Printed in Brazil - ©2003 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
Review
* e-mail: baran@quimica.unlp.edu.ar
Model Studies Related to Vanadium Biochemistry: Recent Advances and Perspectives
Enrique J. Baran
Centro de Química Inorgánica (CEQUINOR/CONICET,UNLP), Facultad de Ciencias Exactas, Universidad Nacional de La Plata, C. Correo 962, 1900-La Plata, Argentina
Nos ultimos anos, vêm se acumulando as evidências sobre a necessidade de vanádio dos organismos mais evoluídos. Estudos de compostos modelos constituem-se numa importante ferramenta para o entendimento dos diferentes aspectos da bioquímica deste bioelemento. Neste artigo, apresentamos os resultados dos nossos estudos, assim como os de outros grupos de pesquisa, sobre compostos modelos relacionados a vários aspectos do metabolismo e detoxificação do vanádio e a haloperoxidasas dependentes do vanádio. São também apresentadas informações sobre as interações do cátion oxovanádio(IV), relevante do ponto de vista biológico, com bioligantes importantes, tais como nucleotideos, carboidratos, fosfatos e outros sistemas correlatos.
Increasing evidence on the need of the higher forms of life for vanadium has accumulated during recent years. Model studies have become a very important tool to attain a better understanding of different aspects of the biochemistry of this bioelement. In this account we present the results of our own studies, as well as those of other research groups, on models related to different aspects of its metabolism and detoxification and to vanadium dependent haloperoxidases. Additional information about the interaction of the biologically relevant oxovanadium(IV) cation with important bioligands, such as nucleotides, carbohydrates, phosphates and related systems are also presented.
Keywords: vanadium, model studies, metabolism, detoxification, haloperoxidases, biologically relevant ligands
1. Introduction
The biological effects, biodistribution and toxicology of vanadium, such as its requirement and pharmacological activity, are areas of increasing research interest. Although numerous biochemical and physiological functions have been suggested for this element, and despite the amount of the knowledge so far accumulated, vanadium still does not have a clearly defined role in the higher forms of life.1-5
The best evidence for a biological role of vanadium comes from bacteria (the so-called alternative nitrogenases in which vanadium replaces molybdenum in the FeMo-cofactor of some Azotobacter species)4-7 and from plants
(vanadium- dependent haloperoxidases found in some algae, lichens and fungi).4,5,8,9
On the other hand, experiments with laboratory animals have shown that vanadium deprivation enhances abortion rates, reduces milk levels during lactation and produces thyroidal disorders. Other evidence points to the possible role of vanadium in the regulation of ATP-ases, phosphoryl
transfer enzymes, adenylate cyclase, and protein kinases. However, many actions of vanadium can be also explained by its having a role similar to, or enhancing recently described growth factors such as the epidermal growth factor, the fibroblast growth factor and even insuline.10
Like molybdenum, vanadium occupies an exceptional position among the bioelements because both, anionic and cationic forms can participate in biological processes.3,5,11,12
In its anionic forms (vanadates(V)), it resembles phosphates, but in its cationic forms - mainly as VO2+ - it behaves like a
typical transition metal ion, which competes with other metal cations in coordination with biogenic ligands or compounds. This duality, together with the facility with which it changes oxidation states and coordination environments, may be responsible for the very peculiar and somewhat unparalleled behavior of this biometal.5,12
Aspects of the coordination chemistry of vanadium relevant to its presence and activity in biological systems have been recently reviewed.2-4,11-14 In this account we will
2. Model Studies Related to Vanadium
Metabolism
Although information about the metabolism of physiological amounts of vanadium in the higher forms of life remains scarce, an increasing amount of data has accumulated during recent years, mainly from animal studies. Some general aspects related to the absorption, transport, biological transformations, toxicity and
excretion of vanadium become understandable.15,16
In Figure 1 we present a summary of this knowledge, briefly summarized, as follows: Dietary vanadium occurs mainly as H2VO4- and enters cells probably through the
phosphate transport mechanism. Most of the ingested vanadium(V) undergoes a rapid one-electron reduction in the gastrointestinal tract, consistent with the fact that vanadium (V) is a rather strong oxidizing agent, especially at low pH. Most of the ingested and reduced vanadium remains unabsorbed and is excreted. Strong association between VO2+ and dietary fiber is postulated. In vivo, all
the vanadium is converted to a common form. The organ distribution is essentially independent of the oxidation state and chemical nature of the form of the element.
Evidently VO2+ undergoes autooxidation to vanadate in
the presence of oxygen, whereas glutathione, ascorbate, cysteine and similar reducing agents can reduce vanadate. This means that endogenous reducing agents and dissolved oxygen ensure that both vanadium(V) and vanadium(IV) species are present in serum. Experimental evidence points to a relation between vanadium and iron metabolism. It has been suggested that the iron- transport protein transferrin may be also involved in vanadium transport. It remains to be determined if ferritin, the storage protein for
iron, is also a useful storage system for vanadium. Interactions between vanadium species and serum albumin are probable, but very little is known about these processes and the possible nature of the complexes generated. Bone seems to be the major sink for vanadium. Final excretion of the small fraction of ingested and not retained vanadium occurs mainly through urine, as low-molecular-weight VO2+
complexes. Biliar excretion seems to be a secondary route. We have performed a series of model studies related to different steps of the metabolic patterns in order to clarify some essential aspects.
2.1. Complexes of glutathione and related ligands
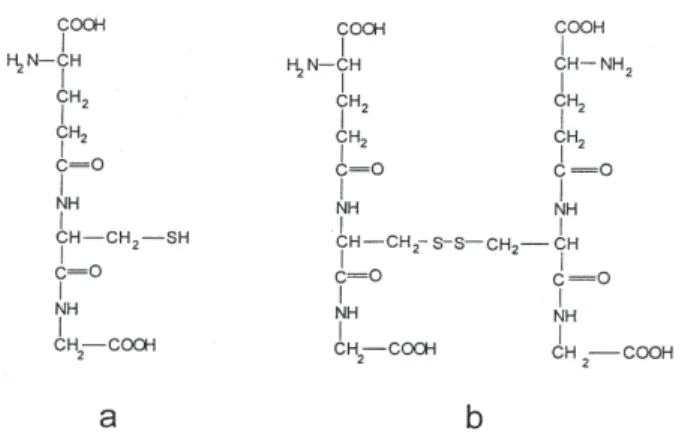
Reduced glutathione (GSH, Figure 2a), the tripeptide
γ-L-glutamyl-L-cysteinylglycine, the major non-protein
thiol present in animal cells, is an extremely important biological reducing agent, involved in detoxification processes of exogenous materials17 and apparently plays a
central role in vanadium metabolism.
