Análise Molecular da Diversidade Bacteriana de Solos do Cerrado Utilizando
Bibliotecas de rDNA 16S - Uma Perspectiva Biotecnológica.
Dissertação apresentada ao Programa de
Pós-Graduação “Strictu Sensu” em Ciências
Genômicas e Biotecnologia da Universidade
Católica de Brasília, como requisito parcial
para a obtenção do título de Mestre em
Ciências Genômicas e Biotecnologia.
Orientador: Prof. Dr. Ricardo Henrique
Krüger
Dedicatória
Agradecimentos
À Deus por ser, sempre, força tão presente nas tribulações.
Aos meus pais e irmãos, que sempre torceram muito pelo meu sucesso.
Ao meu orientador, Ricardo Krüger, pelo apoio e dedicação em seus ensinamentos.
Ao Prof. Dr. Rui de Araújo Caldas, um grande diretor.
Ao William, pelo apoio e conforto nas horas de desespero. Obrigada por me amar e
suportar.
À Alinne, meu braço direito durante todo esse período. Obrigada!!!
À Adriane, recém-chegada, mas já amiga. Obrigada!!!
Aos Profs. Drs. Mercedes Bustamante e Robert Miller pelas valiosas sugestões e críticas.
À Profa. Dra. Betânia Quirino, pela revisão e pela ajuda na finalização da dissertação.
Ao Prof. Dr. Georgios Joannis Pappas Júnior, pela ajuda na análise das seqüências.
Ao Prof. Dr. Tatsuya Nagata, pelos conselhos.
Aos meus colegas de curso que foram muito importantes na minha trajetória e se tornaram
grandes amigos.
À Regina, pelas figuras cedidas. Obrigada!
Ao Fábio, secretário, pelo prestimoso serviço prestado.
Aos técnicos do laboratório André, Willian, Marcinha e Idacuí pela excelente ajuda na
nossa árdua rotina de laboratório.
Aos professores do Curso de Mestrado em Ciências Genômicas e Biotecnologia, pelo
excelente ensino oferecido.
O único homem que está isento de erros, é
aquele que não arrisca acertar.
Abreviaturas
%
Porcentagem
°C
Grau Celsius
µF
Micro-Faraday
µg
Micrograma
µL
MicroLitro
µM
MicroMolar
Lambda
O
Ohms
BSA
Albumina de Soro Bovino
cm
Centímetros
CTAB
Cetiltrimethilammonium Bromido
DNA
Ácido Desoxirribonucléico
dNTPs
Deoxiribonucleosideo Trifosfatos
EDTA
Ácido Etileno Diamono Tetrecético
EMBL
European Molecular Biology Laboratory
g
Grama
GET
Glicose, EDTA, Tris-Cl
IPTG
Isopropil-beta-D-thiogalactopiranosideo
Kb
Kilobase
KV
KiloVolts
M
Molar
Meio LB Meio Luria-Bertani
mg
Miligrama
mM
MiliMolar
OD
Densidade Óptica
pb
Pares de Base
PCR
Polymerase Chain Reaction
pH
Potencial Hidrogeniônico
RNAse
Ribonuclease
rpm
Rotações Por Minuto
Sau
3AI
Staphylococcus aureus
3AI
SDS
Sodium Dodecil Sulfato
Taq
Thermus aquaticus
TAE
Tris-Acetato-EDTA
TBE
Tris-Borato-EDTA
TdT
Terminal Transferase
TE
Tris-EDTA
Tris
Tris-(hidroximetil)-aminometano
U
Unidade
UV
Radiação Ultravioleta
X-Gal
5-bromo-4-cloro-3-indolil-beta-D-galactopiranosideo
Resumo
O cerrado é o segundo maior bioma do Brasil, ocupando cerca de 200 milhões de
hectares. Nas duas últimas décadas, o Cerrado tem sido progressivamente convertido em áreas
de pastagem e agricultura. No entanto, poucos estudos sobre a diversidade microbiana de seus
solos têm sido feitos e não se sabe como sua conversão em áreas de pastagem tem impactado as
comunidades microbianas de seus solos.
O método clássico de estudo da comunidade bacteriana é baseado na cacterização
fenotípica e genotípica de organismos isolados e cultivados em laboratório.
No entanto, tem
sido demonstrado que os organismos cultivados representam apenas uma pequena fração da
diversidade de espécies em comunidades microbianas. Para evitar este problema, uma
abordagem molecular usando 16S rDNA foi utilizada na tentativa de identificar e descrever a
diversidade bacteriana presente em solos de Cerrado
stricto sensu
e cerrado convertido para
pastagem sem necessidade de isolamento e cultivo prévios.
