Faculdade de Ciências
Departamento de Biologia Animal
Pesquisa e caracterização de mutações em genes
relacionados com o metabolismo do ferro em
indivíduos com hiperferritinemia grave
Vera Lúcia Viana Santos
Dissertação
Mestrado em Biologia Humana e Ambiente
2012
Faculdade de Ciências
Departamento de Biologia Animal
Pesquisa e caracterização de mutações em genes
relacionados com o metabolismo do ferro em
indivíduos com hiperferritinemia grave
Dissertação orientada por:
Doutora Maria Paula Duarte Faustino Gonçalves (Departamento de Genética Humana, Instituto Nacional de Saúde Dr. Ricardo Jorge)
Professora Doutora Ana Maria Viegas-Crespo (Departamento de Biologia Animal, Faculdade de Ciências da Universidade de Lisboa)
Vera Lúcia Viana Santos
Dissertação
Mestrado em Biologia Humana e Ambiente
2012
Aos meus avós Florêncio Viana, Alzira Viana e Emília Rosa na esperança que se orgulhem de mim lá onde estão.
i
Gostaria de dirigir as primeiras palavras deste trabalho a todos os que, durante a sua realização, me colocaram desafios e me disponibilizaram diversas formas de apoio, pelo que a todos expresso o meu reconhecido agradecimento:
Ao Professor Doutor José Pereira Miguel, Presidente do Conselho Directivo do INSA, e ao Doutor João Lavinha, coordenador da Unidade de Investigação e Desenvolvimento do Departamento de Genética Humana do INSA, que autorizaram e permitiram a realização deste trabalho.
À Doutora Paula Faustino, pela oportunidade de realizar este trabalho, por toda a sua amabilidade, sabedoria, orientação e por me ter acolhido tão bem no seu grupo.
À Professora Doutora Ana Crespo pela disponibilidade demonstrada na qualidade de orientadora interna.
Ao Bruno, pelas preciosas indicações e ensinamentos, por se ter mostrado sempre disponível e por ter acompanhado o meu trabalho. O maior agradecimento é para ti.
Às minhas colegas de laboratório, à Andreia pela confiança que sempre depositou em mim e à Rute pela disponibilidade para me ajudar.
Aos colegas do grupo de investigação em metabolismo do RNA pelos óptimos momentos que passamos juntos.
Às minhas amigas, à Cláudia pela entreajuda e por estar sempre presente ao longo de todo o trabalho. À Dany por ter estado sempre ao meu lado, nos bons e nos maus momentos, apesar de haver um oceano a separar-nos. À Vanessa pela dedicação sem fim, ajuda e infinita paciência para me escutar. Às três agradeço a amizade e imprescindível apoio. Sem vocês tinha sido mais difícil.
Aos meus amigos Agri, André, Hugo, Mariazinha e Tixa por terem sido tolerantes em relação a todas as minhas ausências.
À minha família em especial ao meu pai pelo incentivo e confiança que sempre depositou em mim, e à minha mãe pelos valores que me incutiu ao longo da vida. A ambos agradeço o esforço e os sonhos de que abdicaram em prol dos meus. Ao Miguel pela disponibilidade, auxílio e amizade.
À minha irmã, por todo o amor, compreensão, amizade, preocupação e infindável apoio ao longo da minha vida e deste trabalho. Obrigada por me fazeres acreditar que eu posso alcançar o universo.
ii
O Ferro desempenha um papel biológico muito importante nos organismos vivos. No entanto, tanto o seu excesso como a sua deficiência no organismo humano estão associados a consequências negativas para a saúde. A Hemocromatose Hereditária (HH) é dos distúrbios genéticos mais comuns em indivíduos de ascendência Norte-Europeia. Caracteriza-se por uma absorção excessiva de ferro a nível intestinal e sua consequente acumulação em vários órgãos. As complicações mais frequentes da doença são cirrose hepática, carcinoma hepatocelular, cardiomiopatias, diabetes, problemas endócrinos, artrite e hiperpigmentação da pele. O gene HFE é o principal associado à patologia (HH clássica, tipo I). Os genótipos mais comuns são a mutação C282Y em homozigotia e a heterozigotia composta desta com a mutação H63D. Têm surgido evidências de que mutações noutros genes possam estar envolvidas nesta patologia. Entre estes encontra-se o gene do Receptor 2 da Transferrina (TfR2) sendo a HH resultante (Hemocromatose do tipo III) caracterizada por sintomas clínicos idênticos aos da forma clássica. A Hemocromatose do tipo II, ou Hemocromatose Juvenil, é a forma mais grave da doença devido ao facto da sobrecarga de ferro ocorrer a um ritmo mais acelerado levando a problemas em idades jovens. Este tipo de HH ocorre devido a mutações no gene da Hemojuvelina, HJV (HH do tipo 2A) ou no da Hepcidina (HAMP) (HH do tipo 2B).
O objectivo principal deste estudo consistiu na pesquisa e caracterização de mutações em genes relacionados com o metabolismo do ferro (HFE, TfR2, HJV, HAMP, BMP-6), em 11 indivíduos com hiperferritinemia muito grave (ferritina sérica > 2500 μg/L), cujos fenótipos não eram justificáveis pelos genótipos HFE apresentados. As sequências génicas foram amplificadas por metodologias de PCR e a análise mutacional dos fragmentos amplificados foi efectuada recorrendo à sequenciação automática. Nesta análise foram detectadas 9 alterações, sendo que 3 delas são identificadas pela primeira vez neste estudo (a P124L no gene HJV, a c.1281-197 T→C e a c.1542+4 G→A no gene BMP-6). Posteriormente foram realizados estudos in silico às sequências proteicas de modo a identificar possíveis alterações a nível estrutural recorrendo ao programa bioinformática PolyPhen-2 e aos modelos HumDiv e HumVar. Para além disso, também se procedeu à análise do impacto das alterações nucleotídicas a nível do splicing utilizando para esse fim o software Human Splicing Finder. Foi possível através desta pesquisa justificar a sobrecarga em ferro em 2 dos indivíduos em estudo. Num dos casos, colocamos a hipótese de que a conjugação da homozigotia para a mutação C282Y em HFE e da alteração R752H em heterozigotia no gene TfR2 seja o motivo da sobrecarga em ferro verificada. No outro caso, encontrou-se a alteração P124L em heterozigotia no gene HJV, que em co-herança com a homozigotia para a alteração C282Y, também poderá estar na origem da hiperferritinemia grave.
A HH não clássica é caracterizada por uma grande heterogeneidade genética, que traduz mecanismos fisiológicos complexos inerentes à homeostase do ferro, ainda não completamente conhecidos. Com este estudo contribuímos para aumentar o conhecimento sobre a fisiopatologia deste tipo de Hemocromatose e esclarecemos as relações genótipo/fenótipo nalguns dos casos estudados.
Palavras-chave: Ferro; Hemocromatose Hereditária; Hiperferritinemia; HFE; TfR2; HJV; HAMP; BMP-6;
iii
Iron plays a very important role in biological organisms. However, its excess and deficiency in the human body are both associated with negative health consequences. Hereditary Haemochromatosis (HH) is an autossomal recessive disorder commonly found in Caucasians of Northern European ancestry. It is characterized by an increased intestinal iron absorption and subsequent accumulation on several organs, resulting in tissue damage. Hepatic cirrhosis, hepatocellular carcinoma, cardiomyopathy, diabetes, endocrine abnormalities, arthritis and skin hyperpigmentation are some examples of the most frequent clinical manifestations. HFE gene is the main gene associated to the disease (HH type I). The most common genotypes found in HH patients are the homozygosity for the C282Y mutation or its compound heterozigosity with the H63D mutation. Some studies have shown that there are HH cases without mutations in the HFE gene, where other iron metabolism related genes are involved. Among these, is the Transferrin Receptor 2 gene (TfR2) which is associated with HH type III, characterized by clinical symptoms similar to the ones observed with the classical HH. On other hand, HH type II, also named juvenile haemochromatosis is a severe form of the disease, where iron overload occurs faster and the patients present cardiomyopathy and/or endocrine abnormalities at young age. This type of HH is the result of mutations in the hemojuveline (HJV) (HH type 2A) or hepcidin (HAMP) (HH type 2B) genes.
The main aim of this study was to perform a mutational screening in 5 genes related with iron metabolism (HFE, TfR2, HJV, HAMP, BMP-6) in 11 individuals presenting severe hyperferritinemia (serum ferritin level > 2500 μg/L), whose phenotypes were not justified by HFE genotypes. It was also intended the characterization of the mutations newly found. Gene sequences were amplified by PCR methodologies and mutational analysis of amplified fragments was performed by automated sequencing. In this analysis we detected 9 DNA alterations, 3 of them reported for the first time in this study (P124L in the HJV gene, c.1281-197 T→C and c.1542+4 G→A in BMP-6 gene). In silico studies were performed in order to identify possible changes at protein structural level using the bioinformatics program PolyPhen-2 and HumDiv e HumVar models. Furthermore, the impact of nucleotide changes at the splicing level was also explored using the Human Splicing Finder software.