Besides its reducing potential GSH can act as a ligand for the stabilization of the VO2+ oxocation.12,18,19
Conse-quently, we have repeatedly investigated the VO2+/GSH
system. Our first studies, using electronic absorption spectroscopy, have shown that the interaction is strongly dependent on the initial metal-to-ligand ratios and the pH of the solution, and at least two different species were identified.20 More detailed speciation studies of this system
have shown an even higher complexity.21,22 In the most
recent and complete study, combining pH-potentiometry with EPR spectroscopy, visible absorption and circular dichroism measurements, it was shown that in the pH range between 5 and 7.5 and at any ligand-to-metal ratio between
10 and 140, the predominant complex, is a 2:1 species in which each GSH molecule coordinates through one O-atom of the deprotonated carboxylate group and the amino N-atom of the glutamyl residue.22 Four other species were
identified in other pH-ranges.22 Previous EPR studies of
this system also support VO2+ interaction through oxygens
of the deprotonated carboxylate groups, and NH2
moieties.23
On the other hand, a series of model compounds with sulfhydryl-containing pseudopeptides, and investigated by a combination of numerous physicochemical techniques, also demonstrates the predominance of the cited 2:1 species in the pH range 5-7.24
The oxidation product of glutathione (GSSG, Figure
2b) can also interact with the VO2+ cation, and model
speciation calculations reveal that in the pH range 6-7, GSSG is a more efficient oxovanadium(IV) binder than GSH.22 Two different 2:1 complexes, easily interconverted
simply by changing the metal-to-ligand concentrations,
are generated in the GSSG/VO2+ system. At low GSSG
concentrations, coordination takes place through carboxylate groups, whereas at higher concentrations, N-donors appear to be principally involved in coordi-nation.25 EPR studies at a 25 : 1 ligand-to-metal ratio
suggest coordination through one or two monodentate
carboxylate groups or through one or two α-amino acid
moieties, i.e., COO– + NH
2 or 2COO – + 2 NH
2. 23
Most recently, Costa Pessoa et al.26 performed a detailed
speciation study of this system. They concluded that in the pH range 6-8 and at a ligand-to-metal ratio = 10, a 1:1 species predominates. Coordination in this complex involves one carboxylate oxygen-atom and the NH2 group of the two terminal glutamyl moieties of GSSG.
These results confirm that both GSH and GSSG may participate in the stabilization and in the transport of VO2+
immediately after the GSH-mediated reduction of vanadate(V) in cellular systems.16,22
The amino acid L-cysteine is another potential reducing agent for vanadate in biological systems. Model studies in the VO3–/cysteine system show that vanadate is
rapidly reduced, irrespective of the pH of the solution. At pH 6.8, reduction is followed by the formation of a purple
complex. In this 2:1 ligand-to-metal species, the VO2+
cation interacts with the amino N-atom and the
deprotonated –SH group of two amino acid molecules.27
This complex seems to be similar to the VO2+ complexes of
cysteine esters of the same stoichiometry, which were isolated in the solid state.28,29 We also demonstrated that
oxovanadium(IV) interacts with cystine, the oxidation product of L-cysteine. In this case, coordination apparently occurs through the carboxylate and amino groups.30 These
results suggest that again both the excess of amino acid or its oxidation product was bind to the VO2+ cation.
2.2. Oxovanadium(IV) complexes of L-ascorbic acid and of its oxidation products
L-ascorbic acid (vitamin C, Figure 3) is another possible natural reducing agent of vanadates(V) to oxovanadium(IV). The reduced species can interact with the acid and with some of its oxidation products.12,31,32
A detailed study of the interaction of L-ascorbic acid with the VO2+ cation showed that different complexes are
generated in solution at different pH-values. Some of them were isolated as powdered solids. As shown by spectroscopic studies, in these complexes the acid acts as a monodentate ligand through its deprotonated 3-hydroxo group, generating species of very low stability, consistent with the absence of chelation, suggesting that these VO2+/
ascorbate complexes are probably not significant in the stabilization of the reduced vanadium.
On the other hand, the VO2+ cation might interact with
some of the species generated after the oxidation of
L-ascorbic acid.32 Dehydroascorbic acid, which is the
primary oxidation product, is very unstable and undergoes a rapid series of transformations, first generating 2,3-diketogulonic acid, which can be further degraded to a
mixture of oxalic and L-threonic acids.12,16,32
Dehydroascorbic acid interacts rapidly with VO2+.
Solutions of these adduct are highly unstable towards the
oxidation of the ligand. The VO2+/dehydroascorbate
complex hydrolyzes irreversibly with opening of the lactone ring, generating a 2:1 ligand-to-metal complex, in which the enolized form of 2,3-diketogulonic acid is bidentate. A sodium salt of this complex anion, of composition Na2[VO(C6H6O7)2].3H2O, has been isolated and characterized.32
Recently, we characterized some other VO2+ -complexes
of this type, obtained as microcrystalline powders, by direct interaction of sodium metavanadate with ascorbic acid.33
2.3. Transferrin and serum albumin complexes
It is well known that in the oxidation states +3, +4 and +5, vanadium binds tightly to transferrin 3,13,34, forming
vanadium-modified transferrins, which are believed to be
involved in vanadium transport in higher organisms.35
Nevertheless, the coordination environment around vanadium in these systems is not yet totally known.
Vanadium(V) interaction with human serum transferrin has been investigated in detail, showing that two equivalents of vanadate are reversibly bound at the two metal-binding sites of the protein.36 The interaction was
also modeled using the hexadentate ligand ethylenebis-(o-hydroxyphenlylglycine) showing that, at pH 9.5, the
vanadium is bonded to phenolic residues as the VO2+
cation.36
In the case of the VO2+-complex of human lactoferrin,
an octahedral structure with O3N equatorial coordination, involving one tyrosinate, one aspartate, one histidine and one monodentate carbonate, with another tyrosinate trans
to the V=O bond has been proposed, on the basis of a computer simulation, using the atomic coordinates of the FeIII and CuII complexes.37 Vanadium(III), as V3+, and
vanadium(V) as VO2+, can also be accommodated in a
similar environment.37
Recently, Neves et al.38,39 have prepared some
interesting model systems for the VO2+-transferrin complex,
using N,O-donor ligands. With some of these models they were able to reproduce UV-Vis and EPR properties of oxovanadium(IV) complexes of human serum transferrin and ovotransferrin and they advanced some new proposals about the coordination sphere of vanadium in these systems. It has also been shown that some of these complexes can be oxidized chemically or electroche-mically to the respective vanadium(V) complexes without changing the coordination sphere of the vanadium.39
Some aspects of the vanadium/albumin interactions are now understood.13,34 In one of the first EPR studies of
the VO2+/bovine serum albumin system, it was found that
the cation binds tightly probably at the specific sites for
CuII, located at the N terminus of the polypeptide chain.
