INVESTIGAÇÃO DE ALTERAÇÕES NOS GENES SHORT
STATURE HOMEOBOX E RECEPTOR DO HORMÔNIO DE
CRESCIMENTO EM PESSOAS COM BAIXA ESTATURA
IDIOPÁTICA NO DISTRITO FEDERAL
Brasília - DF
2012
INVESTIGAÇÃO DE ALTERAÇÕES NOS GENES SHORT STATURE HOMEOBOX E RECEPTOR DO HORMÔNIO DE CRESCIMENTO EM PESSOAS COM BAIXA
ESTATURA IDIOPÁTICA NO DISTRITO FEDERAL
Dissertação apresentada ao Programa de Pós-Graduação Stricto Sensu em Ciências Genômicas e Biotecnologia da Universidade Católica de Brasília, como requisito parcial para obtenção do Título de Mestre em Genética Molecular Humana.
Orientador: Prof. Dr. Robert Pogue.
Coorientador: Prof. Dr. Rinaldo Wellerson.
E RECEPTOR DO HORM
ESTATURA IDIOPÁTICA NO parcial para obtenção do grau de Universidade Católica de Brasília, pela banca examinadora abaixo assi
________________
Ciências Genômicas
________________ Prof
Ciências Genômicas e
________________ Prof
________________ Prof. Dra. Maria Te
HORMÔNIO DE CRESCIMENTO EM PESSOAS
IDIOPÁTICA NO DISTRITO FEDERAL", apresentada como do grau de Mestre em Ciências Genômicas e Biotecnologia
de Brasília, em 30 de março de 2012, defendida pela banca examinadora abaixo assinada:
___________________________________ Prof. Dr. Robert Edward Pogue
Orientador
Ciências Genômicas e Biotecnologia – UCB
___________________________________ Profª. Dra. Rosângela Vieira de Andrade Ciências Genômicas e Biotecnologia - UCB
___________________________________ Profª. Dra. Andréa Barretto Motoyama
Ciências da Saúde - UnB
___________________________________ Prof. Dra. Maria Teresinha de Oliveira Cardoso
Núcleo de Genética - HAB
Brasília 2012
PESSOAS COM BAIXA
Em primeiro lugar agradeço a Deus por estar ao meu lado, mesmo quando eu estive longe.
Agradeço a minha mãe Ivana Frota de Albuquerque, a pessoa mais amável que eu já vi neste mundo. Ela, como farmacêutica, é a grande responsável pelas direções que tomei em minha vida profissional.
Agradeço ao meu pai João Celso Martins Marques, que para mim é a maior referência intelectual e exemplo de profissional. Sem ele nada seria possível, pois nos momentos em que estive cego, ele sempre esteve presente para me mostrar como percorrer a trilha que escolhi.
Agradeço aos meus irmãos (Leonardo e Tarsila) que sempre me deram apoio e serenidade para poder concluir este mestrado.
Agradeço a Karina, o amor da minha vida, que me acompanhou lado a lado nestes dois anos. Escutou todos os meus anseios, frustrações e expectativas em praticamente todos os dias deste período. O seu apoio foi fundamental para que eu fosse fisicamente capaz de superar as adversidades deste trabalho.
À minha tia Sandra e ao meu tio Nicolao, que me acolheram como filho, dando-me o apoio (principalmente no início) necessário para minha vinda e estabelecimento nesta cidade até então desconhecida.
Obrigado aos meus avos Daniel (in memoriam), Oneide, Marina e Arnaldo. A todos os meus familiares, em especial aos meus primos Lucas, Mariana, Natália, Nicole e Dani, que sempre me incentivaram nesta escolha.
Aos professores que me deram a formação necessária para chegar até aqui, em especial, ao Prof. Dr. Rogério Teles, que me apresentou à Química e me fez tão apaixonado por ela, ao amigo Prof. Dr. Raimundo Antônio Gomes Oliveira que influenciou diretamente a minha formação, quando me mostrou a Genética e a Biologia Molecular, ao Prof. Rinaldo Wellerson que me acolheu desde o meu primeiro momento (durante a seleção de mestrado) e ao Prof. Dr. Robert Pogue, hoje um grande amigo e peça fundamental para a concretização deste sonho.
A todas as pessoas da UCB que me aturaram, em especial à Jéssica que foi minha primeira amiga no laboratório, às pessoas que sempre se prontificaram a ajudar, fosse em uma técnica, fosse com insumos (Marcela, Nídia Betúlia, Virgílio, Profª. Rose, Bárbara, e todos os outros que não citei, por favor.. perdoem-me). Quero agradecer ao grupo do Robert, principalmente à Gabriela que sempre me incentivou nos momentos difíceis e ao Ricardo que me ajudou em praticamente todo sequenciamento do SHOX.
transcender as mutações e a morte, e assim seguir a natureza do universo. É a
certeza de que existe uma Verdade fundamental e que a vida é a oportunidade sagrada para evoluir e se aproximar dela”.
A baixa estatura é uma condição que afeta 2,3% da população. Dentro deste grupo, encontra-se a Baixa Estatura Idiopática (BEI), definida como estatura inferior a 2 desvio-padrões (SD) da altura média correspondente a uma determinada idade, sexo e grupo populacional sem nenhuma outra anomalia esquelética ou sistêmica, distúrbios endócrinos, nutricionais ou cromossômicas. Investigações de mutações de ponto e investigações de copy number variations (CNVs) em genes-chave, como Short Stature Homeobox Gene (SHOX), conhecido como maior causa monogênica, e Growth Hormone Receptor (GHR) – peça importante do eixo Growth Hormone/Insulin-like Growth Factor-1 (GH/IGF-1) – são imprescindíveis ao diagnóstico de pacientes com BEI. Esta pesquisa se propôs a sequenciar os genes SHOX e GHR para determinar a frequência de mutações de ponto e inserções/deleções que afetam suas funções; investigar alterações de CNV em regiões regulatórios upstream e downstream do SHOX por Multiplex Ligation-dependent Probe Amplification (MLPA); genotipar por Polymerase Chain Reaction (PCR) o éxon 3 do gene GHR(genótipos GHRd3 e GHRfl) para validar por Real time Polymerase Chain Reaction (qPCR); e comparar a frequência destes alelos nos pacientes com BEI e na população brasileira. Foram encontradas uma deleção SHOX(3,6%) pela técnica de MLPA, uma mutação de ponto no SHOX(3,6%) e uma mutação de ponto no GHR (3,6%) ainda não relatada na literatura. Além disso, a genotipagem do GHR por qPCR se mostrou 100% congruente em comparação com a técnica da PCR. Sua frequência diferiu nos pacientes com BEI da população brasileira, podendo, portanto ter um efeito aditivo ao fenótipo da altura. Este trabalho teve como foco principal execução de um projeto-piloto para um Serviço Diagnóstico Genético-Molecular no Distrito Federal (DF), em que pesquisas semelhantes a esta irão ajudar a compor um centro de excelência que investigue doenças relevantes ao DF. Esta pesquisa conseguiu padronizar diversas técnicas diagnósticas e, mais importante, elas se mostraram bem sucedidas na detecção de mutações. Estes resultados irão constituir um banco de amostras e dados que serão usados em estudos futuros, como alvo de identificação de novas áreas do genoma associados à estatura. Em relação aos princípios farmacogenéticos e saúde pública, o governo brasileiro ainda não apoia tratamento com GH em pacientes com mutações no SHOX. Futuramente, quando este protocolo for aprovado, o trabalho descrito aqui será mais significante, pois servirá de subsídio para indicar o tratamento apropriado para pacientes com BEI.
Idiopathic short stature (ISS) is a condition that affects 2.3% of the population. This clinical diagnosis is defined as at least 2 SD (standard deviations) below the average height controlled for a determined age, gender and populational group, without any other skeletal or systemic anomaly, and without endocrine, nutritional or chromosomal disturbances. Analysis of point mutations and copy number variations (CNV) in key genes as Short Stature Homeobox Gene (SHOX), known as the most frequent monogenic cause, and Growth Hormone Receptor (GHR) - an important component of the GH/IGF-1 (Growth Hormone/Insulin-like Growth Factor-1) axis - are part of the ISS diagnostic strategy. This research has been undertaken with the objectives of: sequencing the SHOX and GHR genes in order to detect point mutations frequency and insertions/deletions that cause ISS; investigating CNV alterations at upstream and downstream SHOX regulatory regions by Multiplex Ligation-dependent Probe Amplification (MLPA); genotyping by Polymerase Chain Reaction (PCR) the exon 3 polymorphism from the GHR gene (GHRd3 and GHRfl genotypes) in order to validate the use of Real time Polymerase Chain Reaction (qRT-PCR); and to compare the frequency of these GHR alleles in the ISS patients and the Brazilian population. The MLPA analysis identified one deletion in the SHOX region (3.6% of patients), and one point mutation was found in each of the SHOX and GHR genes using Sanger sequencing (3.6% of patients for each gene). The GHR mutation was previously unreported in the literature. Genotyping by qRT-PCR of the SNP rs_ showed 100% correlation with direct genotyping of the GHR exon 3 polymorphism, and it was shown that there was a significant difference between the allele frequencies in the ISS patients and the general Brazilian population. This may indicate an influence for this polymorphism in ISS. One of the principal focuses of this work was to initiate the establishment of a molecular genetic diagnostic service for the Federal District (DF), in which diseases relevant to the general health of the DF will be analyzed. This research succeeded in optimizing several diagnostic protocols, and in finding mutations using each of them. The samples collected will also constitute a sample and data bank that can be used in the future for further investigations into the genetic and genomic causes of short stature. Pharmacogenetic research can indicate not only the treatment, but also the therapeutic dose to be used, leading to increased efficiency and success in treatment. These data may in the future serve to help convince the Ministry of Health that growth hormone treatment should be made available to certain patients with ISS.