Through this study was possible to justify the iron overload in 2 of the individuals analyzed. In one case, we put the hypothesis that the combination of homozygosity for the mutation C282Y in HFE and the heterozygosity for the R752H in TfR2 gene is the reason for the observed iron overload. In another case, we found one change in heterozygosity, P124L in HJV gene that co-inheritance with homozygosity for the C282Y in HFE, can also be the cause of severe hyperferritinemia.
The nonclassical HH is characterized by a large genetic heterogeneity, which translates into complex physiological mechanisms involved in iron homeostasis, some of them still unclear. This study was intended to contribute to increase knowledge of the pathophysiology of this type of haemochromatosis.
Keywords: Iron; hereditary hemochromatosis; hyperferritinemia; HFE; TfR2; HJV; HAMP; BMP-6;
iv Páginas Agradecimentos i Resumo ii Abstract iii Índice geral iv Índice de Figuras vi
Índice de Tabelas viii
Abreviaturas e Notações x
I-Introdução 1
I.1-Ferro 2
I.2-Metabolismo do ferro 3
I.2.1-Absorção 3
I.2.2-Transporte, distribuição e armazenamento 4
I.2.3-Homeostase do ferro 6
I.2.3.1-Regulação intracelular 6
I.2.3.2-Regulação sistémica 6
I.3-Alterações patológicas no metabolismo do ferro 9
I.3.1-Diferentes tipos de Hemocromatose Hereditária e sua base molecular 11
I.3.1.1-Hemocromatose do tipo I 12
I.3.1.2-Hemocromatose do tipo II ou Hemocromatose Juvenil 15
I.3.1.3-Hemocromatose do tipo III 17
I.3.1.4-Hemocromatose do tipo IV ou Doença da Ferroportina 18
I.3.2-Diagnóstico 19
I.3.3-Tratamento 20
II-Objectivos 21
III-Materiais e Métodos 23
III.1-População estudada 24
III.1.1-Critérios de selecção da amostra 24
III.1.2-Obtenção de material genético 24
III.2-Reacção em cadeira da polimerase (PCR) 24
III.2.1-Amplificação por PCR de regiões dos genes em estudo 24
III.3-Electroforese em gel de agarose 31
III.4-Purificação dos produtos de amplificação 32
III.5-Sequenciação dos produtos purificados 32
III.6-Análise bioinformática 33
III.6.1-Análise das sequências nucleotídicas 34
III.6.2-Análise das alterações a nível proteico 34
III.6.3-Análise do impacto das alterações nucleotídicas a nível do splicing 34
IV-Resultados 35
IV.1-Pesquisa de mutações em genes relacionados com o metabolismo do ferro 36
IV.2-Pesquisa de alterações no gene HJV 37
IV.2.1-Alteração P124L 37
v
IV.3.1-Alteração c.857+12 C→T 39
IV.3.1.1-Estudo do impacto da alteração a nível do splicing 40
IV.3.2- Alterações c.1029 C→T e c.1104 G→C 41
IV.3.2.1-Estudo do impacto das alterações a nível do splicing 42
IV.3.3-Alterações c.1281+24 T→C e c.1281-197 T→C 43
IV.3.3.1-Estudo do impacto das alterações a nível do splicing 45
IV.3.4-Alteração c.1542+4 G→A 46
IV.3.4.1-Estudo do impacto da alteração a nível do splicing 47
IV.4-Pesquisa de alterações no gene TfR2 47
IV.4.1-Alteração c.1851 C→T 47
IV.4.1.1-Estudo do impacto da alteração a nível do splicing 48
IV.4.2-Alteração R752H 48
VI.4.2.1-Estudo do impacto da alteração R752H na proteína TfR2 49 IV.4.2.2-Estudo do impacto da alteração a nível do splicing 50
V-Discussão dos Resultados 52
V.1-Pesquisa de mutações em genes relacionados com o metabolismo do ferro 53 V.2- Discussão da relação genótipo / fenótipo nos diversos casos analisados 54
V.2.1-Caso 1 54
V.2.2-Caso 2 57
V.2.3-Caso 4 58
V.2.4-Caso 10 59
V.2.5-Caso 11 59
V.2.6-Casos onde não se detectaram alterações nos genes analisados 59
VI-Conclusão 61
VII-Referências Bibliográficas 64
vi
Páginas
Figura I.1: Regulação da absorção intestinal de ferro. 4
Figura I.2: Distribuição do ferro no organismo humano adulto. 5
Figura I.3: Homeostase do ferro. 8
Figura I.4: Base genética e gravidade dos fenótipos comuns de hemocromatose. 12 Figura I.5: Estrutura do gene HFE, respectivo mRNA e proteína. 13 Figura I.6: Representação esquemática de mutações descritas no gene HFE. 15 Figura I.7: Representação esquemática de mutações descritas no gene HJV. 16 Figura I.8: Representação esquemática de mutações descritas no gene HAMP. 17 Figura I.9: Representação esquemática de mutações descritas no gene TfR2. 18 Figura I.10: Representação esquemática de mutações descritas no gene SLC40A1. 18
Figura III.11: Marcadores de massa molecular. 32
Figura IV.12: Detecção e identificação da mutação P124L no exão 3 do gene HJV 37 Figura IV.13: Alinhamento de sequências de aminoácidos da proteína HJV de 10 espécies de mamíferos diferentes, utilizando o software PolyPhen-2.
38
Figura IV.14: Previsão da patogenicidade da alteração P124L na proteína HJV através do programa Polyphen-2.
38
Figura IV.15: Detecção e identificação da alteração c.857+12 C→T a jusante do exão 2 do gene BMP-6.
39
Figura IV.16: Detecção e identificação das alterações c.1029 C→T e c.1104 G→C no gene BMP-6.
41
Figura IV.17: Detecção e identificação das alterações c.1281+24 T→C e c.1281-197 T→C no gene BMP-6.
44
Figura IV.18: Detecção e identificação das alterações c.1542+4 G→A no gene BMP-6. 46 Figura IV.19: Detecção e identificação da alteração c.1851 C→T no exão 16 do gene TfR2.
47 Figura IV.20: Detecção e identificação da alteração R752H no exão 18 do gene TfR2. 49 Figura VI.21: Alinhamento de sequências de aminoácidos da proteína TfR2 de 10 espécies de mamíferos diferentes, utilizando o software PolyPhen-2.
vii do programa Polyphen-2.
Figura V.23: Esquema representativo dos genes estudados e da localização das alterações encontradas.