O solo do cerrado é ácido e possui altas concentrações de ferro e alumínio. Por este
motivo, um protocolo para extração direta de DNA de amostras de solo foi otimizado. Após
uma série de passos de purificação para remoção de acidos húmicos e outros contaminantes,
DNA de alta pureza foi obtido. Os parâmetros da reação em cadeia de polimerase (PCR) foram
otimizadas para amplificação de 16S rDNA utilizando dois pares de
primers
específicos para
Eubacterium
e outro par específico para
Archea
. Os produtos de PCR foram clonados em
vetores e quatro bibliotecas 16S rDNA foram geradas, sendo três destas bibliotacas de Cerrado
stricto sensu
e uma de pastagem. Um total de 373 clones foram sequenciados e foi observado
do grupo dos
Actinomicetos
foram muito freqüentes em todas as bibliotecas, representando 15 a
30% das seqüências 16S rDNA. Outros grupos identificados foram de
Acidobactérias
e
Proteobactérias
. Foi observado o isolamento de um exemplar do reino
Crenarchaeota
,
pertencente ao domínio
Archea
em solo de cerrado
stricto sensu.
As seqüências foram alinhadas e árvores filogenéticas foram construídas. A diversidade
bacteriana dentro dos grandes grupos varia de cerrado
stricto sensu
para pastagem.
Para explorar o potencial biotecnológico dos microrganismos presentes no solo do
cerrado, está sendo construída uma biblioteca metagenômica de expressão destes dois tipos de
solo.
Abstract
The Cerrado is a savanna vegetation and is the second largest biome in Brazil covering
an area of approximately 200 million hectars. Over the past two decades, the Cerrado has been
progressively converted into agricultural and pasture areas. The bacterial soil community in
Cerrado is largely unknown. Furthermore, it is not known how its conversion to pasture areas
has impacted this bacterial community.
The classical microbiological approach for studying a bacterial community is to
characterize phenotypically and genotypically organisms isolated to pure culture. This method is
highly biased and is unable to accurately describe bacterial diversity. To avoid this problem, a
molecular approach using 16S rDNA was taken to identify and describe the bacterial diversity
present in Cerrado
stricto sensu
and cerrado managed to pasture soils without previously
isolation and cultivation.
In general Cerrado soils are acidic and contains high levels of iron and aluminum. For
this reason, a protocol for direct DNA extraction from soil samples was optimized. After a
series of purification steps to remove humic acids and other contaminants, DNA of high purity
was obtained. Parameters of polymerase chain reaction (PCR) were optimized for 16S rDNA
amplification using two primer pairs specific to
Eubacteria
and another primer set specific to
A
rchea
. The PCR amplification products were cloned into vectors to generate four 16S rDNA
libraries. Three of these libraries were from cerrado
stricto sensu
and one from pasture soils.
Sequencing a total of 373 clones was performed. It was observed that the bacterial groups
present in Cerrado as well as pasture soils are similar to those described in soil bacterial
diversity studies from other areas. Representatives from the
Actinomyces
group of bacteria were
groups of bacteria identified were
Acidobacteria
and
Proteobacteria
. Interestingly, one clone
from the cerrado
stricto sensu
identified the presence of a member from the
Crenarchaeota
kingdom that belongs to the
Archea
domain.
All sequences were aligned and phylogenetic trees were constructed. The bacterial
diversity found within the great groups of bacteria differed between cerrado
stricto sensu
and
pasture soils. To explore the biotechnological potential of microorganisms present in cerrado
soil an expression metagenomic library is currently being constructed.
1- Introdução
1.1-
O Cerrado
Sendo o segundo maior bioma do Brasil, o Cerrado ocupa mais de 200 milhões de
hectares, abrangendo os estados de Goiás, Minas Gerais, Tocantins, Bahia, Maranhão, Piauí,
Mato Grosso, Mato Grosso do Sul, Pará, Ceará, Rondônia e Distrito Federal e abriga um rico
patrimônio de recursos naturais. É constituído por árvores de pequeno porte, esparsas,
disseminadas em meio a arbustos, sub-arbustos e uma vegetação baixa constituída, em geral, por
gramíneas, caracterizando, assim, uma savana tropical (Hungria
et al
, 1997).