ANVISA: Agência Nacional de Vigilância Sanitária BEF: Baixa Estatura Familial
BEI: Baixa Estatura Idiopática
BNPP: Brain Natriuretic Peptide Protein
CGH: compartive genomic hybridization CKII: Caseína Kinase II
CNV: copy number variation
dbSNP: database Single Nucleotide Polymorphism DGH: GH deficiente
DLW: Discondroestose de Leri-Weill
SD: desvios–padrão
EHW: Equilíbrio de Hardy-Weinberg EMEA: European Medicines Agency FDA: Food and Drug Administration FISH: Fluorescent in-situ hybridization g: gramas
GH: Growth Hormone GHR: GH Receptor
GHRH: GH releasing hormone GHRHR: GHRH Receptor GHBP: GH Binding Protein
GHRd3ou d3: GHR exon 3 deletado GHRflou fl: GHR full length
HAB: Hospital de Apoio de Brasília IGF-1: Insulin-like Growth Factor-1 IGF-1 R: IGF-1 Receptor
IGFBP-3: IGF Binding Protein 3
mA: miliampere Mb: Mega-base mL: mililitro
MLPA: MultiplexLigase-dependent Probe Amplification
mM: millimolar
mRNA: RNA mensageiro MS: Ministério da Saúde OAR: Opt, Aristaless and Rax OMIM: Medelian inheritance in Man PAR1: Região Pseudo-autossômica1
pb: pares de base PCNT: Pericentria
PCR: Polymerase Chain Reaction pmol: picomol
rpm: rotações por minuto qPCR: PCR quantitativa
SHOX: Short Stature HomeoboxGene
SNP: Single Nucleotide Polymorphism
Stat: Signal transducer and activator of transcription STH: Somatrotopina
Teste (t): Teste tde Student
U.V: ultravioleta V: Volts
X2= Qui-quadrado ng: nanograma nm: nanômetro µg: micrograma
Figura 1: Descrição do SHOX...19
Figura 2: Frequência e a localização das mutações no SHOX...20
Figura 3: Fequência de alinhamento de vários homeodomínios. ...22
Figura 4: Osteogênese e atuação do SHOX. ...24
Figura 5: Estrutura do gene GHR. ...27
Figura 6: Estrutura do GHRe rotação do dímero. ...28
Figura 7: Vias de sinalização do receptor GHR e IGF-1...29
Figura 8: Estrutura de um par de sondas do MLPA...36
Figura 9:Esquema da técnica MLPA ...37
Figura 10: Amplificação multiplexdo éxon 3 do GHR...47
Figura 11: Softwarecoffalyser. ...55
Figura 12: Discriminação alélica do polimorfismo do GHRpor PCR ...60
Figura 13: Discriminação alélica do polimorfismo do GHRpor qPCR ...61
Figura 14: Padrão e intensidade dos picos produzidos pelo MLPA no ABI 3130 ...65
Figura 15: Resultados normais para MLPA ...66
Figura 16: Resultado alterado para o MLPA...67
Figura 17: Mutação de ponto em heterozigose em SHOX ...68
Figura 18: Lisina e treonina. ...69
Figura 19: Mutação de ponto em heterozigose em GHR. ...70
Figura 20: Triptofano e arginina...71
Figura 21: Local de interação do triptofano 187 com GH ...72
Figura 22: Sítio de ligação do trp187...73
Tabela 1: Tamanho (em pares de base) de todos os éxons do gene SHOX...18
Tabela 2: Frequência de alterações no SHOXem pacientes com BEI...20
Tabela 3: Tamanho (em pares de base) de todos os éxons do gene GHR. ...26
Tabela 4: Primers usados na PCR e sequenciamento do gene SHOX. ...45
Tabela 5: Primers usados na PCR e sequenciamento do gene GHR...46
Tabela 6: Sondas utilizadas pelo kit MLPA p018-e1...50
Tabela 7: Dados clínicos dos pacientes com BEI ...59
Tabela 8: Frequência genotípica e e alélica do polimorfismo doGHRem BRA ...61
Tabela 9: Frequencia dos polimorfismos e genótipos do GHRem BRA e BEI. ...63
Tabela 10: Comparação da frequência genotípica de populações com BEI...64
Tabela 11: Comparação da frequência genotípica de populações normais. ...64
Tabela 12: Genótipo do polimorfismo em 105 indivíduos da população brasileira. ..85
Tabela 13: Genotipo discriminado de cada paciente com BEI...87
1.1 BAIXA ESTATURA IDIOPÁTICA ...16
1.2 GENES ENVOLVIDOS EM BEI ...16
1.2.1 SHOX (Short Stature Homeobox Gene)...17
1.2.2 GHR (Growth Hormone Receptor)...26
1.3 FARMACOGENÉTICA...31
1.3.1 Situação do tratamento com rhGH no Brasil...33
1.4 ANÁLISE DE MUTAÇÕES NO SHOXE GHRRELEVANTES À BEI ...34
1.4.1 CNVs e Multiplex Ligation-dependent Probe Amplificaition (MLPA)...35
2 JUSTIFICATIVA...39
3 OBJETIVOS...41
3.1 OBJETIVO GERAL ...41
3.2 OBJETIVOS ESPECÍFICOS...41
4 MATERIAL E MÉTODOS...42
4.1 CASUÍSTICA ...42
4.2 COLETA DE AMOSTRAS...42
4.3 TÉCNICAS MOLECULARES...43
4.3.1 Extração e quantificação de DNA...43
4.3.2 Verificação da integridade do DNA...43
4.3.3 Quantificação do DNA...43
4.3.4 PCR...44
4.3.5 Genotipagem GHR...46
4.3.6 MLPA...48
4.3.7 Sequenciamento de DNA...53
4.4 BIOINFORMÁTICA E ANÁLISE DE DADOS ...54
4.4.1 Genotipagem por qPCR...54
4.4.2 MLPA...54
4.4.3 Sequenciamento...55
4.5 ANÁLISE ESTATÍSTICA...57
4.5.1 Qui-quadrado (X2) ...57
4.5.2 Teste (t) de Student...57
5 RESULTADOS E DISCUSSÃO...59
5.2.2 Comparação Entre Pacientes com BEI e População Brasileira...62
5.3 ANÁLISE SHOX...64
5.3.1 MLPA – SHOX...64
5.3.2 Sequenciamento – SHOX...67
5.4 ANÁLISE –GHR...70
5.4.1 Sequenciamento – GHR...70
6 CONCLUSÃO...75
REFERÊNCIAS...77
APÊNDICE A - Genótipo do grupo de 105 indivíduos da população brasileira. ...85
APÊNDICE B - Genótipo dos pacientes com BEI da população brasileira. ...87
APÊNDICE C – Deleções do MLPA em todos os pacientes. ...88
ANEXO A - Protocolo Clínico e Diretrizes Terapêuticas para o tratamento da Deficiência do Hormônio do Crescimento – Hipopituitarismo Somatropina ...89
ANEXO B – Termo de Consentimento Livre e Esclarecido ...96
1 INTRODUÇÃO
A biologia molecular combinada à genética possibilitou grandes avanços à ciência. Dentro desta curta história, podem se enumerar pontos importantes, que
hoje impactam diretamente nos rumos traçados por elas. Fatos como a elucidação da estrutura do DNA por Watson e Crick (WATSON; CRICK, 1953) e a invenção da Polymerase Chain Reaction (PCR) (MULLIS et al., 1986) são alguns dos exemplos dos vários ‘Big Bangs’deste universo em expansão chamado ciências genômicas.
O desenvolvimento dos alicerces da genômica e biologia molecular
despertaram muito o interesse científico. Utilizando os fundamentos de outras ciências, como Química e Física, a biologia molecular promoveu grandes avanços em áreas que afetam a saúde humana e a investigação científica (AZEVEDO et al., 2003). Isto tem possibilitado o entendimento de diversas doenças, a forma como elas são herdadas ou como podem ser diagnosticadas, proporcionando a melhoria
da qualidade de vida da população (BORGES-OSÓRIO; ROBINSON, 2001).