viii
Páginas
Tabela I.1:Classificação genética da Hemocromatose 10
Tabela III.2: Regiões dos genes estudadas 25
Tabela III.3: Condições laboratoriais utilizadas para cada reacção de amplificação de PCR para os exões do gene HFE
25
Tabela III.4: Condições laboratoriais utilizadas para cada reacção de amplificação de PCR para os exões do gene TfR2
26
Tabela III.5: Condições laboratoriais utilizadas para cada reacção de amplificação de PCR para os exões do gene HJV
26
Tabela III.6: Condições laboratoriais utilizadas para cada reacção de amplificação de PCR para os exões do gene HAMP
27
Tabela III.7: Condições laboratoriais utilizadas para cada reacção de amplificação de PCR para os exões do gene BMP-6
27
Tabela III.8: Sequência dos oligonucleótidos iniciadores usados nos PCRs do gene HFE e respectiva dimensão dos fragmentos de DNA amplificados
28
Tabela III.9: Sequência dos oligonucleótidos iniciadores usados nos PCRs do gene TfR2 e respectiva dimensão dos fragmentos de DNA amplificados
28
Tabela III.10: Sequência dos oligonucleótidos iniciadores usados nos PCRs do gene HJV e respectiva dimensão dos fragmentos de DNA amplificados
29
Tabela III.11: Sequência dos oligonucleótidos iniciadores usados nos PCRs do gene HAMP e respectiva dimensão dos fragmentos de DNA amplificados
29
Tabela III.12: Sequência dos oligonucleótidos iniciadores usados nos PCRs do gene BMP-6 e respectiva dimensão dos fragmentos de DNA amplificados
29
Tabela III.13: Condições de PCR para amplificação dos exões do gene HFE 30 Tabela III.14: Condições de PCR para amplificação dos exões do gene TfR2 30 Tabela III.15: Condições de PCR para amplificação dos exões do gene HJV 30 Tabela III.16: Condições de PCR para amplificação dos exões do gene HAMP 31 Tabela III.17: Condições de PCR para amplificação dos exões do gene BMP-6 31 Tabela III.18: Condições laboratoriais utilizados por cada reacção para a sequenciação automática
ix
Tabela IV.20 Características dos indivíduos em estudo, genótipo HFE e outros genes onde foram detectadas alterações
36
Tabela IV.21: Descrição do efeito da alteração c.371 C→T no splicing do RNA de HJV 39 Tabela IV.22: Descrição do efeito da alteração c.857+12 C→T no splicing do RNA de BMP-6
40
Tabela IV.23: Descrição do efeito das alterações c.1029 C→T e c.1104 G→C no splicing do RNA de BMP-6
43
Tabela IV.24: Descrição do efeito das alterações c.1281+24 T→C e c.1281-197 T→C no splicing do RNA de BMP-6
46
Tabela IV.25: Descrição do efeito da alteração c.1542+4 G→A no splicing do RNA de BMP-6
47
Tabela IV.26: Descrição do efeito da alteração c.1851 C→T no splicing do RNA de TfR2 48 Tabela VI.27: Descrição do efeito da alteração c.2255 G→A no splicing do RNA de TfR2 51 Tabela V.28: Alterações moleculares identificadas neste estudo 54
x
A Adenina
β2-M Beta-2-Microglobulina
BMP-6 Proteína morfogenética do osso-6 (Bone morphogenic protein-6)
C Citosina
Cp Ceruloplasmina
DCYTB Citocromo b duodenal (Duodenal cytochrome b)
DMT1 Transportador de metais divalentes 1 (Divalent metal transporter-1) DNA Ácido desoxirribonucleico
EtBr Brometo de Etídeo
Fe Símbolo químico do ferro Fe2+ Forma ferrosa de ferro Fe3+ Forma férrica de ferro Fe2Tf Transferrina associada ao ferro
FPN-1 Ferroportina 1
Ft Ferritina
G Guanina
HAMP Hepcidina (Hepcidin anti-microbial peptide) HCP Proteína transportadora de ferro hémico HFE High Fe ou Fe elevado
HH Hemocromatose Hereditária HJ Hemocromatose Juvenil
HJV Hemojuvelina
Hp Hefastina
INSA Instituto Nacional de Saúde Dr. Ricardo Jorge
IREs Elemento de resposta ao ferro (Iron Responsive Elements) IRPs Proteína reguladora do ferro (Iron Regultory Proteins)
IVS Intrão
xi mRNA RNA mensageiro
pb Pares de bases
PCR Reacção em cadeia da polimerase (Polymerase chain reaction) p/v Percentagem expressa em peso por volume
ROS Espécies reactivas de oxigénio (Reactive oxygen species)
SLC40A1 Gene da ferroportina (Solute carrier family 40 member 1)
SNP Polimorfismo de um nucleótido (Single-nucleotide polymorphism)
T Timina
Tf Transferrina
TfR1 Receptor 1 da transferrina TfR2 Receptor 2 da transferrina
- 2 - I.1-Ferro
O Ferro (Fe) tem um papel biológico muito importante nos organismos vivos. É um micronutriente essencial mas potencialmente perigoso (Andrews and Schmidt, 2007). Trata-se do metal mais abundante na crosta terrestre, estando tanto o seu excesso como a sua deficiência no organismo humano associados a consequências negativas para a saúde (Aisen et al., 2001).
A maioria das células utiliza o ferro como co-factor em processos bioquímicos. É fundamental no transporte de oxigénio e na síntese de DNA, sendo também essencial no metabolismo celular (Wang and Pantopoulos, 2011). A utilidade deste deve-se a uma coordenação química flexível e à sua capacidade redox, permitindo a associação a proteínas e ligação a moléculas de oxigénio, possibilitando também a transferência de electrões e a mediação de reacções de catálise. O ferro pode encontrar-se em solução na forma ferrosa (Fe2+) ou na forma férrica (Fe3+), sendo a troca de electrões entre estas duas formas o que lhe confere o potencial redox (Aisen et al., 2001).
Apesar de muito abundante a maior parte do ferro encontra-se na forma Fe3+, a qual é praticamente insolúvel em água a pH neutro. Assim sendo, de forma a tornar possível a sua utilização, os organismos vivos evoluíram sistemas complexos de transporte e distribuição, bem como uma homeostase sujeita a um controlo interno (Andrews and Schmidt, 2007). A troca de electrões é essencial para as reacções redox que ocorrem a nível celular. Contudo, esta capacidade de troca electrónica potencia a formação de espécies reactivas de oxigénio (ROS) que aumentam o stresse oxidativo comummente associado a danos celulares (Garrick, 2011), dado estas possuírem a capacidade de danificar moléculas biológicas como os lípidos, carbohidratos, proteínas e ácidos nucleicos (Roy and Enns, 2000).
A alimentação humana fornece duas formas de ferro, o hémico e o não hémico. O primeiro encontra-se na carne e é facilmente absorvido, graças à acção de enzimas pancreáticas que libertam o grupo heme da molécula de globina no lúmen intestinal. O ferro não hémico está presente essencialmente em cereais, grãos e em alguns vegetais, e é menos absorvido que o hémico (Johnson-Wimbley and Graham, 2011). A fracção mais importante do ferro celular encontra-se associada a proteínas na forma hémica. De entre estas, as hemoproteínas mais comuns são a hemoglobina e a mioglobina que transportam oxigénio, respectivamente, no sangue e nos músculos. O ferro não hémico encontra-se sobretudo em metaloproteínas, as quais desempenham diversos papéis funcionais desde a transferência de electrões à catálise (Papanikolaou and Pantopoulos, 2005).
O organismo humano adulto contém cerca de 3 a 4 g de ferro. Diariamente são absorvidas a partir da dieta alimentar entre 1 a 2 mg de modo a compensar as perdas diárias ocorridas pela transpiração, descamação das células epiteliais e por processos hemorrágicos (Zhang and Enns, 2009). Não existe nenhuma via de excreção eficaz deste metal, deste modo um organismo saudável deverá ter a capacidade de avaliar a sua quantidade de ferro interna e de responder adequadamente às suas oscilações, alterando os processos de absorção e de acumulação, pois a ausência de resposta ou respostas indevidas podem levar ao aparecimento de diversas patologias (Roy and Enns, 2000).
- 3 - I.2-Metabolismo do Ferro
I.2.1-Absorção
Em mamíferos, o ferro da dieta é absorvido no duodeno ao nível dos enterócitos (Figura I.1). A absorção ocorre em dois passos principais: absorção pela membrana apical e transferência para a corrente sanguínea através da membrana basolateral (Fleming and Sly, 2002).
O ferro não hémico absorvido a partir dos alimentos encontra-se maioritariamente na forma Fe3+ sendo necessária a sua redução à forma Fe2+. Esta redução ocorre devido à acção de uma reductase férrica denominada citocromo b duodenal (DCYTB) expressa nas vilosidades do lúmen intestinal, na zona do duodeno. Assim sendo, esta reductase é o primeiro elemento importante na absorção do ferro. A sua expressão aumenta em resposta à carência e níveis baixos de ferro e hipoxia (Mckie et al., 2001). Por outro lado, a absorção do ferro hémico parece ser mediada por uma proteína transportadora (HCP) expressa ao nível dos enterócitos intestinais. Este receptor foi descrito recentemente e quando alterado parece modificar a absorção do ferro que se encontra nesta forma. Quando a expressão deste transportador é aumentada como resultado de hipóxia ou deficiência em ferro verifica-se consequentemente um aumento dos níveis de ferro no organismo (Shayeghi et al., 2005; Wang et al., 2010). O transportador de metais divalente 1 (DMT1) é responsável pela entrada dos iões Fe2+ do lúmen intestinal para a mucosa requerendo um pH baixo para funcionar (Gunshin et al., 2005). Após a entrada na célula o Fe2+ pode ser armazenado intracelularmente na forma de ferritina (Ft) (esta acomoda até 4500 átomos de Fe3+ por molécula), ou pode atravessar o enterócito até à zona basolateral onde se localiza a proteína ferroportina-1 (FPN-1), responsável pela saída de ferro da célula e a sua entrada em circulação (Garrick, 2011). O ferro que é retido na Ft endotelial é perdido após 2 ou 3 dias para o lúmen intestinal por processos de descamação (Andrews and Schmidt, 2007).