There are also four or five additional weaker binding sites for VO2+ at carboxylate groups of the protein. However, the
nature of the metal coordination sites could not be
unambiguously established.40
The interaction of VOSO4 and NaVO3 with human serum albumin (HSA) was also investigated in aqueous solution at physiological pH, using gel and capillary electrophoresis and IR spectroscopic techniques. Gel electrophoresis results showed that a maximum of twenty VO2+ cations is
bound per HSA molecule, at two sites with different affinities. Capillary electrophoresis confirmed the existence of two major binding sites for the
oxovanadium(IV) cation, whereas VO3- has only a very
weak binding affinity 41, consistent with previous studies.42
IR spectroscopic analysis showed that, as a
conse-quence of the VO2+/HAS interaction, major structural
changes are produced at the protein secondary structure.41
In a recent comparative study of the binding of vanadate to HSA, human fresh frozen plasma and human transferrin, it was demonstrated that the binding capacity of HSA is about one thousandth of those of the other two systems.43
2.4. Accumulation of vanadium in hard tissues and related systems
Bone seems to be the most active vanadium accumulator. The high skeletal retention of vanadate is probably related to its rapid exchange with bone phosphate, which must be favored by the strong similarities of PO43- and VO
4 3-.
In order to investigate this exchange, calcium hydroxylapatite, Ca10(PO4)6(OH)2, was used as a model for
the inorganic phase of bone.44 Under physiological
conditions, the exchange was only observed with amorphous material. These model studies showed that the incorporation of small amount of vanadium into the phosphate sites only produces weak distortions at macroscopic (crystallographic parameters, crystal ordering) and microscopic (local distortions, weakening of chemical bonds) levels in the apatite lattice.44
Possible competition between VO2+ and CaII in the
hydroxylapatite lattice has also been analyzed. Precipitation of Ca10(PO4)6(OH)2 in the presence of
oxovanadium(IV)45 as well as interaction of apatite
suspensions with the oxocation46 demonstrated that VO2+
is not incorporated into the apatite lattice, but that it is strongly adsorbed on the material surface.46,47
In addition, in vivo experiments, analyzing bone
samples of rats treated with an interesting and promising
antidiabetic drug,48 bis(maltolato)oxovanadium (IV), by
means of electron spin-echo envelope modulation (ESEEM) spectroscopy, suggest that phosphate is involved in this surface interaction of VO2+ with bone.49
As it is known34,50 that VO2+ interacts with
tropocollagen, it was useful to investigate whether it interacts with components of the organic matrix of bone. The interaction of VO2+ with chondroitin sulfate A (CSA),
a well-known muchopolysaccharide present in connective tissues and other mineralized systems, was investigated in aqueous solutions by electron absorption spectroscopy and IR techniques. The generation of a complex species of stoichiometry VO(CSA)2, involving metal coordination to the carboxylate group and the glycosidic oxygen of the
D-glucuronate units of CSA was demonstrated.51 It was
also found that the two isolated components of CSA
(D-glucuronic acid and N-acetylgalactosamine) behave
towards VO2+ in a similar way as they do in the
muchopolysaccharide.52
2.5. Vanadium excretion
As mentioned in the introduction, ingested vanadium is excreted most rapidly fecally. The postulated strong association of VO2+ with dietary fiber must be facilitated
by the high affinity of the oxocation for a number of functional residues such as carboxylate, phosphate or hydroxo groups. However, very little information about the characteristics of these interactions in the gastrointestinal tract is available.
Concerning the final urinary excretion of the fraction of vanadium initially retained there exist a number of conflicting reports. Probably this excretion involves
low-molecular-weight VO2+ complexes.15 However, other
evidence also suggest the simultaneous presence of high
molecular-weight complexes.53
In recent studies the low-molecular-weight vanadium species in urine was identified as a vanadium/ascorbate complex.54,55 But on the basis of our studies with this system,
discussed above (section 2.2.), it is most likely that the ligand may be any of the oxidation products of ascorbic acid, perhaps 2,3-diketogulonic acid.
In the most recent study of this system it was shown that, after intraperitoneally 48V injection, vanadium in urine
is found both as high-(protein-bound) and as low-molecular-weight species. The partition of these forms apparently depends on the time elapsed after vanadium administration. Different high- and low-molecular-weight forms were detected by chromatography depending of the elapsed times. But, after 48 h vanadium is largely excreted
as a low-molecular-weight complex.56
3. Other Models of Biochemical Interest
A great number of other vanadium-containing complexes and systems are also of direct biochemical interest. In this section we restrict the information only to some aspects of systems involving nucleotides, phosphates and carbohydrates and some closely related ligands.
3.1. Interaction of VO2+ with nucleotides and related
materials
The coordination behavior of both vanadates(V) and oxovanadium(IV) with nucleotides and their constituents is of great interest in relation to competitive processes in the regulation of the ATP-ases, ribonucleases and similar systems, as well as with regard to the possible cancerostatic action of vanadates.
As the field of VO2+/nucleotide interactions has been
recently reviewed,57 only the most important conclusions
are summarized here: in acid media all types of phosphate nucleosides (mono-, di- and tri-phosphates) interact with the cation, but only through the phosphate groups; at high pH interaction takes place only through the deprotonated OH-groups of D-ribose; at neutral pH di- and tri-phosphate nucleosides generate VO(nucl)2 complexes at high ligand-to-metal ratios; at lower ratios, participation of N atoms of the nucleic acid bases occurs. The behavior of monophosphate nucleosides is more complex. Important ligand rearrangements take place with increasing pH. Phosphate groups, together with OH-groups of D-ribose, participate in coordination.
Interaction of the VO2+ cation with the nucleic bases58
and with nucleosides59 has also been investigated.
Even though different solid VO2+/nucleotide complexes
have been reported,60 but they have not been well
characterized. Our own experience in these systems has shown that the isolation and purification of such complexes is not easy.12
Simple and complex phosphates deserve special attention, not only to the presence of vanadium in nucleotides but also due to its participation in a wide range of biological systems and processes.12
D-ribose-5-phosphate (Rib-5P) shows a similar solution
behavior to the monophosphate nucleosides61 and three
powdered solid oxovanadium(IV) complexes containing
this species were isolated and characterized.62 The
Na6[VO(Rib-5P)2].6H2O complex, coordination takes place through pairs of two adjacent deprotonated OH-groups of the sugar moiety.