Com isso, surgiu, em meados dos anos 70, uma nova forma de abordagens e conceitos para patologias não esclarecidas: a genética médica. A partir dela, está sendo possível entender melhor a etiologia de algumas doenças, identificá-las precocemente, ou mesmo propor novos tratamentos a partir de estudos genéticos, como por exemplo, na genotipagem dos genes CYP2C9 e VKORC1, que orienta na determinação da dosagem de varfarina em pessoas com problema de coagulação (LIU et al., 2011).
Dentre as várias doenças genéticas, um grupo de doenças esqueléticas é caracterizado por malformação óssea, crescimento desproporcional e deformação do esqueleto (SUPERTI-FURGA; BONAFE; RIMOIN, 2001). Elas podem ser
distinguidas pela expressão temporal de seus genes causativos: as disostoses são definidas como deformações estáticas e ocorrem durante a embriogênese; enquanto as displasias esqueléticas (ou osteocondrodisplasias) resultam de genes que permanecem ativos por toda a vida, ou pelo menos durante a fase de crescimento (HALL, 2002; RIMOIN et al., 2007).
Fenotipicamente, distúrbios do esqueleto podem ser divididos em três grupos:
cartilaginoso, levando à baixa estatura. E, por fim, as disostoses têm sido
caracterizadas como malformação de um osso específico ou um grupo de ossos específicos (RIMOIN et al., 2007), como na Síndrome de Treacher Collins, caracterizada por disostose mandibulofacial.
Até o ano de 1997, estas doenças eram classificadas de acordo com a clínica e dados radiográficos dos pacientes. Entretanto, a partir daquele ano, após os
avanços da genética humana, o critério molecular passou a fazer parte da classificação das doenças ósseas.
Inicialmente, surgiram ambiguidades ao se caracterizar um grupo de doenças, pois pela clínica poderia pertencer a um grupo, mas quando se observara o gene envolvido, classificava-se em outro. Por exemplo, dentro do grupo de baixa estatura, o gene Growth Hormone Receptor (GHR), do ponto de vista molecular, era classificado como pertencente ao Eixo Growth Hormone/Insulin-like Growth Factor-1 (GH/IGF-1), porém, fenotipicamente, poderia ser classificado como causador de
baixa estatura idiopática (BEI), que, por sua vez, era classificada dentro do grupo das osteocondrodisplasias.
Consequentemente, seguiram-se sucessivas revisões do International Nosology and Classification of Constitutional Disorders of Bone. Em 2006, fora realizada a última, a qual qualificou 372 patologias esqueléticas em 37 grupos de acordo com os critérios moleculares, bioquímicos e radiográficos (RIMOIN et al., 2007). No entanto, esta classificação ainda não é ideal, pois existem algumas sobreposições entre os grupos. Para solucionar isto, fatores como vias bioquímicas e morfologia celular também deveriam ser considerados (RIMOIN et al., 2007).
Apesar destas exaustivas classificações de doenças esqueléticas que causam baixa estatura, pode-se subdividi-las, sob a óptica da genética, em três grupos: desordens básicas do crescimento (defeitos intrínsecos ao tecido ósseo ou conjuntivo); desordens secundárias do crescimento (fatores externos ao tecido ósseo ou conjuntivo, como problemas endócrinos); e aquelas sem origem conhecida, ou seja, baixa estatura idiopática (KANT; WIT; BREUNING, 2003).
1.1 BAIXA ESTATURA IDIOPÁTICA
Embora o emprego do termo ‘idiopático’ não seja totalmente adequado, a expressão surgiu diante da necessidade de uma descrição mais específica para um grupo de patologias que se encaixam dentro de um mesmo fenótipo (JORGE et al.,
2008). Com isso, deveria existir outra classificação para os indivíduos com BEI que chegaram a um diagnóstico conclusivo, ou seja, que não pertenceriam mais ao grupo de idiopáticos. Por definição, BEI é caracterizada por altura abaixo de dois desvios–padrão (SD) da média para uma determinada idade, sexo e origem étnica,
que não possua qualquer fenótipo passível de correlação com alguma patologia e de causa não esclarecida (LIFSHITZ; BOTERO, 2007).
No entanto, estes aspectos são arbitrários, pois não se aplicam aos descendentes de pais mais baixos que o normal. Assim, pode-se adotar como critério para o diagnóstico 2 SD abaixo do desvio-padrão parental (SDS) (CHEN et al., 2009). Além disso, a altura de uma população obedece à curva de Gauss, em que 2% das crianças sempre serão consideradas com -2 SD e, se levar-se em consideração que há grande variação nas taxas de maturação de um indivíduo para o outro (de tal forma que a criança possa ter ossos curtos para idade, mas não para maturação óssea), este debate se tornará ainda mais delicado (KUCZMARSKI et al., 2000).
Recentemente, um consenso entre o Growth Hormone Research Society, The Lawson Wilkins Pediatric Endocrine Society e European Society for Paediatric Endocrinology definiu BEI como -2 SD da altura média correspondente de uma determinada idade, sexo e grupo populacional, sem nenhuma outra anomalia esquelética ou sistêmica, distúrbios endócrinos, nutricionais ou cromossômicas e hormônio de crescimento normal (COHEN et al., 2008). Porém, este conceito não é
totalmente ideal, visto que nesta classificação também há sobreposições entre pacientes com atraso constitucional de crescimento e maturação com baixa estatura familial (BEF), sendo tratados da mesma maneira (ROSENBLOOM, 2009).
1.2 GENES ENVOLVIDOS EM BEI
de genes (ELLISON et al., 1997). A baixa estatura por si só compõe um grupo de doenças de difícil diagnóstico devido a sua grande heterogeneidade e uma grande quantidade de genes envolvidos (SAVAGE et al., 2007). Já a investigação de BEI é
ainda mais complexa em razão de não haver um fenótipo que seja correlacionado a um defeito de um gene específico (COHEN et al., 2008).
No entanto, a literatura tem evidenciado que os genes GHR e Short Stature Homeobox Gene (SHOX) são os dois maiores responsáveis pela BEI (BINDER, 2011; BONIOLI et al., 2005; JORGE et al., 2007; KANT; WIT; BREUNING, 2003), representando, pois, uma boa estratégia para se iniciar as investigações moleculares de BEI. Em média, 5% dos pacientes com BEI possuem alteração no SHOX(HUBER et al., 2006; RAO et al., 1997).
O GH, o GHR e o IGF-1 e uma gama de moléculas afins compõem o eixo regulador do crescimento pré e pós-natal (BUZI et al., 2007). Os permanentes avanços nas técnicas de biologia molecular têm facilitado a identificação de defeitos genéticos nos diferentes componentes do eixo GH-IGF-1 em crianças com baixa estatura (KANT; WIT; BREUNING, 2003). Ao todo, mais de 40 mutações já foram descritas no GHR, a maioria em homozigose ou heterozigose composta, porém há casos de heterozigose com efeito dominante negativo (JORGE, 2008). Atualmente,
estima-se que entre 1 a 5% das crianças com BEI possam apresentar defeito no GHR(ATTIE, 2000; SAVAGE et al., 2007)
1.2.1 SHOX (Short Stature Homeobox Gene)
Os primeiros estudos acerca deste gene iniciaram-se em 1961 (JACOBS et al., 1961). Eles relataram relações fenotípicas de baixa estatura e deleção do braço curto do cromossomo X em mulheres com quadro de amenorréia primária. Elas diferiam das pacientes com deleção no braço longo, as quais possuíam estatura normal. Posteriormente, em 1982, Goldman e cols. (colaboradores) mostraram pela primeira vez que havia correlação entre deleção do braço curto do cromossomo X e baixa estatura nas pacientes com síndrome de Turner (GOLDMAN et al., 1982). Assim, surgiam os primeiros indícios de que o braço curto do cromossomo X alocasse algum gene relacionado ao desenvolvimento somático.
pertencentes à região pseudo-autossômica 1 (PAR1) dos cromossomos sexuais. O
PAR1 (Figura 1) possui aproximadamente 2,6 mega-bases (Mb) e está posicionada na extremidade do braço curto dos cromossomos sexuais e ao que parece, todos os genes localizados nesta região não são silenciados com inativação do cromossomo X, ou seja, são expressos bialelicamente (SCHAEFER et al., 1993).
1.2.1.1 Estrutura do gene SHOX
Em 1997, Rao e cols. conseguiram mapear este gene através de estudos moleculares em pacientes que possuíam monossomias parciais e baixa estatura. Ele apresenta 7 éxons (Tabela 1), sendo que o éxon 1, a porção 5’ do éxon 2 (5’UTR) e as regiões 3’ dos éxons 6a e 6b (3’UTR) não são traduzidas, porém todos os outros éxon são (MUNNS et al., 2004; RAO et al., 1997).