A FPN-1 parece ser o único exportador de ferro nas células duodenais, assim como nos macrófagos, hepatócitos e nos trofoblástos sinciciais da placenta. A exportação de ferro pela FPN-1 depende da interacção de duas oxidases: ceruloplasmina (Cp) presente em circulação e hefastina (Hp) localizada na membrana basolateral dos enterócitos. Estas são responsáveis pela conversão de Fe2+ em Fe3+ que será depois incorporado na transferrina (Tf) (Zhang and Enns, 2009). Esta proteína existente em circulação tem elevada afinidade com o ferro, sendo a responsável pelo seu transporte a nível sistémico (Zhang and Enns, 2009; Johnson-Wimbley and Graham, 2011)
- 4 -
Figura I.1: Regulação da absorção intestinal de ferro. O ferro hémico é absorvido pela proteína transportadora de ferro hémico (HCP), sofre endocitose, e o Fe2+ é libertado dentro do endossoma ou lisossoma. A porção de ferro não hémico inclui Fe2+ e Fe3+. O Fe3+ é reduzido a Fe2+ pelo ácido ascórbico no lúmen intestinal ou por ferroreductases, onde se inclui a citocromo b duodenal (DCYTB). Na membrana apical, o meio ácido potencia a passagem do Fe2+ para o enterócito através do transportador de metais divalente 1 (DMT1). Na membrana basolateral, a passagem do ferro para a transferrina (Tf) em circulação é mediado pela ferroportina 1 (FPN-1) em associação com a hefastina (Hp). A hepcidina tem afinidade para a FPN-1, causando a sua internalização e degradação levando a uma diminuição da exportação do ferro para o sangue. O ferro que não é transferido para a circulação é armazenado no enterócito sob a forma de ferritina (Adaptado de Zimmermann and Hurrell, 2007).
I.2.2-Transporte, distribuição e armazenamento
A maioria das células do organismo adquire/liberta ferro recorrendo ao “ciclo da transferrina” (Garrick, 2011). O fluxo deste elemento do interior dos enterócitos para o sangue ocorre através da membrana basolateral e é mediado pela FPN-1. O ferro é, posteriormente, oxidado pela Hp e Cp entrando em circulação ligado à Tf (Fe2Tf). Este complexo é então distribuído pelo
organismo de acordo com as necessidades celulares (Papanikolaou and Pantopoulos, 2005; Zimmermann and Hurrell, 2007). A Tf tem elevada afinidade com o ferro em circunstâncias homeostáticas e é a proteína responsável pelo seu transporte em circulação. A Ft armazena-o em altas concentrações mantendo-o sem acesso a substratos que levem à formação de ROS. Em humanos sem patologias associadas ao metabolismo do ferro, cerca de 30% dos locais da transferrina plasmática com função de ligação ao ferro estão ocupados (Andrews and Schmidt, 2007; Li et al., 2010).
O complexo Fe2Tf é distribuído na corrente sanguínea, ligando-se posteriormente ao receptor
1 da transferrina (TfR1), expresso na superfície celular. O complexo Fe2Tf-TfR1 entra nas
células por endocitose, a acidificação do endossoma provoca a dissociação do Fe3+ do complexo Tf-TfR1 (Garrick, 2011). O ferro é depois libertado para o citoplasma da célula por intermédio do DMT1 presente na membrada do endossoma, enquanto a Tf livre e o TfR1 são recicladas para a superfície celular. O ferro que entra nas células eritróides é direccionado para
- 5 -
as mitocôndrias onde é incorporado na protoporfirina originando grupos heme. Nos outros tipos celulares é armazenado como ferritina e hemosiderina (Andrews, 1999).
Um homem adulto tem normalmente entre 35 a 45 mg de ferro por quilograma de massa corporal. Nas mulheres esse valor é geralmente menor devido às perdas de sangue decorrentes da menstruação. No organismo humano, mais de dois terços do ferro é utilizado pela eritropoiese. As reservas no organismo estão maioritariamente localizadas nos hepatócitos e nos macrófagos reticuloendoteliais. Estes últimos fagocitam os eritrócitos senescentes armazenando o ferro que fica disponível para ser posteriormente reutilizado. Esta reciclagem é muito importante dado que a eritropoiese requer cerca de 20 mg de ferro diariamente, mas só 1 a 2 mg são normalmente absorvidas por dia através do duodeno (Figura I.2) (Roy and Enns, 2000; Frazer et al., 2003).
Figura I.2: Distribuição do ferro no organismo humano adulto. Devido a processos como a descamação da pele é perdido ferro diariamente, sendo a reposição efectuada por absorção diária de cerca de 1 a 2mg proveniente da alimentação. A maior parte do ferro que está em circulação encontra-se ligada à hemoglobina. A reciclagem do ferro presente nos eritrócitos senescentes mediado pelos macrófagos é crucial para manter os níveis de ferro. Os maiores reservatórios de ferro são o fígado, os macrófagos reticuloendoteliais e o tecido muscular (Adaptado de Andrews, 1999).
- 6 - I.2.3-Homeostase do ferro
O facto de não existir uma via de excreção eficiente de ferro do organismo, faz com que a quantidade que é absorvida a partir da alimentação e as perdas que ocorrem estejam sujeitas a um crítico processo de regulação a fim de manter a homeostasia (Andrews, 1999). Em indivíduos saudáveis, o sistema de manutenção da homeostase é responsável pela regulação da requisição, absorção, acumulação e controlo das reservas deste elemento. No entanto, a absorção em humanos parece assumir o papel mais importante na regulação do balanço do ferro do que a sua eliminação (Finch, 1994). A homeostase do ferro é regulada através de dois mecanismos: um intracelular, consoante a quantidade de ferro que a célula possui, e outro sistémico, onde a hepcidina tem um papel muito importante.
I.2.3.1-Regulação intracelular
Relativamente à regulação intracelular, de forma a evitar o excesso ou deficiência de ferro no interior da célula, existem proteínas reguladoras as proteínas reguladoras do ferro (IRPs) que controlam a expressão pós-transcricional de alguns genes envolvidos no metabolismo do ferro através de elementos de resposta ao ferro (IREs). Estes elementos em hairpin estão localizados nas regiões não transcritas (5´-UTR ou 3´-UTR) de vários RNA mensageiros (mRNAs) que codificam proteínas envolvidas no metabolismo do ferro. Os IREs são reconhecidos pelas proteínas IRPs e controlam a transcrição dos genes, a estabilidade ou a tradução dos mRNAs. Assim os genes que possuem estas estruturas são directamente regulados pelo ferro (Campillos et al., 2010; Arora, 2012). As interacções IRE/IRP regulam o nível de expressão de mRNAs que codificam para proteínas necessárias à aquisição (TfR1, DMT1), armazenamento (Ft), utilização e exportação de ferro (Muckenthaler et al., 2008).
Quando existe deficiência deste elemento na célula, as IRPs ligam-se aos IREs que são sequências altamente conservadas e que se encontram nas regiões 3´ ou 5´ do mRNA. Se os IREs estão na extremidade 3´ do mRNA, quando se ligam as IRPs, estas protegem o mRNA da degradação e promovem a síntese proteica (ex. do TfR1), promovendo uma maior absorção de ferro pela célula; simultaneamente a ligação IRPs aos IREs na extremidade 5´ do mRNA (exemplo da ferritina) impedem a sua tradução, diminuindo assim o armazenamento de ferro na célula. Quando existe excesso de ferro no interior das células, as IRPs são inactivadas, a não ligação dos IRPs ao IRE na extremidade 5´ do mRNA favorece a síntese da proteína (exemplo da ferritina), enquanto na extremidade 3´ bloqueia a tradução (ex TfR), diminuindo assim a absorção de ferro pela célula (Muckenthaler et al., 2008; Arora, 2012).
I.2.3.2-Regulação sistémica
O primeiro fenómeno de regulação na absorção do ferro ocorre a nível do duodeno e é o chamado “bloqueio da mucosa”. Após a ingestão de uma grande quantidade de alimentos ricos em ferro e consequente aumento do seu nível nos enterócitos, a absorção deste elemento pelo intestino é activamente reduzida passadas algumas horas. Esta diminuição da absorção ocorre devido a uma rápida diminuição no nível de mRNA do DCYTB e do DMT1. Desta forma, os elementos transportadores de ferro são inversamente regulados pelo nível deste nos enterócitos, podendo existir um bloqueio da absorção directamente no lúmen
- 7 -
intestinal e na membrana basolateral nos enterócitos duodenais, caso seja detectado um excesso de ferro (Frazer et al., 2003).
Existe outro mecanismo de regulação do ferro que é denominado “regulação das reservas”. Neste caso, a absorção deste elemento da dieta ocorre de acordo com as reservas do organismo. Em situações de escassez a absorção pode triplicar, no entanto quando os níveis regularizam, a absorção retorna ao normal em alguns dias, de forma a impedir que venha a ocorrer um excesso de ferro no organismo. O funcionamento desta via de regulação implica a sinalização entre o fígado, músculos e o intestino. Esse mecanismo não está completamente entendido e tem sido proposto que requer a programação de percursores das células das criptas do epitélio intestinal após a detecção da saturação da transferrina plasmática (Taylor et al., 1988; Roy and Enns, 2000).
Finalmente a absorção de ferro pode ainda ser controlada por outro sinal, conhecido como “regulador de eritropoiese”. Este sinal é muito importante na regulação da absorção porque a maioria do ferro no organismo é utilizado neste processo. Assim sendo, a regulação pela eritropoiese é mais eficiente para aumentar a absorção deste elemento do que a “regulação das reservas” (Andrews, 1999; Papanikolaou and Pantopoulos, 2005) acima descrita. A natureza deste mecanismo, ainda não está completamente esclarecido, no entanto, sabe-se que está relacionado com a síntese de hepcidina. Este sinal é assim, sensível a mudanças na taxa de eritropoiese, levando um aumento da taxa a uma diminuição da produção de hepcidina (Fried, 2009).