Phytic acid and thiamine diphosphates are other biologically interesting phosphates. From the nutritional point of view, phytic acid (mio-inositol hexaphosphate) appears especially interesting because of its important effects on the bioavailability of essential trace metals.63
Depending on the pH, the VO2+ cation interacts with
phytate forming both soluble and insoluble complexes.64
Regarding thiamine diphosphate (cocarboxylase, TDP), a coenzyme that catalyzes the decarboxylation of
α-ketoacids, we have found that in the pH-range 3-4 a 1:1
complex with VO2+ is formed, and this involves only
O-phosphate bonds. The participation of the N(1) atom of the pyrimidine ring in bonding has been suggested at
higher pH.65 A solid complex of composition
[VO(TDP)Cl].7H2O, was precipitated with ethanol from an aqueous solution at pH 3.5. In this complex, the terminal PO3 group of TDP is bidentate.66
Another complex, relevant to a better understanding
of the VO2+/phosphate interactions, is the recently
investigated trimer species Na6[(VO)3(P2O7)3].7H2O.67
3.2. VO2+ complexes of carbohydrates
As carbohydrates are the most abundant class of
compounds in the biosphere,12,68 the study of their
interaction with relevant vanadium species is of great interest. It is well known that sugars interact with metal ions either as reductants and/or chlelators.12,68 Most of them
reduce vanadates(V) to oxovanadium(IV) and complex this cation. This field of vanadium biochemistry has also been
recently reviewed69 and therefore we shall reduce the
discussion only to its most relevant aspects.
Due to its strong hydrolytic tendency, the VO2+ cation
usually needs the presence of additional donor groups (e.g. carboxylates) in the sugar molecule. Once bonded, it can easily deprotonate the OH-groups and strongly coordinate up to four of them. Oxovanadium(IV) complexes coordinated by pairs of doubly deprotonated sugar moieties usually display a very characteristic, three band, electronic absorption spectrum.69,70
Oxovanadium(IV) coordination is favored in basic media and only occurs with sugar molecules provided with pairs of adjacent OH-groups.69,71
A great number of VO2+/monosaccharide complexes
have been reported in recent years. Their stoichiometries are summarized in Table 1. All these complexes are green-colored powders and are usually hygroscopic and very soluble in water.
Only five oxovanadium complexes with disaccharides as ligands have been so far reported. These are sucrose,74
turanose,74 maltose73,77 and lactose,78 with the following
stoichiometries:
Na3[VO(D-Suc)2OH].H2O Na3[VO(D-Tur)2OH].3H2O Na5[VO(D-Mal)2OH].10H2O Na5[VO(D-Mal)2].CH3OH Na4[VO(Lact)2].3H2O
Even though a number of oxovanadium(IV) complexes with some carboxylate derivatives of carbohydrates and sugar phosphates, and with polysaccharides, has been also investigated, most of these compounds remains poorly characterized.69
4. Model Studies Related to Vanadium
Detoxification
Contamination of the environment by vanadium has
Table 1. Composition of the known oxovanadium (IV) monosaccharide complexes
Complex Ref. Complex Ref.
[VO(D-Glc)2] 72 Na4[VO(L-Sor)2].3H2O 76
Na2[VO(D-Glc)2].CH3OH 73 Na4[VO(D-Gal)2 ].5H2O 77
Na3[VO(D-Glc)2(OH)].5H2O 74 Na2[VO(D-Gal)2(OH)Cl].0.5H2O 75
Na2[VO(D-Glc)2(OH)Cl].H2O 75 Na2[VO(D-Gal)2].CH3OH 73
Na2[VO(D-Fru)2] 73 Na2[VO(D-Gal)2(H2O)] 76
Na6[(VO)2(D-Fru)5].4H2O 74 Na4[VO(D-Man)2 ].8H2O 77
Na2[VO(D-Fru)2(OH)Cl].5H2O 75 Na3[VO(D-Man)2(OCH3)].4CH3OH 73
Na2[VO(D-Xyl)2].CH3OH 76 Na5[VO(D-Lyx)2(OH)].6H2O 77
Na4[VO(D-Xyl)2].5H2O 77 Na4[VO(D-Lyx)2].2H2O 76
Na2[VO(D-Xyl)2(HO)Cl].CH3OH.1.5H2O 75 Na2[VO(Ino)2] 76
Na4[VO(D-Ara)2].CH3OH.3H2O 76 Na4[VO(D-Rib)2].H2O 76
Na4[VO(D-Ara)2].5H2O 77 Na3[VO(D-Rib)2(OH)].4H2O 62
dramatically increased in recent years, especially in the most developed countries, due to the increasing use of fossil fuels, which liberate V2O5 during combustion.79
Therefore, the toxicology and detoxification of vanadium constitute an area of increasing development.
The degree of toxicity depends on the route of administration, oxidation and chemical form and it is also to some extent species-dependent. Vanadium compounds, especially V2O5, are strong irritants of the airways and the eyes. Acute and chronic exposure gives rise to conjunc-tivitis, rhinitis, reversible irritation of the respiratory tract, and to bronchitis, bronchospasms, and asthma-like diseases in more severe cases. It has shown that it can produce gastrointestinal distress, fatigue, cardiac palpitation, and kidney damage. In humans, acute vanadium toxicity has been observed in vanadium miners, as well as other industrial workers exposed to high concentrations of the element. The classic symptoms of this malady, referred to as “green tongue” syndrome, are a green discoloration of the tongue, accompanied by some of the cited disorders.80,81
It has been usually accepted that vanadium toxicity increases with increasing oxidation state, vanadium(V) being the most toxic.80,81
Living systems have developed defense mechanisms to deal with the reactive and potentially harmful by-products that arise from cellular metabolism and to control the effects of exogenous substances that eventually invade the organism (biological detoxification). Drugs have been
developed to chelate metal ions in vivo, not only to
eliminate excesses of essential metals but also to prevent possible damage caused by nonessential, toxic elements (chemical detoxification).