Tabela 1: Tamanho (em pares de base) de todos os éxons do gene SHOX.
Éxon Tamanho (pb)
Éxon 1 262
Éxon 2 708
Éxon 3 203
Éxon 4 58
Éxon 5 89
Éxon 6a 1.166
Éxon 6b 625
Fonte: Adaptado de NCBI, 2012.
Este gene possui duas regiões promotoras P1 e P2, que estão localizadas upstream do éxon 1 e na porção 5’ do éxon 2, respectivamente. Os transcritos gerados (SHOX1 e SHOX2) diferem apenas quanto ao tamanho da região 5’UTR não traduzida. Como este tamanho é inversamente proporcional à eficiência da tradução, o SHOX1, por apresentar a porção 5’UTR maior que o SHOX2, é menos
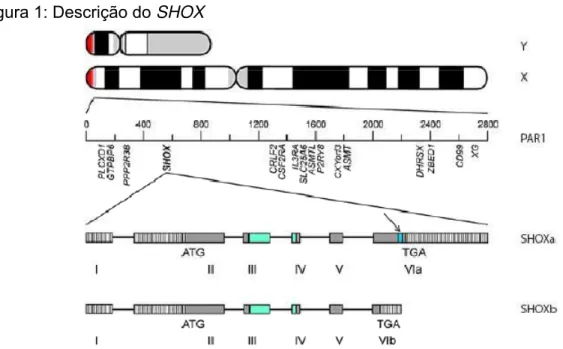
Figura 1: Descrição do SHOX
Fonte: (HELENA MANGS; MORRIS, 2007).
O esquema mostra a localização cromossômica de PAR1 (em vermelho), o conteúdo genético de PAR1 incluindo SHOXe os dois mRNAs diferentes produzidos por splicingalternativo, que contêm os éxons 1 a 6a e 1 a 6b e suas respectivas regiões 5’UTR e 3’UTR representadas por retângulos com linhas verticais. Homeobox
indicado em azul e o opt aristaless and rax (OAR), presente apenas no SHOXa, indicado pela seta.
1.2.1.2 Mutações no SHOX
A maioria das mutações neste gene é determinada por troca de bases (80%) (Figura 2), que podem originar proteínas truncadas, com códons de parada prematuros ou mesmo permutas não-conservativas, ou seja, de aminoácidos com
propriedades químicas diferentes (polar para apolar, por exemplo), fazendo com que a estrutura terciária da proteína se modifique. Elas são encontradas por todo gene, mas a grande maioria delas se concentra nos éxons 3 e 4, que, por sua vez, alocam o homeodomínio (domínio da proteína que atua como fator de transcrição; Figura 1) (SABHERWAL et al., 2004), prevê-se, assim, que a conservação dele seja essencial
Figura 2: Frequência e a localização das mutações no SHOX.
Fonte: Adaptado de (BLASCHKE; RAPPOLD, 2001).
A figura mostra a distribuição de mutações missense (azul escuro), nonsense (laranja), framshift(amarelo) e pequenas inserções/deleções (azul claro) de cada éxon do SHOX.
Diferentes trabalhos já mostraram a frequência de alterações no SHOX em pacientes com BEI (Tabela 2). Estes estudos apontam diferentes frequências de mutações de ponto e deleções citogenéticas (HUBER et al., 2006; RAO et al., 1997)
(Tabela 2). A recorrência de mutações de ponto pode variar de 1 a 5%. Estas diferenças podem ser causadas por diversos fatores, tais como: tamanho do grupo estudado, critério de seleção dos pacientes e técnica molecular utilizada.
Tabela 2: Frequência de alterações no SHOX em pacientes com BEI.
Autor/Ano Deleções citogenéticas Mutações de ponto
Rao e col., 1997 0/91 1/91
Musebeck e col., 2001 0/35 NA
Binder e col., 2000 1/68 0/68
Rappold e col., 2002 3/150 3/150
Stuppia e col., 2003 4/56 3/56
Binder e col., 2003 3/140 NA
Morizio e col., 2003 4/56 NA
Huber e col., 2006 14/84 4/84
Rappold e col., 2007 25/1534 9/1534
Jorge e col., 2007 0/63 2/63
Tabela adaptada de (FUNARI, 2009). NA: não avaliada.
Kilo-bases (Kb), localizada downstream do SHOX, importante para a estatura e aparentemente envolvida na patogênese da discondroestose de Leri-Weill (DLW) (BENITO-SANZ et al., 2005). Estudos dessa região denominada de "área do shox", em pacientes ainda sem diagnostico molecular, resultaram na definição de uma nova classe de deleções do PAR1, cuja análise de Copy Number Variations (CNV) passou a ser incluída na avaliação dos pacientes com BEI (BENITO-SANZ et al., 2005).
A origem disto está em parte relacionada à elevada taxa de crossing overda
região PAR1 (ROUYER et al., 1986). Os padrões de segregação entre X/Y resultam em um evento de recombinação único e obrigatório durante a meiose masculina. Nas regiões terminais do braço curto dos cromossomos X e Y, há 10 vezes mais recombinações que entre as mesmas regiões nas células germinativas femininas,
isto se deve a diferença de tamanho entre as regiões PAR1 do cromossomo X e Y (ROUYER et al., 1986). Assim, o aumento na taxa de crossing over favorece o aumento da frequência de deleções no SHOX, e como esta região não sofre o processo de inativação do X, as mutações ali são herdadas de forma mendeliana.
1.2.1.3 Estrutura Protéica do SHOX
Esta proteína possui dois motivos: homeobox, codificado pelos éxons 3 e 4 (MARCHINI et al., 2004), presente tanto no SHOXa, como no SHOXb; e o domínio OAR, codificado pela porção C-terminal do éxon 6a (Figura 1) (SABHERWAL et al., 2004), portanto presente apenas no SHOXa (RAO et al., 1997). Estes domínios estão envolvidos na função de fator transcricional e transativação, respectivamente.
Por isso, acredita-se que o SHOXb (225 aminoácidos) atue modulado negativamente a atividade de SHOXa (292 aminoácidos) (Figura 1) (RAO et al., 2001), uma vez que no momento em que a maquinaria de transcrição sintetiza o SHOXb, esta deixa automaticamente de produzir o SHOXa.
A conservação do homeodomínio não é só relevante para o SHOX. Ele é extremamente conservado entre os diversos genes que atuam como fatores
Figura 3: Sequência de alinhamento de vários homeodomínios.
Fonte: (SABHERWAL et al., 2004)
Os aminoácidos destacados fazem parte do sinal de localização nuclear relacionados aos homeodomínios. As setas na parte superior indicam os aminoácidos conservados em todas as proteínas deste gênero.
1.2.1.4 Mecanismo de ação do SHOX
Pelo fato da atividade protéica do SHOX ser dosagem-dependente, a perda de uma das cópias do gene (haploinsuficiência) determina o surgimento de doenças que vão de BEI à DLW (JORGE et al., 2008). Diferentemente da BEI, que possui um fenótipo variável, a DLW apresenta baixa estatura desproporcional, além do
encurtamento mesomélico dos membros (perna e antebraço) e deformidade de Madelung (luxação na porção distal da ulna) (RAO et al., 2001). Somado a estes fenótipos, existem situações mais raras, em que ambas as cópias de SHOX estão ausentes (ou inativas), resultando em uma doença mais grave conhecida como displasia mesomélica de Langer (BELIN et al., 1998).
A haploinsuficiência explica o porquê de a BEI aparecer em pacientes com mutações em heterozigose (inativação de uma das cópias do gene) ou em pacientes que apresentam o SHOXem hemizigose (ausência de uma das cópias do gene) e o porquê de mais de 2/3 das mulheres com Síndrome de Turner (45, X) ou com deleções parciais (ou totais) do braço curto possuírem baixa estatura, característica marcante desta aneuploidia (BINDER et al., 2004).
durante condrogênese (formação de cartilagem. Clement-Jones e cols. mostraram
que a partir da condensação do mesênquima, forma-se a estrutura pré-cartilaginosa do osso para iniciar sua ossificação (CLEMENT-JONES et al., 2000), ou seja, SHOX
está expresso nos elementos esqueléticos desde as fases mais iniciais do desenvolvimento.
A placa (ou núcleo) de crescimento localizada na epífise óssea é composta
por um único tipo de células, chamada de condrócitos. Uma camada de condrócitos de reserva constitui um ‘pool’ de células, que quando estimuladas entram na fase de
proliferação celular, aumentando o volume de matriz disponível para posteriormente ser substituído pelo tecido ósseo. Eventualmente, a proliferação acaba quando as células entram na fase de diferenciação, que é caracterizada pelo crescimento celular, seguida de apoptose dos condrócitos hipertróficos, fundamentais para o crescimento ósseo (MARCHINI et al., 2007).
