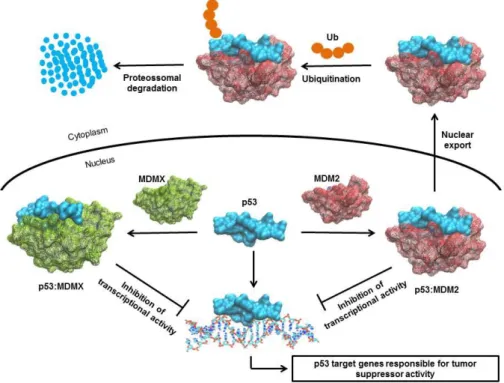
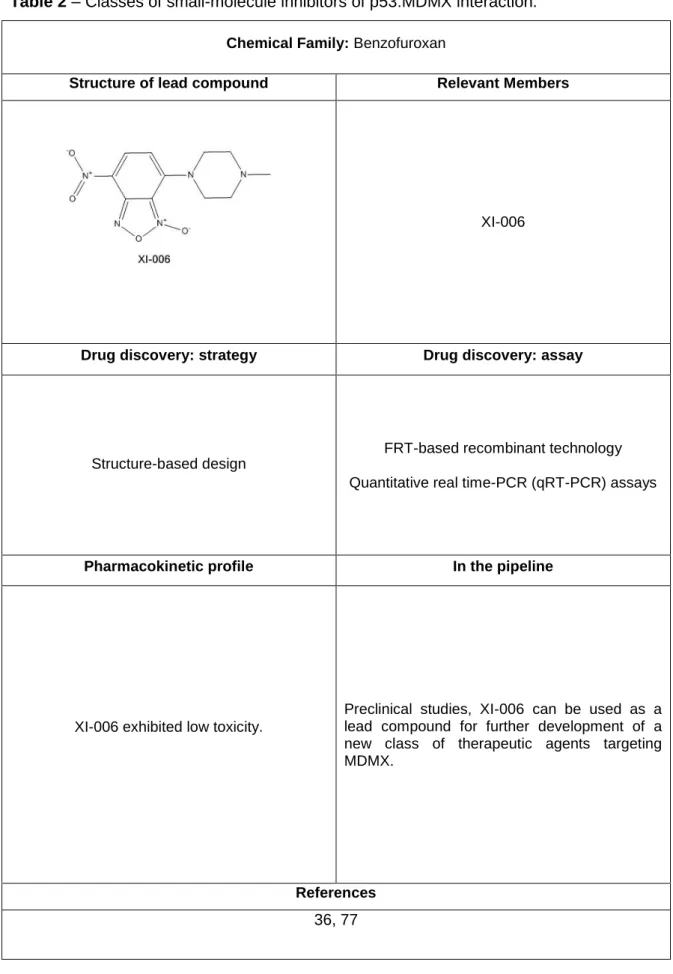
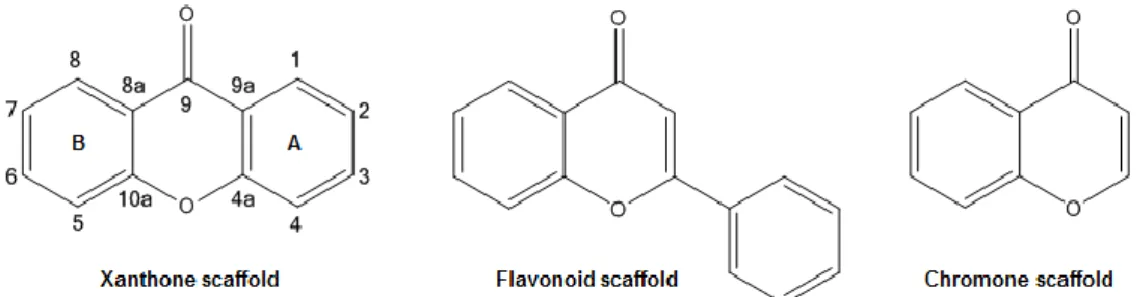
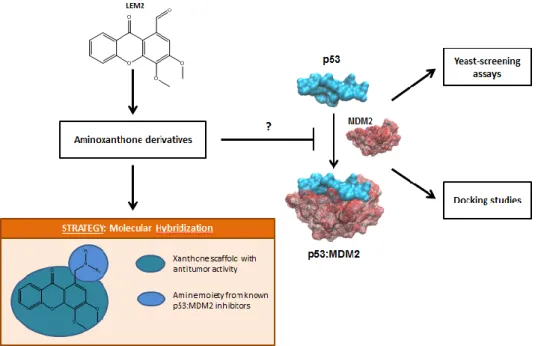
Design and synthesis of aminoxanthone derivatives as promising disruptors of p53:MDM2 interaction
134
0
0
Texto
Imagem



+7



Documentos relacionados