Some of the systems mentioned in this account are evidently relevant to the toxicity and detoxification of vanadium. Some of the metabolic processes (glutathione-, ascorbate- or cysteine- mediated reduction of vanadates(V);
complexation of VO2+ by different biomolecules;
accumulation of vanadium in hard tissues) must play an important role in biological vanadium detoxification.81
Most of the systems assayed for chemical detoxification contain chelating or reducing/chelating agents.81 Animal
studies have demonstrated that the best detoxification agent may be L-ascorbic acid.81-83 Its action is related to
the facility with which it reduces vanadium(V) to VO2+
and to the possibility that the oxocation generated may be complexed by its oxidation products, as discussed above. Better knowledge of the fundamental metabolic steps and a thorough characterization of new vanadium species with chelating or reducing agents may be useful for the development of more potent and specific detoxification agents for vanadium.
5. Model Studies Related to Haloperoxidases
Haloperoxidases are enzymes which catalyze the oxidation of halides (Cl-, Br-, I-) by hydrogen peroxide,
resulting in the halogenation of appropriate organic substrates.84 The presence of vanadium as an essential
component for a haloperoxidase was discovered in the red
algae Ascophyllum nodosum in 1984.85 Within these
vanadium-dependent haloperoxidases, both vanadium bromoperoxidases, isolated mainly from algae, and vanadium chloroperoxidases, found essentially in fungi, have been subsequently identified.9
Recently, the crystal structure of a vanadium chloro-peroxidase isolated from the fungus Curvularia inaequalis, was reported by Messerschmidt and Wever.86 The structural
features of the active vanadium center seems to be characteristic of all such systems.9 The metal, in the V
oxidation state, has a trigonal bipyramidal geometry, ligated by azide (a result of the azide-containing crystallization buffer), three non-protein oxygen atoms, and a histidine N atom. In the native structure the azide ligand, located in trans position to the histidine N atom, is apparently replaced by an OH group.9, 86
The finding of a specific function of vanadium in haloperoxidases allows new speculations, on its possible functions in higher organisms. Thyroid peroxidase is one of the best known animal haloperoxidases.84 As mentioned
previously, vanadium deprivation increases thyroid weight and also affects the response of thyroid peroxidase activity.5,10 Perhaps, vanadium plays some role in the
halogenating activity of this enzyme.
A great number of model studies have been performed recently, in an attempt to understand better the structural characteristics of the metal site as well as to elucidate the reaction mechanisms the vanadium-dependent halo-peroxidases.
All the information so far accumulated shows that vanadium remains in the V oxidation state during the entire catalytic cycle and it has also been demonstrated that during the process one peroxide group is bonded to the
metal.4,87 A variety of mechanistic studies has been
performed using different vanadium/peroxide complexes
as model systems.4,88-91 Some vanadium-based
semi-synthetic and biomimetic models have also been assayed as catalysts for enantioselective oxidations.92
Recently, we have also initiated some model studies related to these natural systems, investigating the kinetics of the bromination of phenol red by the peroxo-vanadium(V) species generated by acid decomposition of [VO(O2)2(NH3)]- and [O{VO(O
2)2}2] 4-.93
of both the active vanadium (V) site and the catalytically inactive reduced oxovanadium(IV) site, have been also performed.4,14,94-97
One interesting aspect of the structure of the active site is the simultaneous presence of N- and O-donors and of V=O and V-OH groups. The ligand 8-hydroxyquinoline,
the well-known analytical reagent oxine (HQ), is
particularly interesting for model studies related to these systems and to other biologically relevant vanadium centers. It normally stabilizes chelate complexes of the types MQ2 and MQ3, generating MN2O2 or MN3O3 and, in certain cases, also MN2O3(OH) environments.
In spite of the fact that the simplest oxovanadium(IV) complex of oxine, VOQ2, has been widely investigated, several contradictory reports, mainly derived from its easily oxidability, are found in the literature. 12 In order to extend
these studies, we have synthesized and characterized a series of VO2+ complexes with different derivatives of oxine.
Those derived from 5,7-dihalogenated oxine are stable in air but show a very complex solution behavior that includes oxidation phenomena, ligand loss and
interactions with solvents. 98 On the other hand, the
presence of halogen atoms on the oxine ring has a
negligible effect on the thermal stability of the complexes.99
A detailed study of the magnetic behavior of these complexes shows that a ferromagnetic interaction between the VO2+ groups becomes operative at temperatures below
40 K with an exchange integral J 2.73 cm-1. 100 These results
constitute the first direct evidence of the formation of
...V=O...V=O... ferromagnetic chains in these and in similar
oxovanadium(IV) complexes.12,100
Other related complexes that were also investigated in detail are the bis-chelated VO2+ species derived from
8-hydroxyquinoline-N-oxide.101 and from
7-iodo-8-hydroxyquinoline-5-sulfonic acid (the analytical reagent
ferron).102
Some vanadium(V) species containing oxine or its
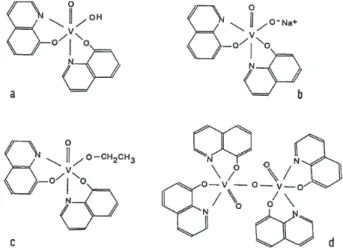
derivatives as ligands are much more interesting in relation to the active site of haloperoxidases. The complex hydroxobis(8-hydroxyquinolinato)oxovanadium(V) (Figure 4a) can be considered as an “inorganic analog” of a carboxylic acid.12,103 Accordingly, it is possible to prepare
salts (Figure 4b), esters (Figure 4c) and dimer anhydrides (Figure 4d). We obtained a great number of complexes of these types and characterized them thoroughly by different physicochemical methods,98,101,102,104,105 including detailed
electrochemical studies.106,107
Another class of model system related to this biological site, in its reduced (inactive) form are the recently reported species cis-[VO(OH)(bipy)2]+ and cis-[VO(OH)(o-phen)
2] +
which structural analysis, as the BF4- salts, showed the
presence of a severely distorted octahedral coordination with the vanadium(IV) above the mean equatorial plane defined by three bipy (o-phen) N-atoms and the OH-group. The oxo group and the remaining N-atom of one of the organic ligands occupy the apical positions.108
Also, Schiff-base complexes of vanadium(V) and of oxovanadium(IV) have often been investigated as useful models for this and other biological systems containing the metal.109-115
Finally, it is worthy commenting that numerous model compounds developed for a better understanding of the activity and action of vanadium-dependent halopero-xidases and other biological systems containing this element are also reagents in modern organic synthesis.116
It is pertinent to emphasize that a number of model
compounds investigated in relation to Amavadine, the
natural vanadium complex present in the fungus Amanita
muscaria (cf.117,118 and references therein), also have very
interesting catalytic capabilities.119
6. Conclusions and Perspectives
We hope the above discussion has clearly shown the importance of model studies for a deeper understanding of some fundamental aspects related to the relatively complex biochemistry of vanadium. These studies have revealed valuable information concerning vanadium metabolism, toxicity, detoxification and catalytic activity. They have also extended the coordination chemistry of vanadium, especially that of the vanadium(IV) and vanadium(V) oxocations, as a direct consequence of the use of a variety of new ligand types. Nevertheless, there are a number of very important and interesting problems that remain open. A better understanding of vanadium metabolism requires new efforts to comprehend its transport in both
anionic and cationic forms, and for the initial reduction processes of vanadium(V) to VO2+ in the gastrointestinal
tract. It would also be interesting to obtain crystalline VO2+
complexes with glutathione and related ligands and to explore further the interaction of this oxocation with serum albumin, transferrin and ferritin. A better chemical characterization of excreted vanadium species also seems very important.