Figura 4: Osteogênese e atuação do SHOX.
A atuação SHOX se dá na zona vermelha (à esquerda). A placa de crescimento (à direita) é composta pela (A) camada de condrócitos de reserva, (B) zona de
proliferação, (C) zona de diferenciação, (D) zona de crescimento e (E) apoptose de condrócitos hipertróficos. O SHOX atua entre as zonas B e C de forma a atrasar a diferenciação dos condrócitos e, consequentemente, a entrada na fase de
crescimento e apoptose.
De forma molecular, o SHOX pode atuar como fator de transcrição se ligando ao DNA, preferencialmente a determinadas regiões palindrômicas (5’-TAAT(N)ATTA-3’) (MARCHINI et al., 2004); ou como ligante de proteínas que atuam como fatores de transcrição em células de origem osteogênicas (SABHERWAL et al., 2004). Mas, para isso, a proteína deve ser primeiramente ativada, atravessar a carioteca para
atuar no núcleo celular. Uma vez fosforilada (ativada) em sítios de serina, a proteína pode ser transportada para o núcleo com o auxílio do sinalizador de localização nuclear (NLS), ambos presentes no domínio homeobox(Figura 1) (MARCHINI et al., 2006).
O primeiro alvo do SHOX identificado foi o gene Brain Natriuretic Peptide Protein(BNPP), um importante regulador da ossificação endocondral (MARCHINI et al., 2007). Além de atuar sobre oBNPP, o SHOX também pode agir como inibidor de expressão quando se liga a múltiplas sequências promotoras do gene Fibroblast Growth Factor Receptor 3 (FGFR3), que por sua vez atua negativamente sobre outros alvos: COL2A1 e AGC1 (DECKER et al., 2011). Estes codificam componentes da matriz cartilaginosa de colágeno tipo II e aggrecan,
(ALA-KOKKO, 2002). Por isso, a hiperativação de FGFR3 pode levar a
acondroplasia, o tipo mais comum de baixa estatura, uma vez que este gene desempenha um papel fundamental no desenvolvimento dos membros (DECKER et al., 2011).
Além de se ligar diretamente ao DNA, o SHOX pode atuar sobre proteínas que também são fatores de transcrição. Os domínios homeboxe OAR, presentes no
SHOX, dimerizam duas proteínas (SOX5/SOX6). Após esta etapa o SOX9 pode se ligar ao dímero e formar o SOX trio (SOX5/SOX6/SOX9), principal complexo da condrogênese, expresso principalmente durante o desenvolvimento fetal humano, sendo responsável pela ativação dos principais componentes da matriz da cartilagem de colágeno II (AZA-CARMONA et al., 2011; IKEDA et al., 2004).
Assim, mutações nos domínios homeobox, OAR ou em outras regiões, que alteram sua estrutura terciária, causam sua perda de função ou diminuem quase que totalmente a capacidade de formar o SOX trio, além de interferir na a expressão de os genes citados: BNPP, COL2A1e AGC1 (ALA-KOKKO, 2002; AZA-CARMONA et al., 2011; DECKER et al.; MARCHINI et al., 2007). Portanto, a integridade destes domínios é essencial para garantir a interação entre os diversos alvos do SHOX, quer sejam eles proteínas ou genes (LEFEBVRE, 2002).
Por isso, a identificação precoce de mutações no SHOXcomo causa da baixa
estatura evita a realização de uma exaustiva bateria de exames, tais como investigação de outros genes que podem ocasionar a baixa estatura como PCNT, FGFR3, IGF-1, GH, GHRHR etc. Poupa o paciente de exames relacionados a distúrbios endócrinos, que podem depender da administração de diversos
medicamentos, conforme protocolos específicos, como insulina, clonidina, levodopa, glucagon entre outros, levando, na maioria das vezes, a um diagnóstico inconclusivo.
Por outro lado, uma vez que a doença seja identificada por mutação no SHOX, é possível tomar uma série de medidas que visam à melhoria da qualidade de vida do paciente e de seus familiares, como: traçar uma projeção da altura final
1.2.2 GHR (Growth Hormone Receptor)
As primeiras investigações do receptor do hormônio do crescimento (GHR) se iniciaram no ano de 1966. Na ocasião uma síndrome com fenótipos bem definidos,
semelhante à deficiência isolada de GH, se diferenciava por apresentar altos níveis de GH e baixos níveis de IGF-1 no sangue. O que mais tarde ficou conhecida como síndrome de Laron, em homenagem ao seu descobridor (LARON, 1999). Mas foram Godowski e cols. em 1989 que mostraram, pela primeira vez, a associação do GHR à doenças genéticas, após diagnosticar um paciente com uma grande deleção em
homozigose em pacietnes com baixa estatura (BOGUSZEWSKI, 2001; GODOWSKI et al., 1989).
1.2.2.1 Estrutura do gene GHR
A estrutura do gene GHR possui um total de 10 éxons, em que o éxon 1 e parte do éxon 2 formam a porção 5’UTR (não traduzida) do mRNA, os éxons 2 e 8 codificam o domínio transmembrana, porém o éxon 2 codifica também uma sequência sinal de 18 aminoácidos que é clivada quando na proteína madura, os éxons 3-7 codificam o domínio extracelular do receptor, ou seja, a parte ligante ao GH e os éxons 9 e 10 codificam o domínio citoplasmático de transdução de sinal e a região 3’UTR (não traduzida) (GODOWSKI et al., 1989) (Figura 5; Tabela 3).
Tabela 3: Tamanho (em pares de base) de todos os éxons do gene GHR.
Éxon Tamanho (pb)
1 181
2 81
3 66
4 130
5 173
6 179
7 166
8 91
9 70
10 3426
Figura 5: Estrutura do Gene GHR.
Fonte: Adaptado de (ROSENBLOOM, 2000)
A linha horizontal em preto representa as sequências intrônicas; Promotores (em amarelo), éxon 1 (em amarelo) e éxon 2 (vermelho) formam a região 5’UTR , éxons 3-7 (azul claro) compõem do domínio extracelular do receptor, éxon 8 (em verde) faz parte do domínio transmembrana, os éxons 9 e parte do 10 (azul escuro) são
responsáveis pela transdução de sinal, enquanto a extremidade do éxon 10 (amarelo escuro) faz parte da região 5’ UTR.
1.2.2.2 Mutações no GHR
Mutações no GHR podem causar insensibilidade ao GH (BARTON et al., 1989), que por sua vez podem ocasionar fenótipos que vão de Síndrome de Laron à BEI. Diferentemente da baixa estatura idiopática, a Síndrome de Laron possui um conjunto de alterações fenotípicas características que permitem seu reconhecimento: retardo de crescimento desde o nascimento, desproporção
craniofacial, hipoplasia da parte central da face, cabelos ralos, alterações de arcada dentária, extremidades curtas, obesidade em tronco, atraso de idade óssea e hipoglicemia (BOGUSZEWSKI, 2001).
Estima-se que cerca de 1 a 5% das crianças com baixa estatura idiopática possam apresentar algum defeito no GHR (ATTIE, 2000). Este gene possui várias mutações já descritas, totalizando pelo menos 40 delas (BOGUSZEWSKI, 2001). A
maioria é homozigota recessiva ou heterozigota composta. Há casos em que o gene possa ter uma mutação heterozigota com efeito dominante negativo. Nesta situação as moléculas de GHRs defeituosos se acumulam na superfície da membrana, formando dímeros com as moléculas de GHRs normais, impossibilitando o GH circulante de ancorar (ROSS et al., 1997).
O gene GHRcodifica um receptor que se apresenta em forma de dímero na
membrana plasmática. A existência de duas folhas betas antiparalelas em sua estrutura garante a sua funcionalidade como receptor de citocinas em um único domínio transmembrana (CLACKSON et al., 1998).
Quando elas se combinam ao GH, formam dois sanduíches: um superior em Trp122 e um inferior em Trp187 (CLACKSON et al., 1998) e estas interações de triptofanos, pontes de hidrogênio entre outros aminoácidos e o reposicionamento estérico do GHR contribuem para a energia de ligação de GH-GHR e GHR-GHR (Figura 6) (CLACKSON et al., 1998).
Figura 6: Estrutura do GHR e rotação do dímero.
Fonte: Adaptado (CLACKSON et al., 1998).
A ação do GHR baseia-se no mecanismo de ativação do receptor. O GH (azul) liga-se a duas moléculas de GHR (vermelho), possibilitando a rotação do dímero.