Tal como referido anteriormente, a ausência de um mecanismo específico para eliminar o excesso de ferro exige que exista uma fina comunicação entre os locais de absorção, utilização e armazenamento. O elemento primordial desta regulação sistémica é a hormona hepcidina produzida no fígado (Hentze et al., 2010). Esta é codificada pelo gene HAMP localizado no braço longo do cromossoma 19 (19q13.1) (Ganz, 2003) e é secretada a partir do fígado pelos hepatócitos (Peslova et al., 2009).
A hepcidina é o regulador central do ferro e a sua produção nos hepatócitos varia em resposta ao aumento da quantidade deste elemento no organismo, assim como em caso de inflamação, infecção, anemia, hipóxia e aumento da taxa de eritropoiese (Camaschella and Silvestri, 2011). A deficiência na quantidade desta hormona é responsável por sobrecarga de ferro, enquanto o seu excesso leva ao aparecimento de anemia (Babitt et al., 2006).
Quando os níveis de ferro no organismo são elevados, o aumento de hepcidina em circulação promove a sua interacção com a FPN-1, levando primeiro à internalização desta e à sua posterior degradação proteolítica. Desta forma é impedida a libertação de ferro dos hepatócitos, macrófagos e enterócitos (Dunn et al., 2007). Essa perda de FPN-1 na superfície celular conduz a uma redução da exportação de ferro das células, aumentando a sua concentração intracelular (Zhang and Enns, 2009). Assim, a hepcidina controla a absorção intestinal e a libertação de ferro dos macrófagos para o plasma num processo de retroacção negativa (Dunn et al., 2007). Para além da acção sobre a FPN-1, a hepcidina regula ainda negativamente o DMT1 duodenal, impedindo a entrada de ferro para os enterócitos (Mena et al., 2008) (Figura I.3).
- 8 -
Em condições fisiológicas, a expressão de hepcidina nos hepatócitos é regulada por um conjunto de proteínas também expressas nestas células como a HFE (High Fe ou Fe elevado), receptor 2 da transferrina (TfR2), hemojuvelina (HJV), proteínas morfogénicas do osso (BMP), matriptase-2, entre outras (Zhang and Enns, 2009). Estas proteínas, expressas à superfície celular, têm a capacidade de activar várias vias de transdução de sinal, incluindo as vias BMP-SMAD, JAK-STAT e HIF1 que culminam com a alteração da transcrição do gene HAMP. Destas a BMP-SMAD parece ser particularmente importante e perturbações nestas vias vão abolir a resposta da hepcidina a diversos estímulos. A HJV é membro de uma família de proteínas que funciona como co-receptor para as proteínas BMP. Esta liga-se a receptores do tipo BMP-1 e por estimulação com outras BMPs que melhoram a fosforilação da SMAD1/5/8. Essas SMADs activadas formam complexos que se movem até ao núcleo das células onde podem estimular a expressão de hepcidina (Darshan and Anderson, 2009) (Figura I.3).
Figura I.3: Homeostase do ferro. Quando os níveis de ferro no organismo são elevados, proteínas como a HFE, Hemojuvelina (HJV) e o Receptor 2 da Transferrina (TfR2) aumentam a expressão hepática de hepcidina. A HJV actua como co-receptor para os ligandos BMP que activam a via de sinalização SMAD induzindo a expressão da hepcidina. A via pela qual a HFE e a TfR2 induzem a expressão da hepcidina continua por esclarecer (marcada na figura a tracejado). A infecção e inflamação aumentam o nível de citocinas, como a interleucina-6 (IL-6) que estimulam a expressão de hepcidina. Esta liga-se posteriormente à FPN1 na superfície de macrófagos, enterócitos e hepatócitos. O complexo é depois internalizado e degradado, diminuindo a libertação de ferro das células e reduzindo a sua absorção no intestino. A hepcidina também diminui a expressão de proteínas envolvidas na absorção de ferro no intestino, como o DCYTB, o DMT1. Em contraste, o aumento da actividade eritropoiética suprime a expressão da hepcidina, assim como estados de anemia e hipóxia. Na figura, as linhas sólidas indicam as vias de transdução de sinal conhecidas, a tracejado estão indicadas as vias ainda não completamente esclarecidas (Adaptado de Dunn et al., 2007)
Tal como referido, a expressão de hepcidina é regulada em parte por BMPs, (Babitt et al., 2006) tendo sido os níveis de mRNA do BMP-6 associados a alterações nas concentrações de mRNA do gene HAMP (Kautz et al., 2008). Esta associação foi confirmada através da observação de ratinhos em que o gene BMP-6 foi desactivado e onde se verificou um aumento significativo da quantidade de ferro no fígado e noutros órgãos. Nesses casos, verificou-se a
- 9 -
presença de outras moléculas BMPs, mas que não compensaram a ausência de BMP-6, sugerindo assim que a função regulatória do ferro é exclusiva da proteína BMP-6, entre o grupo das BMPs (Andriopoulos et al., 2009). A observação de que o excesso de ferro em ratinhos com alterações no BMP-6 é significativamente maior do que em ratinhos com deficiência ao nível do gene HFE, fez levantar a hipótese de que, em humanos, mutações no gene BMP-6 possam causar sobrecarga grave em ferro, como por exemplo alguns casos de hemocromatose juvenil, para a qual a base genética ainda não foi caracterizada (Meynard et al., 2009).
Um outro factor regulador da expressão da hepcidina é, como referido, o TfR2. Este é predominantemente expresso em hepatócitos. Tal como o TfR1, o TfR2 é uma proteína membranar, com cerca de 45% de aminoácidos idênticos ao TfR1. A diferença fundamental entre estas duas proteínas encontra-se no domínio citoplasmático. A presença de mutações no TfR2 causa uma forma recessiva de Hemocromatose Hereditária, o que levanta a hipótese que este receptor seja, também ele, sensível aos níveis de ferro no organismo (Zhang and Enns, 2009).
I.3-Alterações patológicas no metabolismo do ferro
O mecanismo de regulação do metabolismo do ferro é muito complexo e existem vários factores genéticos e ambientais que podem causar a desregulação deste sistema levando a um aumento ou a uma diminuição da quantidade deste elemento no organismo, sendo que ambos os fenómenos comprometem a saúde e podem originar doenças de gravidade variável (Andrews, 1999).
Os efeitos clínicos da carência de ferro estão descritos na literatura médica desde o século XVI, em relatos de um distúrbio denominado clorose que afectava raparigas adolescentes (Guggenheim, 1995). Actualmente, a deficiência neste elemento é a doença nutritiva mais comum no mundo afectando cerca de 2 biliões de pessoas (Zimmermann and Hurrell, 2007). Apesar de ser mais comum em países em desenvolvimento, verifica-se uma elevada prevalência desta carência em determinadas populações de países industrializados como os Estados Unidos (CDC, 2002). Os sintomas e sinais da deficiência em ferro são parcialmente evidenciados pela presença de anemia. Estes incluem palidez, fadiga, fraca resistência ao exercício físico e diminuição da capacidade de trabalho (Andrews, 1999). Para além disso, vários estudos indicam que existe uma relação entre a carência deste elemento e problemas cognitivos e comportamentais em crianças (Grantham-mcgregor and Ani, 2001). A falta de ferro resulta na maioria dos casos da deficiente ingestão deste metal ou numa absorção ineficaz, contudo, hemorragias, tumores e parasitoses também podem provocar a sua diminuição no organismo (Andrews, 1999).