More detailed speciation studies, including the determination of stability constants, are necessary for a wider characterization of the VO2+/nucleotide systems.
Other efforts should be directed to structural information on oxovanadium(IV) saccharide complexes. It has not yet been possible to obtain single crystals adequate for crystallographic studies,69 therefore EXAFS
studies, like those recently performed on iron-saccharide
complexes,120 should be attempted. Studies on the
interaction of the VO2+ cation with polysaccharides should
also be expanded, as these systems are of particular biological importance.
The possible use of phosphonates and related ligands, as well as the combination of appropriate reducing/ chelating systems for use in vanadium detoxification merits further exploration.
Most of the model systems so far investigated, are also good candidates for theoretical studies, which may be very useful for a better understanding of stability, electronic structures, and reactivity. Some recent examples of this type of work are the molecular modeling of vanadium peroxides121 and studies of the electronic structures of Amavadine models.122
Acknowledgements
It is a great pleasure to acknowledge the contributions of the colleagues and collaborators whose names appear in the references. Work from our laboratory reported here was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas de la República Argentina (CONICET), the Comisión de Investigaciones Científicas de la Provincia de Buenos Aires and the Agencia Nacional de Promoción Científica y Tecnológica. The author is a member of the Research Career of CONICET.
References
1. Chasteen, N.D., ed.; Vanadium in Biological Systems, Kluwer: Dordrecht, 1995.
2. Sigel, H.; Sigel, A., eds.; Metal Ions in Biological Systems,
vol.31:Vanadium andits Role in Life, Marcel Dekker: New
York, 1995.
3. Rehder, D.; Angew. Chem. Int. Ed. Engl. 1991, 30, 148. 4. Slebodnick, C.; Hamstra, B. J.; Pecoraro, V. L.; Struct.
Bond-ing1997, 89, 51.
5. Baran, E. J.; An. Soc. Científ. Argent.1998, 228, 61. 6. Eady, R. R.; Leight, G. F.; J. Chem. Soc., Dalton Trans. 1994,
2739.
7. Eady, R. R.; Chem. Rev. 1996, 96, 3013.
8. Wever, R.; Kustin, K.; Adv. Inorg. Chem. 1990, 35, 81. 9. Butler, A.; Baldwin, A. H.; Struct. Bonding 1997, 89, 109. 10. Nielsen, F. H.; FASEB J. 1991, 5, 2661.
11. Rehder, D.; Biometals 1992, 5, 3.
12. Baran, E. J.; J. Inorg. Biochem. 2000, 80, 1.
13. Butler, A.; Carrano, C.J.; Coord. Chem. Rev. 1991, 109, 61. 14. Rehder, D.; Coord. Chem. Rev.1999, 182, 297.
15. Chasteen, N. D.; Lord, M. E.; Thompson H. J. In Frontiers in
Bioinorganic Chemistry;Xavier, A. V., ed., Verlag Chemie:
Weinheim, 1986, pp.133-141.
16. Baran, E. J.; Bol. Soc. Chil. Quim. 1997, 42, 247.
17. Rabenstein, D.L. In Glutathione; Dolphin, D.; Avramovic, O.; Poulson, R., eds.; J. Wiley: New York, 1989, part A, pp.147-186.
18. Macara, I. G.; Kustin, K.; Cantley,jr., L. C.; Biochim. Biophys. Acta1980, 629, 95.
19. Delfini, M.; Gaggeli, E.; Lepri, A.; Valensin, G.; Inorg. Chim. Acta1985, 107, 87.
20. Ferrer, E.G.; Williams, P. A. M.; Baran, E.J.; Biol. Trace Elem. Res.1991, 30, 175.
21. Armas, M. T.; Mederos, A.; Gili, P.; Domínguez, S.; Hernández-Molina, R.; Lorenzo, P.; Baran, E. J.; Araujo, M. L.; Brito, F.;
Polyhedron2001, 20, 799.
22. Costa Pessoa, J.; Tomaz, I.; Kiss, T; Kiss, E.; Buglyó, P.; J.
Biol. Inorg. Chem.2002, 7, 225.
23. Dessi, A.; Micera, G.; Sanna, D.; J. Inorg. Biochem. 1993, 52, 275.
24. Tasiopoulos, A. J.; Troganis, A. N.; Evangelou, A.; Raptopoulou, C. P.; Terzis, A.; Deligiannakis, Y.; Kabanos, T.
A.; Chem. Eur. J. 1999, 5, 910.
25. Ferrer, E. G.; Williams, P. A. M.; Baran, E. J.; J. Inorg. Biochem.
1993, 50, 253.
26. Costa Pessoa, J.; Tomaz, I.; Kiss, T.; Buglyó, P.; J. Inorg.
Biochem.2001, 84, 259.
27. Sakurai, H.; Shimomura, K.; Ishizu, K.; Inorg. Chim. Acta
1981, 55, L67.
28. Sakurai, H.; Hamada, Y.; Shimomura, S.; Yamashita, S.; Ishizu,
K.; Inorg.Chim. Acta 1980, 46, L119.
29. Ferrer, E. G.; Baran, E. J.; An. Asoc. Quim. Argent. 1992, 80, 429.
30. Ferrer, E. G.; Williams, P. A. M.; Baran, E. J.; J. Trace Elem.
Med. Biol.1998, 12, 56.
31. Baran, E.J.; Ferrer, E.G.; Williams, P. A. M.; J. Inorg. Biochem.
32. Ferrer, E. G.; Williams, P. A. M.; Baran, E. J.; Z. Naturforsch.
1998, 53b, 256.
33. Ferrer, E. G.; Baran, E. J.; Biol. Trace Elem. Res.2001, 83, 111.
34. Chasteen, N. D. In Metal Ions in Biological Systems; Sigel, H.; Sigel, A., eds.; Marcel Dekker: New York, 1995, vol.31, pp.231-247.