1.2.2.4 Eixo GH/IGF-1
Para compreender o funcionamento completo da proteína e como as
mutações genéticas podem causar a baixa estatura, é necessário compreender desde o Eixo GH/IGF-1 até a sua via de sinalização. O GH é um hormônio de origem peptídica (191 aminoácidos) secretado pela glândula anterior da pituitária (adenohipófise) (DAVIDSON, 1987). Ele é controlado pela ação de dois fatores hipotalâmicos: o GHRH, que estimula a secreção de GH; e somatostatina, que inibe secreção de GH (GIUSTINA; MAZZIOTTI; CANALIS, 2008). O hormônio codificado
pelo GH age diretamente em vários tecidos do corpo, quer seja no crescimento somático, quer seja no metabolismo (HUGHES; FRIESEN, 1985).
fígado sob estímulo do GH é quem atua na placa de crescimento, levando as células
a diferenciações maturativas e proliferativas, estimulando o seu amadurecimento na placa de crescimento (Figura 7) (LIST; COSCHIGANO; KOPCHICK, 2001).
Por outro lado, a teoria do efeito duplo (mais aceita) dita que, somado à ação descrita, o GH pode atuar na placa de crescimento agindo diretamente sobre seu receptor genuíno em tecidos periféricos (SAVAGE et al., 2007), principalmente no tecido ósseo, para ativar uma cascata de reações provocando a diferenciação dos pré-condrócitos em condrócitos jovens. Após esta etapa, os condrócitos jovens iniciam a produção de IGF-1 que, através de efeitos parácrinos, estimulam a expansão clonal e a maturação dos condrócitos na placa de crescimento (SAVAGE et al., 2007).
Figura 7: Vias de sinalização do receptor GHR e IGF-1.
Fonte: Adaptado de (SAVAGE et al., 2007).
A esquerda está esquematizado a ‘teoria do efeito clássico’: o GH oriundo da pituitária se liga ao receptor genuíno no fígado, e assim, estimula a produção de IGF-1, que por sua vez é transportado (principalmente) sob a forma de IGFBP-3, para em seguida se ligar ao seu receptor e promover o crescimento; Ao lado direito, a ‘teoria do efeito duplo’: somado à ‘teoria do efeito clássico’, a teoria do efeito duplo dita que o GH pode agir de forma local em tecidos periféricos.
1.2.2.5 Mecanismo de ação do GHR
Para tais respostas, é necessária a ligação do GH ao receptor GHR que se encontra pré-dimerizado. Inicialmente o sítio 1 do GH se liga ao GHR (complexo 1:1, inativo); e em seguida, o sítio 2 se liga a uma outra molécula de GHR (complexo 2:1,
espacial da porção intracitoplasmática do GHR, desencadeando uma sinalização
celular interna de um domínio citoplasmático com as proteínas da família da Janus Kinase (JAK) (CARTER-SU; SCHWARTZ; SMIT, 1996).
Após as fosforilações das JAKs, estas passam a fosforilar sítios de tirosina no próprio GHR, criando assim, novos sítios de acoplamento para outras moléculas sinalizadoras (CARTER-SU; SCHWARTZ; SMIT, 1996; KOPCHICK; ANDRY, 2000).
As mais importantes (em relação aos efeitos de regulação de IGF-1) são as transdutoras de sinal e ativadoras de transcrição (STAT), quando ativadas formam um dímero de translocação nuclear de fatores de transcrição para a regulação de expressão genes alvos relacionados ao metabolismo e ao crescimento (LANNING; CARTER-SU, 2006).
1.2.2.6 Polimorfismo do éxon 3 no GHR
Em 1989, na elucidação do gene GHR, Godowski e cols. também descreveram a existência de diferentes variações do gene, entre elas, a deleção do éxon 3, fruto da recombinação existente entre duas sequências repetidas de retrovírus que flanqueiam-no (GODOWSKI et al., 1989; PANTEL et al., 2000). Isto resultou em dois polimorfismos: GHRfl (GHR full length; fl) e o GHRd3 (GHR éxon 3 deletado; d3).
Inicialmente, acreditava-se que este gene originava duas isoformas protéicas por splicing alternativo. Porém, hoje se sabe que estas duas isoformas são polimorfismos distintos transmitidos por segregação independente (RB et al., 1995). A determinação do polimorfismo foi proposta inicialmente por uma PCR multiplex, com um primer direto e dois reversos. A diferença de 403 pares de bases entre os dois produtos pode ser distinguida por uma eletroforese em agarose a 2% (PANTEL et al., 2000). Mais recentemente um estudo conseguiu determinar (em uma população suéca) que o SNP rs6873545 estava associado a estes polimorfismos (GLAD et al., 2010)
independe dos 22 resíduos codificados pelo éxon 3, já que eles não estão na superfície de ancoragem do GH (BASS; MULKERRIN; WELLS, 1991; PANTEL et al., 2003; SOBRIER et al., 1993).
Posteriormente, surgiram vários questionamentos: qual o real papel do GHRd3?; ele traz alguma vantagem na resposta ao GH? Alguns experimentos mostraram não haver diferença de resposta e outros sim. Pilotta e cols. e Blum e
cols. apontaram que crianças GH deficientes (GHD), tratadas com GH humano recombinante (rhGH) possuíam a mesma velocidade de crescimento quando se compararam os genótiposGHRd3/GHRd3e GHRd3/GHRfl, ou quando se comparou GHRd3/GHRd3 e GHRd3/GHRfl ao genótipo GHRflGHR/fl (BLUM et al., 2006; PILOTTA et al., 2006). Em contrapartida, outro estudo apontou o papel positivo do GHRd3na resposta ao GH (JORGE et al., 2006).
Um estudo com mais de 170 crianças européias tratadas com rhGH concluiu que crianças com genótipo GHRd3 responderam duas vezes mais ao GH que o grupo cujo genótipo era GHRfl/GHRfl (DOS SANTOS et al., 2004). No Brasil, crianças com o genótipo GHRd3 apresentaram capacidade de produzir mais IGF-1 que o grupo de crianças GHRfl, após ambos terem sido estimulados por rhGH (TOYOSHIMA et al., 2007). Recentemente, um estudo de meta-análise (de dezoito
artigos) forneceu dados que reafirmaram a hipótese de que o genótipo GHRd3 favorece o aumento da velocidade de crescimento de crianças GHD ou sem-GH, tratadas com rhGH (WASSENAAR et al., 2009).
Correlacionar polimorfismos (e SNPs) ao eixo GH/IGF-1 não é inédito: o SNP rs4988492 do gene GH Releasing Hormone (GHRH) mostrou ter correlação com a
densidade óssea (DENNISON et al., 2009), de forma semelhante, o polimorfismo GH1 do gene GH predispõe à osteoporose (DENNISON et al., 2004). Recentemente, um grupo chinês mostrou correlação entre o metabolismo e este polimorfismo em crianças (GAO et al., 2011). Portanto, é possível, que a produção de IGF-1 seja influenciada pelos diferentes polimorfismos GHRfle GHRd3.
1.3 FARMACOGENÉTICA
indivíduo e letais para outro. Nos Estados Unidos, mais de 2 milhões de
hospitalizações e 100.000 mortes por ano são decorrentes de reações adversas (LAZAROU; POMERANZ; COREY, 1998).
Resposta a medicamentos são determinados por vários fatores, entre eles, alimentação, saúde, influência ambiental e carga genética. Esta última é estudada pela Farmacogenética e Farmacogenômica, que têm suas pesquisas voltadas para
as variações do genoma humano, que possui aproximadamente 23.800 genes, 3 bilhões de pares de bases e milhões de polimorfismos (principalmente SNPs) (MROZIEWICZ; TYNDALE, 2010), para poderem propor tratamentos individualizados. Por exemplo, polimorfismos em íntrons dos canais de sódio tipo 1 da subunidade alfa (SCN1A) podem alterar o seu receptor, os quais estão relacionados com diferentes respostas à carbamazepina - um anticonvulsivamente usado em pacientes epiléticos (TATE et al., 2005).
O critério tradicionalmente usado para administração de rhGH é baseado no peso ou superfície corporal, com alguns pequenos ajustes feitos para o diagnóstico subjacente e, ocasionalmente, para pacientes na puberdade (COHEN et al., 2002). No entanto, esta abordagem não prevê grandes variações na capacidade de resposta em pacientes rotulados GHD (COHEN et al., 2002). Este problema pode ser ainda mais relevante em crianças com diagnóstico de baixa estatura, pois estudos mostram clara aceleração do crescimento durante a infância nos pacientes tratados com GH. Porém, o tratamento com GH só é benéfico a certos grupos de baixa estatura, entre eles GHD, mas tem sido limitado em pacientes com BEI (WIT; REKERS-MOMBARG, 2002).
Portanto, não se pode desprezar a múltipla etiologia de BEI. A possibilidade de pacientes não responderem ao tratamento deve ser considerada, pois tratamentos com rhGH demandam tempo e dinheiro, e a seu sucesso está diretamente ligado ao diagnóstico correto (KANT; WIT; BREUNING, 2003). Assim, a individualização da terapia de rhGH e a origem da doença poderiam ser utilizados para otimizar as respostas do paciente, diminuindo os efeitos colaterais, bem como,
reduzindo os custos do tratamento (JOHNSTON, 2008).