O Cerrado possui solos antigos, profundos e bem drenados. Apresentam alta acidez e
baixa fertilidade, com níveis elevados de ferro e alumínio. O clima é estacional com
precipitação média anual de 1.500 mm e grandes variações intra-regionais (Klink
et al,
www.icb.ufmg.br/~peld/port_site03.pdf
).
O equilíbrio da microbiota do solo pode ser alterado de várias formas dependendo do
tipo que manejo que se imprime a ele, sendo que o desmatamento, a aração, a monocultura, o
uso indiscriminado de agroquímicos e o fogo são as formas que mais afetam a microbiota do
solo. No Brasil, cerca de 40 milhões de hectares do cerrado são utilizados como pastagens. Esta
área representa cerca de 80% da área total utilizada para agricultura (Lilienfein
et al
, 2003). Este
fato pode ter contribuído para a mudança da microbiota local.
1.2- Diversidade Microbiana
Torsvik
et al.
(1998), a diversidade de espécies é formada por dois componentes: riqueza da
espécie e eqüitabilidade.
Até recentemente, a diversidade taxonômica e genômica de microrganismos era avaliada
principalmente através da caracterização fenotípica e/ou genotípica de organismos isolados e
cultivados em laboratório. Esses métodos de cultivo microbiano têm contribuído para nosso
conhecimento sobre a presença e diversidade das comunidades naturais. No entanto, tem sido
demonstrado que os organismos cultivados representam apenas uma fração pequena da
diversidade de espécies em comunidades microbianas.
Torsvik
et al
. (1998) observaram em microscópio de fluorescência que apenas um grama
de solo ou sedimento corado com corante fluorescente, pode conter mais de 10
10bactérias. No
entanto, quando crescidos em meios de cultura, somente 1% destes microrganismos pode ser
visualizado (Figura 1). Isto faz com que a diversidade dos microrganismos investigados,
baseada apenas em cultivo dos mesmos seja altamente subestimada. Conseqüentemente, os
métodos clássicos de estudo da microbiota não levam a uma representação da composição e
diversidade destas comunidades e não são adequados para a exploração do conteúdo genético
presente no meio ambiente.
Dois fatores podem estar colaborando para a discrepância entre estes dois métodos de
análise (observação direta e cultivo). A primeira hipótese é a diferença de estados fisiológicos
entre os microrganismos cultiváveis e “não cultiváveis’, onde determinados estados fisiológicos
seriam responsáveis pela impossibilidade de cultivo de alguns microrganismos, tornando-os que
“não-cultiváveis”, apesar da sua similaridade filogenética. Esta hipótese é baseada no fato de
que certos microrganismos “cultiváveis” tornam-se “não-cultiváveis” quando expostos a
condições adversas (Borneman, 1999). Outra hipótese é de que os microrganismos
“não-cultiváveis” presentes no solo são filogenéticamente distintos dos ““não-cultiváveis” e seus
requerimentos para cultura em laboratório ainda não foram determinados. Esta hipótese tem
sido apoiada por trabalhos indicando que a diversidade de microrganismos presentes em
diversos ambientes é maior que aquela antecipada pela análise de seqüência de microrganismos
cultiváveis (Rondon
et al
., 1999). Em estudos de diversidade genética de organismos não
cultiváveis de fonte termais, ambientes marinhos e ambientes terrestres, a maioria das
seqüências analisadas pertencia a organismos sem relação com aqueles cultiváveis e já
seqüenciados (Hugenholtz
et al
., 1998).
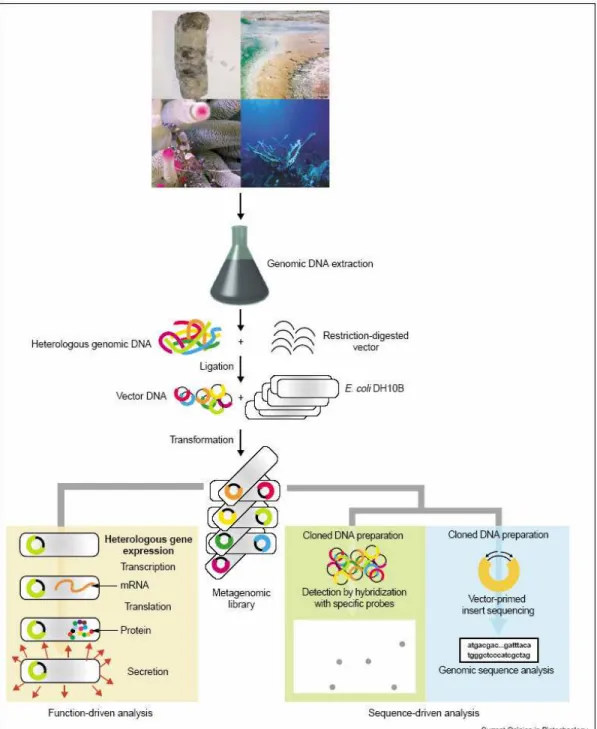
Figura 2: Estratégia para análise da composição da microbiota de solos de Cerrado atravès da clonagem e seqüenciamento de genes 16S rDNA. Esquema gentilmente cedido pelo Prof. Dr. Ricardo Henrique Krüger.