35. Cantley, L. C.; Resh, M.; Guigotti, G.; Nature 1978, 272, 552. 36. Harris, W. R.; Carrano, C. J.; J. Inorg. Biochem. 1984, 22, 201. 37. Smith, C. A.; Ainscough, E. W.; Brodie, A. M.; J. Chem. Soc.,
Dalton Trans.1995, 1121.
38. Neves, A.; Ceccato, A. S.; Erasmus-Buhr, C.; Gehring, S.; Haase, W.; Paulus, H.; Nascimento, O. R.; Batista, A. A.; J.
Chem. Soc. Chem.Comm. 1993, 23, 1782.
39. Neves, A.; de Moraes Romanowski, S. M.; Bortoluzzi, A. J.; Mangrich, A. S.; Inorg. Chim. Acta2001, 313, 137. 40. Chasteen, D. N.; Francavilla, J.; J. Phys. Chem. 1976, 80, 867. 41. Purcell, M.; Neault, J. F.; Malonga, H.; Arakawa, H.;
Tajmir-Riahi, H. A.; Can. J. Chem. 2001, 79, 1415.
42. Chasteen, N. D.; Grady, J. K.; Holloway, C. E.; Inorg. Chem.
1986, 25, 2754.
43. Heinemann, G.; Fichtl, B.; Mentler, M.; Vogt, W.; J. Inorg.
Biochem.2002, 90, 38.
44. Etcheverry, S. B.; Apella, M. C.; Baran, E. J.; J. Inorg. Biochem.
1984, 20, 269.
45. Oniki, T.; Doi, Y.; Calc. Tiss. Internat. 1983, 35, 538. 46. Narda, G. E.; Vega, E. D.; Pedregosa, J. C.; Etcheverry, S. B.;
Baran, E. J.; Z. Naturforsch. 1992, 47b, 395.
47. Vega, E. D.; Pedregosa, J. C.; Narda, G. E.; J. Phys. Chem.
Solids 1999, 60, 759.
48. Baran, E.J.; Acta Farm. Bonaerense 1997, 16, 43.
49. Dikanov, S. A.; Liboirn, B. D.; Thompson, K. H.; Vera, E.; Yuen, V. G.; McNeill, J. H.; Orvig, C.; J. Am. Chem. Soc.
1999, 121, 11004.
50. Ferrari, R. P. Inorg. Chim. Acta 1990, 176, 83.
51. Etcheverry, S. B.; Williams, P. A. M.; Baran, E. J.; Biol. Trace
Elem. Res. 1994, 42, 43.
52. Etcheverry, S. B.; Williams, P. A. M.; Baran, E. J.; Biol. Trace
Elem. Res. 1996, 51, 169.
53. Sabbioni, E.; Marafante, E.; Bioinorg. Chem.1978, 9, 389. 54. Kramer, H. J.; Backer, A.; Meyer-Lehnert, H.; Am. J.
Hypertens.1998, 11, 1208.
55. Kramer, H. J.; Krampitz, G.; Backer, A.; Meyer-Lehnert, H.;
Clin. Exper. Hypertens. 1998, 20, 557.
56. De Cremer, K.; Cornelis, R.; Strickmans, K.; Dams, R.; Lameire, N.; Vanholder, R.; J. Inorg. Biochem. 2002, 90, 71. 57. Baran, E.J. In Metal Ions in Biological Systems; Sigel, H.; Sigel, A., eds.; Marcel Dekker: New York, 1995, vol.31, pp.129-146.
58. Williams, P. A. M.; Etcheverry, S. B.; Baran, E. J.; Z.Naturforsch.
1993, 48b, 1845.
59. Williams, P. A. M.; Etcheverry, S. B.; Baran, E. J.; An. Asoc.
Quim. Argent. 1994, 82, 13.
60. Katsaros, N.; Transit. Met. Chem. 1982, 7, 62.
61. Williams, P. A. M.; Baran, E. J. ;J. Inorg. Biochem. 1993, 50, 101.
62. Williams, P. A. M.; Etcheverry, S. B.; Baran, E. J.; J. Inorg.
Biochem.1997, 65, 133.
63. Cosgrove, D. J.; Inositol Phosphates: Their Chemistry,
Bio-chemistry and Physiology; Elsevier: New York, 1980.
64. Williams, P. A. M.; Baran, E. J.; Biol. Trace Elem. Res. 1993,
36, 43.
65. Williams, P. A. M.; Baran, E. J.; J. Inorg. Biochem. 1990, 38, 101.
66. Ferrer, E. G.; Etcheverry, S. B.; Baran, E. J.; An. Asoc. Quim.
Argent.1998, 86, 146.
67. Muglia, C. I.; Ferrer, E. G.; Baran, E. J.; J. Thermal Anal.
Calorim. 2001, 65, 177.
68. Whitfield, D. M.; Stojkovski, S,; Sarkar, B.; Coord. Chem. Rev. 1993, 122, 171.
69. Baran, E. J.; J. Carbohydr. Chem. 2001, 20, 769. 70. Baran, E. J.; J. Coord. Chem.2001, 54, 215.
71. Branca, M.; Micera, A.; Dessi, A.; Sanna, D.; J. Inorg. Biochem.
1992, 45, 169.
72. Rao, C. P.; Kaiwar, S. P.; Inorg. Chim. Acta 1991, 186, L11. 73. Sreedhara, A.; Raghavan, M. S. S.; Rao, C. P.; Carbohyd. Res.
1994, 264, 227.
74. Etcheverry, S. B.; Williams, P. A. M.; Baran, E. J.; Carbohyd. Res.1997, 302, 131.
75. Bandwar, R. P,.; Rao, C. P.; J. Inorg. Biochem.1997, 68, 1. 76. Sreedhara, A.; Rao, C. P.; Rao, B. J.; Carbohyd. Res. 1996,
289, 39.
77. Williams, P. A. M.; Etcheverry, S. B.; Baran, E. J.; Carbohyd. Res. 2000, 329, 41.
78. Etcheverry, S. B.; Barrio, D. A.; Williams, P. A. M.; Baran, E.
J.; Biol. Trace Elem. Res. 2001, 84, 227.
79. Mamane, Y.; Pirrone, N. In Vanadium in the Environment; Nriagu, J. O., ed.; J. Wiley: New York, 1998, vol.1, pp. 37-71. 80. Faulkner-Hudson, T.G.; Vanadium: Toxicology and
Biologi-cal Significance ; Elsevier: Amsterdam, 1964.