Contrariamente, a sobrecarga em ferro conduz à sua deposição e acumulação em vários órgãos, como no fígado e no coração. O seu excesso apresenta, geralmente, uma de duas características padrão ao nível do armazenamento do ferro. Quando a eritropoiese é normal mas o ferro plasmático excede a capacidade da Tf se ligar a ele (por exemplo no caso da hemocromatose) este é depositado nas células parênquimais do fígado e do coração. Em contraste, quando o excesso se deve ao aumento do catabolismo dos eritrócitos (por exemplo em casos de transfusão de sangue), a acumulação ocorre primeiro nos macrófagos
- 10 -
reticuloendoteliais e só depois nas células parênquimais. O armazenamento nestas últimas é particularmente perigoso pois pode causar danos nos tecidos e fibrose (Andrews, 1999; Deugnier and Turlin, 2011). Para além disso, o excesso de ferro está associado a casos de cancro, como o carcinoma hepático, cancro do esófago, melanoma e leucemias (Huang, 2003). Conforme a origem da sobrecarga em ferro as anomalias podem ser classificadas como genéticas ou adquiridas. De entre as genéticas, existem quatro tipos de desregulações transmitidas de modo autossómico recessivo e uma de modo autossómico dominante (Tabela I.1) (Deugnier and Turlin, 2011). A hemocromatose do tipo I é a mais prevalente, sendo responsável por cerca de 90% dos casos de sobrecarga em ferro e resulta de mutações do gene HFE que levam a alterações funcionais na proteína codificada pelo gene referido. A Hemocromatose do tipo IIA e IIB ou Hemocromatose Juvenil é caracterizada pela acumulação de ferro no organismo numa fase precoce da vida que leva a manifestação de sintomas clínicos antes dos 30 anos de idade, sendo que resultam de mutações associadas, respectivamente, aos genes da HJV ou HAMP (Roetto et al., 2003). A Hemocromatose do tipo III está relacionada com mutações no gene TfR2 (Papanikolaou and Pantopoulos, 2005). Uma forma distinta de doença hereditária transmitida de modo autossómico dominante é a doença da ferroportina que está relacionada com mutações patogénicas no gene SLC40A1 que codifica a proteína exportadora de ferro FPN-1 (Montosi et al., 2001; Sebastiani and Walker, 2007). Ainda dentro das anomalias genéticas relacionadas com o metabolismo do ferro, encontramos as doenças da ferroportina do tipo A, aceruloplasminemia hereditária, atransferrinemia hereditária entre outras. As anomalias adquiridas são aquelas que, não estando directamente implicadas no metabolismo do ferro, têm a capacidade de gerar uma sobrecarga deste elemento no organismo. Incluem-se aqui as doenças hematológicas como as talassémias e a anemia falciforme, que levam a uma sobrecarga em ferro, quer pela produção de um factor eritróide inibidor da expressão de hepcidina quer pela necessidade de múltiplas transfusões sanguíneas. Algumas doenças crónicas no fígado, como por exemplo, a inflamação necrótica dos hepatócitos, a infecção com o vírus da hepatite C e até o próprio consumo de álcool mostrou poder inibir a expressão de hepcidina (Deugnier and Turlin, 2011).
Tabela I.1:Classificação genética da Hemocromatose Tipo de
Hemocromatose
Gene Cromossoma Proteína Hereditariedade NºOMIM
Tipo I ou clássica HFE 6p HFE Autossómica
Recessiva 235 200 Tipo II A ou juvenil HJV 1q Hemojuvelina Autossómica Recessiva 602 390 Tipo II B ou juvenil
HAMP 19q Hepcidina Autossómica
Recessiva
606 464
Tipo III TfR2 7q Receptor 2 da
transferrina Autossómica Recessiva 604 250 Tipo IV ou doença da ferroportina
SCL40A1 2q Ferroportina 1 Autossómica Dominante
- 11 -
I.3.1-Diferentes tipos de Hemocromatose Hereditária e sua base molecular
A Hemocromatose Hereditária (HH) é a forma mais comum de sobrecarga primária de ferro. A sua associação à deposição hepática deste elemento foi a base para o termo Hemocromatose, dos étimos gregos haima para sangue e chromatos para cor, o qual foi cunhado em 1889 por Von Recklinghausen. Foi descrita pela primeira vez por Trousseau em 1865 como uma patologia caracterizada pela presença de diabetes, pigmentação da pele de cor bronze e por cirrose. A origem familiar da Hemocromatose foi postulada por Shiedon em 1935, este classificou a doença como um erro no metabolismo (Andrews, 1999). À semelhança de outras patologias genéticas, inicialmente esta doença foi descrita apenas fenotipicamente e só recentemente, em 1996, é que a base genética molecular foi estabelecida(Feder et al., 1996; Lyon and Frank, 2001).
Trata-se de uma doença hereditária do metabolismo do ferro em que a absorção intestinal é excessiva relativamente às reservas do organismo. Ao longo do tempo o excesso de ferro absorvido leva à saturação da Tf no plasma e à deposição deste em vários tecidos (Fleming and Sly, 2002). A HH é a doença genética mais comum em indivíduos descendentes de populações do Norte da Europa afectando 1 em cada 200 a 300 indivíduos sendo mais prevalente em indivíduos do sexo masculino do que feminino (Lyon and Frank, 2001). Apesar das manifestações clínicas da HH variarem muito, os sintomas mais característicos são artrite, arritmia e falha cardíaca, diabetes, cirrose hepática, fadiga, hiperpigmentação da pele, hipotiroidismo, hipogonadismo e carcinoma hepatocelular (Samuel et al., 2010).Os sinais da doença são mais frequentes em homens do que em mulheres devido ao efeito protector das perdas de sangue decorrentes da menstruação e da gravidez em mulheres pré-menopáusicas. A sintomatologia apresentada por homens e mulheres também tende a diferir sendo os sintomas mais comuns no sexo feminino a fadiga e a hiperpigmentação e nos homens a cirrose e diabetes (Lyon and Frank, 2001). Com o aumento da concentração de ferro no fígado, a prevalência de cirrose, diabetes e doença cardíaca é maior, o que tem implicações directas na taxa de mortalidade. Em geral, pessoas com HH sintomática têm menor taxa de sobrevivência do que indivíduos da mesma idade e sexo sem esta patologia (Hanson et al., 2001).
O fenótipo de HH é determinado em primeiro lugar pela taxa e pela magnitude de sobrecarga em ferro que é dependente das proteínas alteradas e da sua interacção com a hepcidina. Um rápido influxo de ferro no plasma causa rapidamente problemas cardíacos e insuficiência endócrina. Em contraste, o aumento gradual de ferro leva a um fenótipo mais suave e o aparecimento de sintomas é mais tardio. No entanto fenótipos intermédios também estão descritos (Figura I.4) (Pietrangelo, 2010).
- 12 -
Figura I.4: Base genética e gravidade dos fenótipos comuns de hemocromatose. As características básicas da HH são produto de mutações em genes do metabolismo do ferro (HJV, HAMP, TfR2, FPN e HFE). Dependendo do gene envolvido e da sua interferência com a hepcidina, o fenótipo da hemocromatose varia. Se o gene tem um papel importante na síntese de hepcidina (HAMP ou HJV), a sobrecarga de ferro acontece rapidamente e atinge níveis muito elevados, nesse caso o quadro clínico será grave com o aparecimento de sintomas nas primeiras décadas de vida, afectando principalmente o coração e as glândulas endócrinas. Se o gene mutado não afectar tão criticamente este processo (como o HFE) o fenótipo irá manifestar-se mais tardiamente. Existem ainda, fenótipos intermédios (mutações no TfR2 ou combinações raras de mutações) (Adaptado de Pietrangelo, 2010).
I.3.1.1-Hemocromatose do tipo I
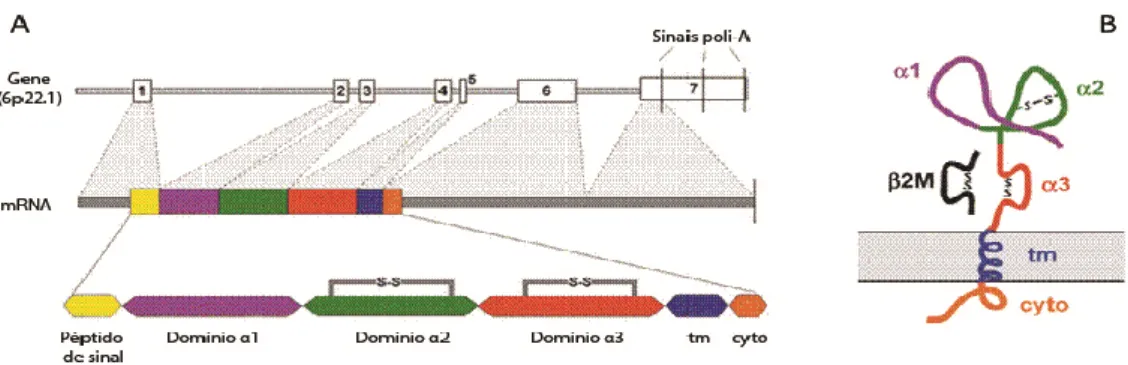
A principal causa da HH são mutações no gene HFE (High Fe ou Fe elevado) localizado no braço curto do cromossoma 6 (6p21.2) (Feder et al., 1996; Sebastiani and Walker, 2007). Este gene é constituído por sete exões, os primeiros seis codificam os domínios que constituem a proteína (péptido de sinal, domínios α1, α2 e α3, domínio transmembranar e o citoplasmático), o sétimo exão não é codificante (Figura I.5) (Fleming and Sly, 2002).