1.3- Análise da Diversidade de Microrganismos
As comunidades microbianas do solo são uma das mais difíceis de se caracterizar devido
a sua imensa diversidade fenotípica e genotípica. Muitos métodos têm sido desenvolvidos para
analisar a estrutura e diversidade destas comunidades (Ovreas
et al
, 1998). Métodos moleculares
ambientais, sem a necessidade de isolamento e cultivo prévios (Lane
et al
., 1985; Giovannoni
et
al
., 1990; Handelsman
et al
., 1998)
A caracterização de amostras ambientais utilizando-se rDNA já vem sendo utilizada a
algum tempo. As primeiras pesquisas foram feitas a aproximadamente uma década, quando
eram utilizadas moléculas de rDNA 5S que eram extraídas diretamente de amostras misturadas
(Lane
et al
., 1985; Stahl
et al
.; 1985). As moléculas encontradas, e que pertenciam a diferentes
comunidades, eram separadas por eletroforese. A análise filogenética era feita por análise
comparativa das seqüências. No entanto, a informação contida no rDNA 5S é relativamente
pequena - apenas 120 nucleotídeos - o que dificultava a separação por eletroforese.
Para tentar solucionar este problema, Olsen
et al
(1986) utilizaram moléculas 16S e 23S
rDNA para seus estudos da ecologia microbiana. Uma molécula de rDNA 16S bacteriano tem
um tamanho médio de 1.500 nucleotídeos e a molécula de rDNA 23S tem aproximadamente
3.000 nucleotídeos. Quando totalmente ou quase totalmente analisados, ambas as moléculas
contêm informação suficiente para análises filogenéticas confiáveis (Amann
et al
., 1995). Além
disso, as moléculas 16S rDNA possuem grande capacidade de armazenamento de informação,
são conservadas e sua distribuição é universal (Lane
et al
., 1985).
Os estudos iniciais de Schmidt
et al
(1991) utilizando rDNA 16S e 23S eram feitos
partindo-se da extração de DNA da comunidade total, preparação de uma biblioteca de DNA em
um bacteriófago lambda, seleção por hibridização com uma seqüência específica de rDNA 16S,
determinação das seqüências dos clones contendo 16S rDNA e análise comparativa das
seqüências encontradas.
Com o advento da PCR foi possível facilitar o trabalho e fragmentos gênicos contendo
rDNA 16 S podiam ser amplificados seletivamente de DNA misturado (Amann
et al
., 1995). Os
seqüenciamento, digestão com enzimas de restrição, análise por eletroforese em gel com
gradiente desnaturante (DGGE) (Ovreas
et al
., 1997; Silva, 2004), eletroforese em gel com
gradiente de temperatura (TGGE) (Felske
et al
., 1999); (Heuer
et al
., 1997), hibridação
DNA/DNA e DNA/RNA (Valinsky
et al
., 2002), T-RFLP (
terminal restriction fragment length
polymorphism
) (Britschgi
et al
, 1991, Liu
et al
., 1997) e análise de seqüências ribossomais
espaçadoras (RISA) (Borneman
et al
, 1997; Fisher, 1999; Ranjard
et al
., 2000).
O primeiro artigo utilizando PCR neste tipo de análise foi publicado por Giovannoni
et
al
. (1990), onde foi descrita uma análise de picoplanctons do Mar Sargasso. Os resultados
indicaram presença de agrupamentos originados de proteobactérias e cianobactérias. Hoje, esta
abordagem vem sendo largamente empregada e tem permitido estudar a microbiota de
ambientes complexos sem que haja a necessidade de cultivo prévio dos microrganismos,
revelando uma grande diversidade microbiana nos mais diversos tipos de ambientes (Barns
et
al
., 1994; Borneman
et al
, 1997; Dunbar
et al
., 1999; Liesack
et al
, 1992; Ovreas
et al
., 1997;
Stackebrandt
et al
., 1993; Uphoff
et al
., 2001; Weidner
et al
., 2000).