(BOGUSZEWSKI, 2001). A saída diante desta situação é o tratamento com IGF-1
recombinante, que acelera o crescimento da criança nos dois primeiros anos, mas não promove a mesma resposta do tratamento com rhGH em pacientes com baixa estatura de origem hipofisária (LARON, 1999) ou por mutações presentes no SHOX.
1.3.1 Situação do tratamento com rhGH no Brasil
Estas drogas não chegam gratuitamente para todos os pacientes brasileiros. Não existe sequer um critério molecular para o tratamento com rhGH (apesar do seu grande uso nos países desenvolvidos). Com conhecimento existente sobre o SHOX como principal causa de baixa estatura em mulheres com Síndrome de Turner, e dos grandes resultados obtidos no tratamento destas pacientes com rhGH, pesquisadores de outros países testaram-no em pacientes com BEI e mutações no SHOX (BINDER, 2011). O sucesso foi tamanho que instantaneamente este tratamento foi aprovado pelo Foods and Drugs Administration (FDA) e European Medicines Agency(EMEA) para este grupo de pacientes (BINDER, 2011).
O FDA também aprovou em 2007 o uso de IGF-1 exógeno para o tratamento de pacientes com deficiência preliminar severa de IGF (IGFD severa), sendo uma alternativa extremamente valiosa aos pacientes que não respondem ao GH, que é a
terapêutica de primeira escolha em virtude de sua resposta mais eficiente. Estes resultados precisam ser avaliados pela Agência Nacional de Vigilância Sanitária (ANVISA) juntamente com o Ministério da Saúde, para elaboração de diretrizes que possam beneficiar pessoas com BEI por mutações no SHOX.
De acordo com o Ministério da Saúde, o diagnóstico para o tratamento com
rhGH pode ser feito de três formas distintas: diagnóstico por exames de imagem, diagnóstico clínico e diagnóstico laboratorial (Anexo A). Nenhum critério contempla modelos moleculares, que podem ir desde citogenética clássica até sequenciamento e análise de CNV. Além disso, este protocolo só contempla pacientes GHD e mulheres com Síndrome de Turner. Portanto, se, porventura, um paciente tem seu diagnóstico de BEI por mutação no SHOX, identificado por uma técnica molecular, o
mesmo não terá disponibilizado o tratamento com rhGH gratuitamente, mesmo sabendo que a origem de sua baixa estatura é a mesma das pacientes com Síndrome de Turner.
o SHOX atua atrasando a fase de diferenciação celular (MUNNS et al., 2004).. A sua
ausência determina o fim prematuro da fase de proliferação (MUNNS et al., 2004). Com isso, o GH promove uma grande proliferação celular e, após o encerramento prematuro desta fase, existirão mais células para sofrerem diferenciação, crescimento e apoptose, resultando no aumento ósseo.
O estudo molecular surgiu como uma informação adicional ao estudo da baixa
estatura. Porém, a relação entre genótipo, fenótipo e resposta ao tratamento não é linear, e a identificação de mutações ajudaria a optar pelo melhor tratamento (SAVAGE et al., 2007), não fazendo dele uma loteria. Portanto, é extremamente necessária a continuidade da investigação genética em relação a este tema.
1.4 ANÁLISE DE MUTAÇÕES NO SHOXE GHRRELEVANTES À BEI
Para se propor um diagnóstico, deve-se ter em mente a escolha das técnicas a serem utilizadas. Para isso, é preciso saber o tipo de gene a ser investigado, bem como as alterações descritas na literatura. Como o SHOX se encontra flanqueado por sequências repetitivas ‘Alu’, há um aumento na probabilidade de recombinação de elementos não homólogos, além da já mencionada alta frequência de crossing over (FUKAMI et al., 2008; ROUYER et al., 1986). Ambos os casos predispõe o surgimento de deleções ou alterações de CNV.
A descoberta de CNVs, que podem não ser identificáveis por bandeamento cromossômico convencional (REDON et al., 2006) ou mesmo sequenciamento, trouxe um novo olhar sobre como enxergar possíveis mutações no genoma. Estes rearranjos genômicos possibilitaram esclarecer a etiologia de patologias até então
obscuras, como doenças mendelianas, alterações comportamentais, ou simplesmente alterações polimórficas benignas (LUPSKI et al., 1992). Qualquer variação (duplicações, triplicações, deleções ou inserções) em elementos essenciais a regulação da expressão gênica podem levar a um fenótipo anormal.
Assim, uma boa investigação do SHOX requer não apenas um tipo de técnica molecular, mas um conjunto de técnicas que se complementem. Desta
regiões intragênicas não codificantes; e sequenciamento, para se identificar
mutações de ponto.
De acordo com a literatura, não há relatos de grandes deleções esporádicas no cromossomo que aloca o GHRe tampouco é uma região suscetível a variações de CNV, portanto, a técnica mais indicada para se realizar investigações à procura de mutações é o sequenciamento.
1.4.1 CNVs e Multiplex Ligation-dependent Probe Amplificaition (MLPA)
Os estudos sobre CNVs possibilita hipotetizar que os nucleotídeos presentes em suas regiões estão mais suscetíveis a variações (polimórficas ou não) que nucleotídeos fora destas regiões, com uma diferença entre 1.000 e 10.000 vezes (LUPSKI, 2007; STANKIEWICZ; LUPSKI, 2010). Portanto, o genoma humano é bem
mais variável do que se pensava e os CNVs são os grandes responsáveis pela evolução e diversidade genética entre inidivíduos. (STANKIEWICZ; LUPSKI, 2010). Dessa forma, estudos adicionais poderão auxiliar significativamente no esclarecimento da herança de outras doenças.
Os CNVs na região do gene são essenciais para a estratégia de screening (DEN DUNNEN; WHITE, 2006). A região do SHOX, que responde por 500 Kb downstream do gene é composta por sequências específicas que estão diretamente relacionadas à sua expressão. Dentre as várias técnicas conhecidas para identificar estas regiões regulatórias no SHOX, podemos citar algumas: Fluorescent in situ hybridization(FISH), Análise de Microssatélite e Multiplex Ligation-Dependent Probe Amplification(MLPA). Quando se comparam estas técnicas entre si, percebe-se uma grande vantagem do MLPA sobre as demais.
O FISH não permite a detecção de pequenas deleções em virtude do tamanho das sondas utilizadas, além disso, é uma técnica extremamente laboriosa e de alto custo. Já a Análise de Microssatélite, apesar de muito utilizada, não detecta mutações de ponto e deleções fora da região dos microssatélites. Assim como FISH, também não identifica deleções intragênicas (FUNARI et al., 2008). O surgimento do
Para compreender o seu funcionamento, é necessário primeiro entender
como as suas sondas são desenhadas. Elas são oligonucleotídeos de DNA divididas em 3 elementos: sequência dos primers; sequência stuffer; e sequência de anelamento (Figura 8). A sequência de anelamento determina a região com a qual a sonda vai se hibridizar, sendo única para cada sonda. Já nas sequências stuffer pouco importa a sequência em si, sendo relevante apenas o seu tamanho. Ela é
variável de uma sonda para outra e tem como objetivo diferenciar os fragmentos pelo número de bases durante a eletroforese do sequenciador. Este mecanismo é importante para o perfeito funcionamento de uma eletroforese com mais de 50 produtos distintos. Enquanto a sequência de primers é exatamente a mesma em todas as sondas, pois na última etapa é feita uma PCR multiplex por um único par de primerscomplementares a estas duas sequências.
Figura 8: Estrutura de um par de sondas do MLPA.
Primer PCR (preto): sequência de anelamento dos primers, igual em todas as sondas do kit; Sequência Stuffer(vermelho): sequência com a função de deixar os fragmentos com diferentes tamanhos para serem distinguidos e identificados na eletroforese do sequenciador; e Sequência de anelamento (azul): sequência
complementar a região a ser investidagada, específica de cada sonda. Cada par de sonda se hibridiza com uma distância de 1 base, com isso, apenas as sondas que sofrerem a reação da ligase é que amplificam.
O MLPA se fundamenta na hibridização adjacente de pares de sondas, seguida por sua ligação e amplificação. O primeiro passo da técnica é promover a hibridização dos pares de sondas ao DNA (cada sonda dista uma base do seu respectivo par). A próxima etapa constitui-se da ligação dessas sondas, assim,
somente quando os pares de oligonucleotídeos estão hibridizados lado a lado é que podem ser unidos pela reação da ligase e, em seguida, amplificados por um par de primers universal, já que a extremidade de todas as sondas possui a mesma sequência nucleotídica.
distingui-las através de uma eletroforese capilar, comparando o padrão de picos
obtidos e os resultados das amostras de referência. O nível de amplificação é proporcional à quantidade de cópias do fragmento, e como cada primer possui um fluoróforo, é possível avaliar pela intensidade do sinal da fluorescência se aquela região investigada pela sonda está duplicada, triplicada ou mesmo deletada (Figura 9).