81. Baran, E. J. In Vanadium in the Environment; Nriagu, J. O., ed.; J. Wiley: New York, 1998, vol.2, pp. 317-345. 82. Jones, M. M.; Basinger, M. A.; J. Toxicol. Enviorn. Health
1983, 12, 749.
83. Domingo, J. L.; Llobet, J. M.; Corbella, J.; Toxicol. Lett. 1985,
26, 95.
84. Baran, E. J.; Química Bioinorgánica; McGraw-Hill Interamericana de España: Madrid, 1995.
85. Vilter, H.; Phytochemistry 1984, 23, 1387.
86. Messerschmidt, A.; Wever, R.; Proc. Natl. Acad. Sci. USA
1996, 93, 392.
88. Butler, A.; Clague, M. J.; Meister, G. E.; Chem. Rev. 1994, 94, 625.
89. Butler, A.; Clague, M. J. In Mechanistic Bioinorganic Chemis-try; Thorp, H. H.;Pecoraro, V. L., eds.; Am. Chem. Soc.: Wash-ington, 1995, pp. 329-349.
90. Guevara-García, J. A.; Barba-Behrens, N.; Contreras, R.; Mendoza-Díaz, G. In Vanadium Compounds: Chemistry,
Bio-chemistry, and Therapeutic Applications; Tracey, A. S.; Crans,
D. C., eds., Am. Chem. Soc.: Washington, 1998, pp. 126-135. 91. Pecoraro, V. L.; Slebodnick, Hamstra, B. In Vanadium Com-pounds: Chemistry, Biochemistry, and Therapeutic
Applica-tions; Tracey, A. S.; Crans, D. C., eds., Am. Chem. Soc.:
Wash-ington, 1998, pp.157-167.
92. van de Velde, F.; Arends, I. W. C. E.; Sheldon, R. A.; J. Inorg.
Biochem. 2000, 80, 81.
93. Tótaro, R. M.; Williams, P. A. M.; Apella, M. C.; Blesa, M. A.; Baran, E. J.; J. Chem. Soc. Dalton Trans. 2000, 4403. 94. Plass, W.; Z. Anorg. Allg. Chem. 1994, 620, 1635. 95. Plass, W.: Inorg. Chim. Acta1996, 244, 221.
96. Hamstra, B. J.; Colpas, G. J.; Pecoraro, V. L.; Inorg. Chem.
1998, 37, 949.
97. Kimblin, C.; Bu, X.; Butler, A.: Inorg. Chem. 2002, 41, 161. 98. González-Baró, A. C.; Baran, E. J.; Monatsh. Chem. 1997,
128, 323.
99. Ferrer, E. G.; González-Baró, A. C.; Baran, E. J.; J. Thermal
Anal. Calorim. 1999, 57, 595.
100. Sáez-Puche, R.; Romero, J.; González-Baró, A. C.; Baran, E.
J.; Chem. Phys. Lett. 1998, 282, 273.
101. González-Baró, A. C.; Baran, E. J.; J. Coord. Chem. 1998, 43, 335.
102. González-Baró, A. C.; Baran, E. J.; J. Braz. Chem. Soc. 2001,
12, 208.
103. Giacomelli, A.; Floriani, C.; de Souza Duarte, A. O.; Chiesi-Villa, A.; Guastini, C.; Inorg. Chem. 1982, 21, 3310. 104. Jubert, A. H.; González-Baró, A. C.; Pis Diez, R.; Baran, E. J.;
J. Raman Spectr.1992, 23, 273.
105. González-Baró, A. C.; Piro, O. E.; Parajón-Costa, B. S.; Baran, E. J.; Castellano, E. E.; Monatsh. Chem. 1998, 129, 31.
106. Parajón-Costa, B. S.; González-Baró, A. C.; Baran, E. J.; J.
Coord Chem.1999, 47, 417.
107. Parajón-Costa, B. S.; González-Baró, A. C.; Baran, E. J.; J.
Coord.Chem. 1999, 49, 17.
108. Tolis, E. J.; Manos, M. J.; Tasiopoulos, A. J.; Raptopoulou, C. P.; Terzis, A.; Sigalas, M. P.; Deligiannakis, Y.; Kabanos, T. A.;
Angew. Chem. Int. Ed. Engl. 2002, 41, 2797.
109. Cornman, C. R.; Kampf, J.; Pecoraro, V. L.; Inorg. Chem.
1992, 31, 1981.
110. Cornman, C. R.; Kampf, J.; Lah, H. S.; Pecoraro, V. L.; Inorg.
Chem. 1992, 31, 2035.
111. Costa Pessoa, J.; Silva, A. L.; Viera, A. L. Vilas-Boas, L. F.; O’Brien, P.; Thornton, P.; J. Chem. Soc. Dalton Trans. 1992, 1745.
112. Vergopoulos, V.; Priebsch, W.; Fritsche, M.; Rehder, D.; Inorg.
Chem. 1993, 32, 1844.
113. Cavaco, J.; Costa Pessoa, J.; Costa, D.; Duarte, M. T.; Gillard, R.D.; Matias, P.; J. Chem. Soc. Dalton Trans. 1994, 149. 114. Plass, W.; Coord. Chem. Rev. 2003, 237, 205.
115. Tsuchida, E.; Oyaizu, K.; Coord. Chem. Rev. 2003, 237, 213. 116. Hirao, T.; Chem. Rev. 1997, 97, 2707.
117. Bayer, E. In Metal Ions in Biological Systems; Sigel, H.; Sigel, A., eds.; M. Dekker: New York, 1995, Vol.31, pp.407-421. 118. Garner, C. D.; Armstrong, E. M.; Berry, R. E.; Beddoes, R. L.;
Collison, D.; Cooney, J. J. A.; Ertok, S. N.; Helliwell, M.; J.
Inorg. Biochem. 2000, 80, 17.
119. Reis, P. M.; Silva, J. A. L.; Fraústo da Silva, J. R.; Pombeiro, A. J. L.; J.Chem. Soc. Chem. Comm. 2000, 1845.
120. Rao, C. P.; Geetha, K.; Raghavan, M. S. S.; Sreedhara, A.; Tokunaga, K.; Yamaguchi, T.; Jadhav, V.; Ganesh, K. N.; Krishnamoorthy, T.; Ramaiah, K. V. A.; Bhattacharyya, R. K.;
Inorg. Chim. Acta2000, 297, 373.
121. Cundari, T. R.; Sisterhen, L. L.; Stylianopoulos, C.; Inorg.
Chem. 1997, 36, 4029.
122. Armstrong, E. M.; Collison, D.; Deeth, R. J.; Garner, C. D.; J.
Chem. Soc.Dalton Trans. 1995, 191.