1.4- Análise Metagenômica
Metagenoma é a análise genômica de microrganismos de uma amostra ambiental que na
sua maioria não pode ser isolada e crescida em laboratório (Schloss
et al
, 2003). A análise
aleatória de seqüências de rDNA a partir de uma amostra ambiental pode levar à identificação
dos principais organismos presentes naquela amostra. No entanto, não é possível identificar uma
possível função destes (Borneman, 1999) e este talvez seja o maior desafio da microbiologia
hoje em dia: ligação entre filogenia e função dos microrganismos no ambiente analisado.
microrganismos responsável por ela. Análises genômicas comparativas e tecnologias de
microarranjos podem ser usadas para se determinar padrões de expressão gênica e se detectar
novas vias metabólicas (Torsvik
et al
, 2002). Outra forma de ligar um microrganismo
identificado à sua função seria através da comparação com seus parentes já estudados (Pace,
1997).
Os genomas de organismos não cultiváveis também podem ser acessados através da
clonagem em vetores de expressão. Da mesma forma, o DNA é isolado diretamente de uma
amostra ambiental, digerido e clonado em um plasmídeo de expressão multi-cópias (Rondon
et
al
., 1999). As amostras colhidas podem ser analisadas em sua função através da análise de
seqüências, que serão comparadas às seqüências já depositadas em banco de dados ou através de
análise por identificação dos clones que expressam uma função procurada, caracterizando os
clones ativos e em seguida, feita a análise bioquímica e de seqüência, como demonstrado na
figura 3 (Schloss
et al
, 2003).
Estudos recentes têm confirmado que esta forma de análise é uma fonte vasta de recursos
para a prospecção de novos produtos biotecnológicos (Courtois
et al
., 2003; MacNeil
et al
.,
2001; Rondon
et al
., 1999; Short, 1997). Novos genes e novos produtos gênicos têm sido
2- Objetivos
2.1- Isolar DNA de comunidades microbianas de solos de cerrado submetidos à
diferentes manejos.
2.2- Amplificação de rDNA 16S de DNA de solo de cerrado
stricto sensu
e pastagem e
posterior seqüenciamento para identificação da comunidade bacteriana presente nestes dois
tipos de solo.
2.3- Comparar a biodiversidade bacteriana presente no solo do cerrado
stricto sensu
e
manejado para pastagens.
3- Hipóteses
3.1- Mudança na microbiota local quando há a conversão do solo de cerrado nativo para
pastagem.
3.2- Existem áreas do cerrado com potenciais diferentes para a bioprospecção de genes
de interesse biotecnológico.
4- Metodologia
4.1- Descrição das Áreas de Coleta do Solo
4.1.1- Cerrado Stricto Sensu
Amostras de solo de cerrado
stricto sensu
foram coletadas em área não perturbada
localizada na Reserva Ecológica do Instituto Brasileiro de Geografia e Estatística (RECOR -
IBGE) localizada em Brasília, DF (figura 4). A Reserva Ecológica do Instituto Brasileiro de
Geografia e Estatística (RECOR-IBGE) está localizada a 35 km ao sul do centro de Brasília,
DF, na BR-251, km 0 (15°55´S, 47° 51´ W) a 1.100 metros de altitude, ocupando uma área de
1.350 hectares. O solo desta área é denominado latossolo vermelho (Silva, 2004).
Figura 4: Cerrado stricto sensu. Reserva Ecológica do Instituto Brasileiro de Geografia e Estatística (RECOR - IBGE). Foto gentilmente cedida por Regina Silva – Ecologia, UnB.
4.1.2- Pastagem
As amostras de solo de cerrado submetido à conversação para pastagem foram obtidas
na Fazenda Rio de Janeiro (figura 5), localizada em Planaltina, GO (15° 14’ S, 47° 42’ W) a 826
m de altitude. O solo desta área é denominado latossolo vermelho-amarelo. Após o
desmatamento da vegetação nativa existente, foi introduzida gramínea. Desde 1990, a área
vinha sendo cultivada com
Brachiaria brizanta
cv. Marandu (Silva, 2004).