- 13 -
Figura I.5: Estrutura do gene HFE, respectivo mRNA e proteína. A). O gene HFE consiste em 7 exões (topo) dos quais apenas os seis primeiros codificam o mRNA da HFE (centro). Cada um desses exões codifica um domínio independente da proteína HFE (abaixo). tm- domínio transmembranar; cyto-domínio citoplasmático. B) Proteína HFE associada com a β2-microglobulina na superfície celular. Os três domínios extracelulares da HFE são designados por α1, α2 e α3. A β2-microglobulina (β2-M) está associada ao domínio α3 (Adaptado de Fleming and Sly, 2002).
O transcrito HFE principal tem cerca de 4,2 kb, no entanto foram descritos outros com diferentes dimensões (Jeffrey et al., 1999; Thénié et al., 2000; Sánchez et al., 2001). Estes são atribuídos a mecanismos de splicing alternativo, sendo o seu significado biológico ainda desconhecido na maioria dos casos (Fleming and Sly, 2002; Martins et al., 2011).
Existem duas mutações mais frequentes no gene da HFE, a C282Y e a H63D (Sebastiani and Walker, 2007). A maioria dos doentes com Hemocromatose do tipo I tem a transição de G→A no nucleótido 845 (exão 4) do gene HFE, que resulta na substituição de uma cisteína por uma tirosina no aminoácido 282 da proteína (C282Y). Esta mutação em homozigotia impede a interacção da proteína HFE com o seu chaperone β2-microglobulina (β2-M) o que leva a que não haja o seu direccionamento para a membrana, pelo que a HFE fica predominantemente localizada no reticulo endoplasmático das células e é posteriormente degradada. Actualmente ainda não é totalmente conhecida a fisiopatologia da doença associada à HFE. De acordo com o modelo da programação dos enterócitos, da falta da HFE funcional resultará numa redução da entrada de Fe2-Tf para as células das criptas duodenais, o que levará a uma falsa avaliação
de carência em ferro no duodeno, pois a quantidade deste elemento armazenado está a aumentar. O resultado é uma regulação positiva da expressão de proteínas do metabolismo do ferro e um aumento na absorção e transporte deste do lúmen duodenal para a circulação. Por outro lado, é colocada também a hipótese de que, devido ao papel que a HFE tem na síntese da hepcidina, a existência da forma mutada possa de alguma forma alterar os sinais ou factores apropriados para a expressão desta hormona crucial na homeostase do ferro. Desta forma o organismo não terá a capacidade de regular a absorção de ferro (Fleming and Sly, 2002).
A maioria dos indivíduos com Hemocromatose Hereditária são descendentes de um ancestral comum Celta que viveu há cerca de 60 ou 70 gerações, portador de uma única mutação missense a C282Y (Ajioka et al., 1997). Dado que esta mutação não confere um obstáculo à
- 14 -
reprodução e pode até ter sido vantajosa no passado aos seus portadores, foi transmitida à descendência e espalhada através de migrações, sendo em geral mais frequente em indivíduos do Norte da Europa (Merryweather-Clarke et al., 1997). Em Portugal existem diferenças regionais nas frequências alélicas desta mutação, sendo que a norte do país a frequência é idêntica à encontrada em alguns países do Norte da Europa com um valor de 5,8 % enquanto a sul a frequência é de 0,9 % (Cardoso et al., 2001).
A outra mutação comum no gene HFE é a H63D, que corresponde a uma transversão de G→C no nucleótido 187 do gene, resultando na substituição de histidina por aspartato no aminoácido 63 da proteína (H63D). Esta é encontrada em 15-40% da população caucasiana (Fleming and Sly, 2002), e pensa-se que impede a formação de uma ponte salina, alterando a conformação proteica daquela região. Esta variante da proteína HFE ao contrário da resultante da C282Y é processada, transportada e expressa à superfície. No entanto, não tem a mesma afinidade pelo TfR1 que a proteína não alterada. Como consequência dá-se uma maior deposição de ferro nas células (Waheed et al., 1997). Esta mutação é mais prevalente que a anterior (Merryweather-Clarke et al., 1997). Em Portugal, ao contrário do que acontece com a mutação C282Y, a mutação H63D apresenta uma distribuição mais homogénea (17-19 %)(Cardoso et al., 2001).Em termos evolutivos a mutação H63D é anterior à C282Y. A primeira poderá ter surgido em vários locais diferentes como resultado de pressões selectivas sobre o gene HFE. Estas pressões podem ter acontecido devido à presença de doenças infecciosas ou condições ambientais de indisponibilidade de ferro (Rochette et al., 1999).
Verificou-se que alguns dos indivíduos compostos heterozigoticamente para a C282Y e para a H63D podem desenvolver excesso de ferro, no entanto a penetrância deste genótipo é menor do que para a homozigotia da C282Y. Aos indivíduos homozigóticos para a H63D estão também associados ao aumento de ferro. A penetrância clinica deste genótipo é menor, no entanto têm vindo a ser reportados uma grande variedade de fenótipos (Gobbi et al., 2004). Outras mutações no gene HFE têm sido descritas, sobretudo em heterozigotia composta com a C282Y, e associadas a casos de hemocromatose do tipo I mas de carácter menos grave. É o caso da mutação S65C, que corresponde a uma transversão de A→T no nucleótido 193 do gene HFE, resultando na substituição de uma serina por uma cisteína (Trifa et al., 2012). Ainda, outras mutações raras na proteína HFE têm sido identificadas, como por exemplo a G93R, I105T e a Q283P (Figura I.6)(Lyon and Frank, 2001).
- 15 -
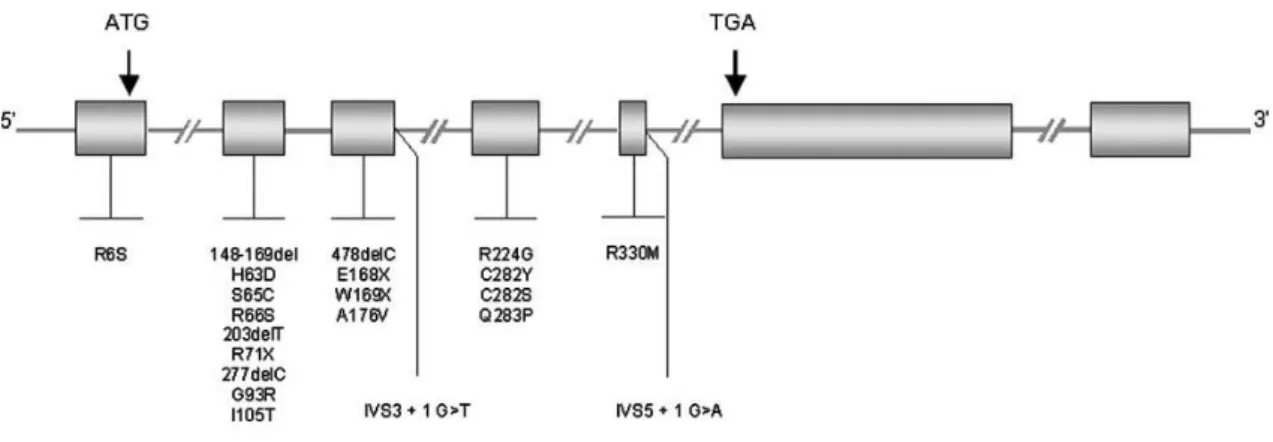
Figura I.6: Representação esquemática de mutações descritas no gene HFE. A mutações mais frequentemente associadas a HH encontram-se no exão 2 (H63D) e no exão 4 (C282Y). As caixas a cinzento representam os exões do gene (Adaptado de Le Gac and Férec, 2005) .
I.3.1.2-Hemocromatose do tipo II ou Hemocromatose Juvenil
A Hemocromatose do tipo II ou Hemocromatose juvenil (HJ) é o tipo mais grave desta patologia podendo levar à morte de indivíduos jovens (Gobbi et al., 2002). Trata-se de uma doença hereditária com transmissão autossómica recessiva que normalmente surge antes dos 30 anos. Enquanto na HH o depósito e acumulação de ferro evolui lentamente e afecta mais os homens, no tipo juvenil a evolução/gravidade dá-se mais rapidamente e afecta os dois sexos em igual proporção. Tal como a HH clássica as principais complicações clínicas são: hipogonadismo, doença cardíaca, cirrose hepática, diabetes, artropatias e pigmentação da pele, mas neste tipo com maior gravidade. No entanto, neste caso os sintomas mais comuns são o hipogonadismo e as cardiomiopatias. O depósito rápido de ferro pode ser fatal na grande maioria dos doentes com HJ, devido a falha cardíaca (Andrews, 1999; Roetto et al., 2003). A HJ é geneticamente heterogénea e está relacionada com alterações em um de dois genes. A Hemocromatose Juvenil do tipo 2A é a forma mais frequente (cerca de 90% dos casos de HJ) e está associada ao locus 1q21 (Roetto et al., 1999). O gene responsável foi identificado em 2004 como sendo o da HJV (Papanikolaou et al., 2004). Mutações neste gene causam sobrecarga grave em ferro o que, correlacionado com baixos níveis de hepcidina, sugere que a HJV afecta positivamente a cascata que regula a expressão desta hormona ao nível do fígado (Figura I.3)(Babitt et al., 2006).