Figura 9: Esquema da técnica MLPA.
Fonte: MRC Holland.
Após a hibridização das sondas com a sequência alvo, os oligonucleotídeos são enzimaticamente ligados. Cada sonda contém uma sequênciastufferde
Por isso, técnica do MLPA foi escolhida para compor esta pesquisa no que diz
respeito à investigação de CNVs. Ela é relativamente nova e possui poucos trabalhos publicados no Brasil. O grande sucesso desta técnica está em sua execução, que apesar de buscar por CNVs, necessita apenas de um termociclador e um sequenciador (o que as difere das demais, que necessitam de uma grande estrutura laboratorial).
2 JUSTIFICATIVA
Dentre os vários distúrbios relacionados ao esqueleto, patologias que causam baixa estatura merecem um destaque especial por serem motivo de preocupação
constante em consultórios pediátricos e possuírem uma incidência média alta (3/100) (RAPPOLD, G. et al., 2007), sendo uma das alterações mais frequentes da infância. Assim, avaliar previsões para estatura sempre foi motivo de preocupação aos pais. O diagnóstico precoce é importantíssimo nestes casos, a fim de propor ao paciente o tratamento correto, visando à melhoria de sua qualidade de vida, tanto
nos aspectos fisiológicos quanto nos aspectos sociais.
Para isso, é imprescindível a criação de um serviço diagnóstico genético-molecular no Distrito Federal (DF). Desta forma seria possível identificar mutações genéticas (não somente para doenças do esqueleto) na população do DF, bem como estimar suas frequências e, inclusive, propor o tratamento mais adequado.
Futuramente, estatísticas geradas por este centro, poderiam servir como indicadores para investimentos mais direcionados à Saúde nesta região. Isto teria um impacto direto na qualidade de vida desta população.
Para isso, faz-se necessário o uso de projetos-pilotos, que visam estabelecer e padronizar técnicas diagnósticas, avaliar suas capacidades de identificar mutações ao longo do genoma e, posteriormente, estimar a frequência de seus achados para
cada grupo de doenças, com o intuito de avaliá-las se deverão (ou não) ser incorporadas ao serviço de diagnóstico.
Em BEI, a literatura tem evidenciado que mutações de ponto nos genes GHR e SHOX são seus maiores responsáveis (em média, 5%) (BINDER, 2011; BONIOLI et al., 2005; JORGE et al., 2007; KANT; WIT; BREUNING, 2003). Além disso, já existem evidências de pacientes com BEI e variações de CNV em regiões
regulatórias próximas ao gene SHOX. A frequência deste último tipo de mutação não é conhecida, tampouco a frequência dos diferentes tipos de mutações em pacientes com BEI na região Centro-Oeste.
Os resultados deste projeto irão constituir um banco de amostras e dados que serão usados em estudos futuros, como alvo de identificação de novas áreas do
Somado a isso, estas informações poderão servir de subsídio para mudanças
no protocolo do Ministério da Saúde quanto à disponibilização de tratamento gratuito para pessoas com baixa estatura, visando duas alterações básicas: inclusão dos pacientes com BEI como possíveis beneficiários ao tratamento com GH ou IGF-1, bem como a inserção de critérios moleculares no diagnóstico desta patologia.
A exemplo da FDA e EMEA, a inserção dos critérios moleculares poderá
3 OBJETIVOS
3.1 OBJETIVO GERAL
Investigar alterações nos genes SHOX e GHRem pessoas com Baixa Estatura Idiopática no Distrito Federal.
3.2 OBJETIVOS ESPECÍFICOS
Buscar alterações de CNVs no gene SHOX utilizando a técnica do MLPA;
Pesquisar mutações de ponto nos genes SHOX e GHR pela técnica de sequenciamento do tipo Sanger;
Genotipar o polimorfismo do gene GHR por PCR na população brasileira (n>100);
Validar o uso do SNP rs6873545 por PCR quantitativa (qPCR) como substituto da técnica de PCR para inferir os genótipos do polimorfismo do GHR; e
Genotipar o GHR dos indivíduos com Baixa Estatura Idiopática; e
4 MATERIAL E MÉTODOS
Esta pesquisa faz parte do projeto ‘Investigação de alterações gênicas como guia em tratamento de pessoas com baixa estatura idiopática no DF’cujo
protocolo 248/2009 foi submetido à apreciação pelo Comitê de Ética e Pesquisa da UCB no dia 15 de julho de 2009 e aprovado em sua 87ª reunião, no dia 17 de agosto de 2009 conforme as diretrizes descritas no Ofício de n° 248/2009 e com parecer consubstanciado N.° CEP/UCB 111/2009 (Anexo C).
4.1 CASUÍSTICA
Trinta e dois pacientes foram pré-selecionados pelo Serviço de Genética Clínica da Secretaria de Saúde do DF (NuGen), com estatura (proporcional) abaixo do 3° percentil para idade, sexo e população, com análise cromossômica normal e avaliação clínica-radiológica que não sugira osteodisplasia ou outra síndrome
genética reconhecível, relacionada ao SHOX ou GHR. Destes, quatro foram eliminados: três por possuírem hipotiroidismo e outro por ter sido diagnosticado com deficiência de IGF-1.
Em um total de vinte e oito pacientes, sete eram do sexo masculino e vinte e um eram do sexo feminino. Esta diferença refletiu o vício da captação preferencial por mulheres, pois o NuGen é um centro de referência em diagnóstico de Síndrome
de Turner para o DF. Todos os pacientes e seus representantes legais foram informados sobre a pesquisa e assinaram o Termo de Consentimento Livre e Esclarecido (Anexo B).
4.2 COLETA DE AMOSTRAS
4.3 TÉCNICAS MOLECULARES
4.3.1 Extração e quantificação de DNA
Isolou-se o DNA genômico a partir de leucócito de sangue periférico utilizando o The Wizard Genomic DNA Purification Kit de acordo com as instruções do frabicante (Promega, Madison, WI, EUA).
4.3.2 Verificação da integridade do DNA
Aplicou-se as amostras de DNA em gel de agarose a 1% durante 15 minutos a 90 V e 150 mA (Bio-Rad, Hercules, CA, EUA). Posteriormente, corou-se o gel em brometo de etídio, o qual foi visualizado sob luz ultravioleta (U.V.) para verificar a
integridade das amostras de DNA (SAMBROOK; RUSSELL, 2001).
4.3.3 Quantificação do DNA
Para a quantificação das amostras de DNA e verificação da pureza, utilizou-se o Espectrofotômetro Nanodrop 2000c (ThermoScientific, Wilmington, DE, EUA). Mensurou-se a concentração das amostras pelo produto da absorbância em comprimento de onda a 260 nm (A260), fator de diluição e por 50, como segue a
equação abaixo (AZEVEDO et al., 2003):
[DNAdupla fita(µg/mL)]= 50 x A260 x diluição
Já a pureza de uma preparação de DNA pode ser facilmente avaliada por espectrofotometria na região do U.V. Resquícios de proteínas, que podem interferir
em uma série de procedimentos metodológicos são co-purificadas, principalmente nesse tipo de preparação. Um teste simples é aproveitar o padrão de absorção típico da proteína a 280 nm e determinar a relação entre A260/A280. A razão ótima entre
A260/A280 para o DNA situa-se na faixa de 1,8 a 2,0. A presença de proteínas tende a
reduzir esses valores (SAMBROOK; RUSSELL, 2001).
Outros compostos aromáticos (como os fenóis) utilizados na purificação de
DNA também podem interferir no grau de pureza. Portanto, essa relação (A260/A280),
4.3.4 PCR
Para a realização da PCR, utilizou-se 20 ng de DNA genômico adicionada a um mixde PCR, cuja concentração dos reagentes utilizada foi:
Tampão: 1x;
dNTP: 0,2 mM;
primers: 0,25 μM;
MgCl2 1,5 mM;
Taq polimerase recombinante: 1,0 unidades/reação (Phoneutria, Belo
Horizonte, MG, Brasil); Água Mili-Q q.s.p: 12μL/reação
Em seguida, levou-se as amostras para o termociclador Veriti (Applied Biosystems, Foster City, CA, EUA), usando a programação genérica:
SHOX:
Desnaturação inicial: 95°C por 5 minutos; Desnaturação: 95°C por 30 segundos;
Anelamento: 1 minuto (temperatura de acordo com o primer)
(repetir 35 vezes as etapas de 2 e 3)
Elongação final: 72°C por 10 minutos Armazenamento: 10 °C∞.
GHR:
Desnaturação inicial: 95°C por 5 minutos; Desnaturação: 95°C por 30 segundos;
Anelamento: 30 segundos (temperatura de acordo com o primer);