 e scytalone desidratase.
A seta cinza indica o sítio ativo (pdb: 8cho, 1oun, 1std). Adaptado de Petsko e Ringe, 2003
1.6. Alinhamento e comparação de seqüências
A comparação de uma seqüência de nucleotídeos ou aminoácidos com outra a fim de
encontrar um grau de similaridade entre elas é uma técnica chave na biologia dos dias atuais.
Uma similaridade marcante entre duas seqüências de genes ou proteínas pode refletir o fato
delas serem derivadas por evolução da mesma seqüência ancestral. Seqüências relacionadas
dessa forma são chamadas de homólogas e a similaridade evolutiva entre elas é conhecida
como homologia (vide item “Termos usados para comparar seqüências e estruturas de
proteínas” na página 29). Seqüências da mesma proteína mas de espécies diferentes também
podem ser comparadas a fim de deduzir relações evolutivas. Dois genes que evoluíram
recentemente de um gene ancestral comum ainda terão relativa semelhança na seqüência.
Aqueles que têm um ancestral comum mais distante terão acumulado muito mais mutações, e
sua relação evolutiva será menos óbvia, ou até impossível de deduzir somente através da
Os casos práticos, abaixo listados, mostram a grande importância e utilidade da
comparação de seqüências para a biologia (Setúbal e Meidanis, 1997):
1. Temos duas seqüências do mesmo alfabeto e ambas têm aproximadamente o
mesmo comprimento (dezenas de milhares de caracteres). Sabemos que as duas
seqüências são praticamente iguais, com poucas e isoladas diferenças tais como
inserções, remoções e substituições de caracteres. A freqüência média dessas
diferenças é baixa, digamos, uma a cada cem caracteres. Queremos localizar os
lugares onde essas diferenças ocorrem. Problemas desse tipo ocorrem quando, por
exemplo, o mesmo gene é seqüenciado por dois laboratórios diferentes e queremos
comparar os resultados.
2. Temos duas seqüências com algumas centenas de caracteres cada uma. Queremos
saber se existe um prefixo de uma que seja similar a um sufixo de outra. Se a
resposta for sim, o prefixo e o sufixo envolvidos devem ser mostrados.
3. Temos o mesmo problema que na situação (2), mas dessa vez temos muitas
centenas de seqüências que devem ser comparadas, uma contra todas. Além disso,
sabemos a maioria desses pares de seqüências não estão relacionados, ou seja, têm
baixa similaridade. Problemas do tipo (2) e (3) surgem no contexto de montagem
de fragmentos em programas que auxiliam seqüenciamento de DNA em larga
escala.
4. Temos duas seqüências com poucas centenas de caracteres cada. Queremos saber
se existem duas sub-seqüências, uma de cada seqüência, que sejam similares.
5. Temos a mesma situação que em (4), mas, ao invés de duas seqüências, temos uma
seqüência e queremos compará-la com milhares de outras. Problemas do tipo (4) e
(5) ocorrem no contexto de busca por similaridades locais usando grandes bancos
Para todos esses casos, uma idéia única e básica de algoritmo pode ser usada para
resolver todos os problemas acima expostos. Algumas vezes, métodos menos generalistas,
porém mais rápidos, são melhores adaptados para cada caso.
1.6.1. Alinhamento global e local
Para se inferir o quanto duas seqüências são semelhantes utilizam-se métodos de
comparação de cadeias de símbolos, e que genericamente são conhecidos pela designação de
alinhamento de seqüência. O objetivo final é o de maximizar o número de coincidências entre
as duas seqüências de acordo com um sistema de pontuação (Setubal e Meidanis, 1997).
Existem dois tipos de alinhamento: global e local (Mount, 2001). No alinhamento global, é
feita uma tentativa de alinhar as seqüências completamente, usando o máximo de caracteres
possíveis em toda a extensão dessas. Seqüências que possuem alguma similaridade e
aproximadamente o mesmo tamanho são candidatas convenientes para alinhamento global.
Um dos algoritmos mais usados nesse caso é o algoritmo de Needleman e Wunsch
(Needleman e Wunsch, 1970), que é um algoritmo rigoroso utilizando a técnica de
programação dinâmica (Eddy, 2004), a qual tenta resolver problemas complexos através da
resolução de sub-conjuntos do problema inicial. A figura 7 mostra um pequeno exemplo de
um resultado desse alinhamento.
Figura 7: Exemplo de alinhamento global entre duas seqüências
Já em alinhamentos locais, trechos das seqüências com as mais altas densidades de
coincidências são alinhados, gerando uma ou mais ilhas de sub alinhamentos nas seqüências.
Alinhamentos locais são mais apropriados para alinhar seqüências que são similares ao longo
de um trecho de suas seqüências mas dissimilares em outros trechos, ou então, seqüências que
algoritmos mais conhecidos é o algoritmo de Smith-Waterman (Smith e Waterman, 1981).
Esse algoritmo também usa programação dinâmica e é matematicamente desenhado para
prover o melhor alinhamento local entre duas seqüências. A figura 8 ilustra um exemplo em
que o alinhamento local é mais apropriado. Na prática, muitas proteínas não apresentam um
padrão global de similaridade, mas apresentam-se como mosaicos de domínios modulares.
Alinhamentos globais não detectam esse aspecto. Em função desses aspectos, alinhamentos
locais são mais complexos que os globais e, portanto, seus algoritmos são mais lentos.
F2 E F1 E K Catalítico
F1 E K K Catalítico
F12
PLAT
F1, F2: Repetições de Fibronectin E:domínio similar EGF
K: domínio Kringle Catalitico: atividade de serine protease
F12: fator de coagulação XII PLAT: ativador de tecido plasminogen
Figura 8: Exemplo de alinhamento local entre duas seqüências
1.7. Ferramentas de bioinformática para busca em banco de dados
1.7.1. FASTA e BLAST
O programa FASTA (Pearson e Lipman, 1988) foi a primeira ferramenta usada
largamente para busca em banco de dados de nucleotídeos e proteínas. Já o programa BLAST
(“Basic Local Alignment Search Tool”) (Altschul et al., 1997) passou a ser mais utilizado
pois provê um método mais rápido de procura. BLAST é sem dúvida a ferramenta
computacional de referência para a busca de similaridade em bancos de dados de seqüências e
por conseqüência extensivamente utilizado na anotação genômica.
FASTA e BLAST também são algoritmos de alinhamento local. Pelo fato de usarem
heurísticas, eles são em torno de 50 a 100 vezes mais rápidos do que o algoritmo de