O gene HJV tem cerca de 4,2kb e foi identificado por Papanikolaou e colaboradores (Papanikolaou et al., 2004). É transcrito e processado num mRNA maduro contendo a totalidade dos exões e em quatro variantes de splicing adicionais, que codificam três isoformas proteicas de 200, 314 e 426 aminoácidos. A HJV é essencialmente expressa no fígado adulto e fetal, no coração e no músculo (Martinez et al., 2004; Papanikolaou et al., 2004). A proteína codificada pelo transcrito principal, a variante que não sofre splicing, contém múltiplos domínios incluindo um péptido sinal na zona N-terminal, um domínio de três aminoácidos repulsive guidance domain (RGD), um factor de Von Willebrand (vWf) e um domínio transmembranar na zona C-terminal característico de uma âncora de glicosilfosfatidilinositol
- 16 -
(GPI-anchor) (Papanikolaou et al., 2004). A remoção dessa âncora poderá gerar uma isoforma solúvel (Lin et al., 2005). A HJV revela uma considerável homologia na sequência de aminoácidos com os factores proteicos repulsive guindance molecules (RGM) que actuam como co-receptores de BMPs, que são factores de crescimento com papéis importantes em várias actividades biológicas através de ligações a determinados receptores, desencadeando vias de sinalização que regulam a transcrição de genes específicos. A HJV actua como co-receptor de BMP para modular a transcrição do gene HAMP, tendo assim um papel importante no metabolismo do ferro através da regulação da expressão da hepcidina (Papanikolaou et al., 2004). Encontram-se descritas várias mutações (Figura I.7), essencialmente localizadas nos exões 3 e 4 deste gene que codificam a zona mais conservada da estrutura proteica, nomeadamente o domínio RGD, vWf e o domínio transmembranar (Lanzara et al., 2004) .
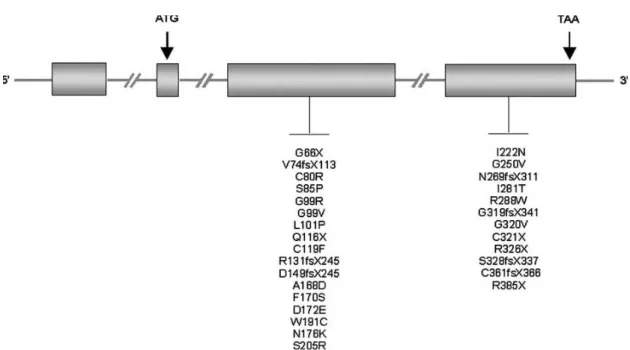
Figura 7: Representação esquemática de mutações descritas no gene HJV. A maior parte das mutações encontram-se no exão 3 e no 4, sendo neste último que se encontra a G320V que está presente na maioria dos indivíduos com HJ subtipo 2A. As caixas a cinzento representam os exões do gene (Adaptado de Le Gac and Férec, 2005) .
Várias mutações no gene da HJV têm sido descritas nos últimos anos em diversas populações. A maioria dos indivíduos com HJ do tipo 2A apresenta uma transversão de G→T no nucleótido 959 do gene da HJV, que resulta na substituição de uma Glicina por uma Valina na posição 320 da proteína (G320V). Casos de HJ relacionados com mutações no gene da HJV não parecem ser agravados quando existem mutações no gene HFE, nomeadamente no caso da mutação H63D (Lanzara et al., 2004; Lee et al., 2004; Papanikolaou et al., 2004).
A Hemocromatose Juvenil do tipo 2B é menos frequente (correspondendo a cerca de 10% dos casos de HJ) e está associada ao locus 19q13.1 onde está localizado o gene HAMP, constituído por 3 exões e que codifica para a hepcidina (Park et al., 2001). Caracteriza-se por uma sobrecarga em ferro particularmente mais grave que a do subtipo 2A. Os seus portadores tendem a apresentar precocemente sintomas cardíacos (Santos et al., 2009). Existem várias mutações descritas no gene HAMP (Figura I.8) que parecem estar relacionadas com este tipo
- 17 -
de HJ, nomeadamente a R56X e a C70R (Roetto et al., 2003). Esta última corresponde a uma troca de T→C, que resulta na substituição de uma cisteína por uma arginina. Esta alteração de um aminoácido neutro por um básico leva a alteração do péptido, uma vez que uma das oito císteinas conservadas e que são criticas para a estabilidade do polipéptido é modificada (Roetto et al., 2004). São vários os estudos que indicam que estados homozigóticos para as mutações referidas estão associados a sobrecarga de ferro (Gobbi et al., 2002; Lanzara et al., 2004).
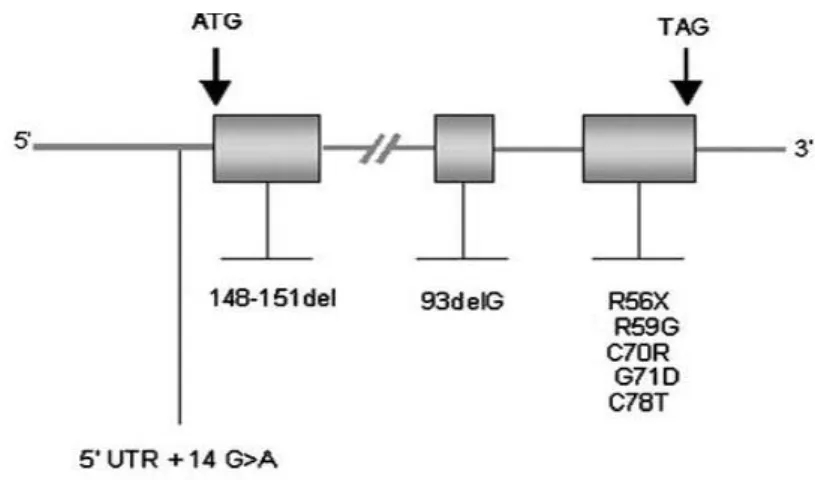
Figura I.8: Representação esquemática de mutações descritas no gene HAMP. No caso das mutações no gene HAMP estão descritas algumas mutações, não existindo nenhuma frequentemente associada à HJ subtipo 2B. As caixas a cinzento representam os exões do gene (Adaptado de Le Gac and Férec, 2005).
I.3.1.3-Hemocromatose do tipo III
A Hemocromatose do tipo III está relacionada com alterações no TfR2. A primeira mutação identificada foi a Y250X, esta é uma mutação nonsense que foi descrita em 6 indivíduos de 2 famílias Italianas (Camaschella et al., 2000). O gene TfR2 é constituído por 18 exões, localiza-se em 7q22 e codifica para a proteína TfR2 que está envolvida na captura de Tf pelos hepatócitos e também na síntese de hepcidina (Santos et al., 2012).
O fenótipo apresentado por estes doentes é idêntico ao dos indivíduos com HH do tipo I, no entanto também é detectado em indivíduos jovens à semelhança do que acontece no caso da HJ (Le Gac and Férec, 2005). Esta patologia é rara e têm sido descritos poucos casos de mutações no TfR2 (Figura I.9), no entanto, tanto em modelos animais como em doentes com HH do tipo III verifica-se um decréscimo dos níveis de hepcidina (Majore et al., 2006).
- 18 -
Figura I.9: Representação esquemática de mutações descritas no gene TfR2. A Y250X foi a primeira mutação a ser descrita neste gene e está localizada no exão 6. As caixas a cinzento representam os exões do gene (Adaptado de Le Gac and Férec, 2005).
I.3.1.4-Hemocromatose do tipo IV ou Doença da Ferroportina
A Hemocromatose do tipo IV também conhecida por doença da ferroportina foi reconhecida clinicamente em 1999 (Pietrangelo et al., 1999) e ligada ao gene SLC40A1 em 2001. Este gene localizado na região 2q32 é constituído por 8 exões (Figura I.10), codifica a ferroportina que é uma proteína envolvida na exportação celular de ferro (Montosi et al., 2001)
Anomalias no funcionamento da FPN-1 provocam uma retenção e acumulação anormal de ferro predominantemente nos macrófagos reticuloendoteliais no fígado e baço. Esta retenção de ferro intracelular provoca uma diminuição nos níveis plasmáticos deste elemento e como tal há uma redução da disponibilidade deste para se ligar à Tf. Este facto explica porque é que os níveis de saturação da Tf são normais ou baixos em fases mais avançadas da doença. A maior parte dos casos relatados são fenotipicamente mais suaves do que os que envolvem a HH clássica, possivelmente porque a deposição reticuloendotelial de ferro é menos prejudicial do que a deposição parenquimatosa (Pietrangelo, 2004).
Figura 10: Representação esquemática de mutações descritas no gene SLC40A1. A maioria das mutações descritas encontra-se no exão 5 e no exão 6