Figura 5: Cerrado submetido a manejo para pastagem. Fazenda Rio de Janeiro - GO. Foto gentilmente cedida por Regina Sartori – Ecologia, UnB.
4.2- Coleta das Amostras
Foram retiradas 10 amostras de solo de cerrado
strictu sensu
de 0-5 cm de profundidade
de diversos pontos da mesma região para compor três amostras compostas e 6 amostras de
pastagem para a composição de uma amostra composta. A camada superficial do solo foi
escolhida pelo fato da maior intensidade de atividade biológica ocorrer na camada superficial do
solo, e uma vez que isso acontece, qualquer uso ou manejo provocado neste solo, poderá mudar
a sua qualidade (Alvarenga, 1999).
As amostras de cerrado
stricto sensu
foram coletadas em julho de 2002 e as de pastagem
foram coletadas em setembro de 2002. Ambos as datas de coletas correspondem a períodos de
seca nas regiões.
4.4- Extração de DNA de Solo
O DNA genômico foi extraído de acordo com protocolo pré-estabelecido baseado em
Yeates
et al
. (1998) e modificado da seguinte forma: 5 g de solo e 5 g de pó de vidro (“
glass
beads
” - Sigma) foram misturados em 14 ml de tampão A (tampão de extração). Em seguida, a
mistura foi agitada no aparelho Vortex Genie 2 (Fisher Scientific) por 4,5 minutos com
intervalos de 10 segundos a cada 90 segundos. Foram, então, adicionados 1,5 ml de SDS 20%,
misturando-se gentilmente. A solução foi incubada por 1 hora a 65°C com leve agitação a cada
15 minutos. Após o período de incubação, a solução foi centrifugada por 15 minutos a 5.000
rotações por minuto em centrífuga Eppendorf 5804. O sobrenadante foi removido e separado em
vários eppendorfs contendo 1 ml de solução em cada e precipitado com 1 volume de
isopropanol 100%, incubado por 15 minutos a temperatura ambiente e centrifugado por 15
minutos a 12.000 rpm. O sobrenadante foi removido e o precipitado foi ressuspendido em 800
µl de TE. Em seguida foi adicionado 1 ml da solução de PEG 8000 e NaCl 1,6 M e incubado 2
horas a temperatura ambiente. A solução foi centrifugada por 20 minutos a 12.000 rpm, o
sobrenadante removido e o precipitado ressuspendido em 400 µl de TE. Foi adicionado KAc
para a concentração final de 0,5 M, colocado em gelo por 5 minutos e centrifugado por 20
minutos a 12.000 rpm. O sobrenadante foi extraído 4 vezes adicionando-se um volume de fenol,
agitando levemente e centrifugando 5 minutos a 12000 rpm, retirando-se a fase aquosa. Em
seguida foram extraídos 2 vezes adicionando-se um volume de clorofórmio: álcool isoamílico
(na proporção 24:1), agitando-se levemente e centrifugando por 5 minutos a 12.000 rpm,
retirando-se a fase aquosa.
foi centrifugada por 20 minutos a 12.000 rpm. O sobrenadante foi removido e o precipitado
lavado com etanol 70% e em seguida centrifugado por 3 minutos. O precipitado foi seco em
Speed Vac (Eppendorf) por 5 minutos e ressuspendido em 200 µl de água milli-Q. Em seguida
este DNA foi purificado utilizando-se o kit UltraCleanTM15 DNA Purification (MOBIO
Laboratories Inc.) seguindo as recomendações do fabricante.
A integridade do DNA foi verificado em gel de agarose 1% corado com brometo de
etídeo (2 µg/ml) e o seu tamanho, estimado por comparação com marcador 1kb plus ladder
(USB - EUA).
4.5- Preparo de Células Eletrocompetentes.
Uma colônia de
E. coli
DH5
α
foi crescida em 5 ml de meio LB sob agitação de 240 rpm
a 37°C por 16 horas. Após este período, os 5 ml de cultura foram inoculados em 400 ml de meio
LB e incubados a 37°C a 240 rpm até que atingisse O.D.600 = 0,8. Em seguida, as células foram
centrifugadas a 5000 rpm por 10 minutos, lavadas duas vezes em água destilada gelada e estéril
e duas vezes em glicerol 10% gelado e estéril. Finalmente, as células foram ressuspendidas em
glicerol 10% estéril. As células foram divididas em alíquotas de 100 µl e congeladas a -80°C.
A eficiência de transformação das células foi medida usando protocolo de transformação
por eletroporação com um plasmídeo comercial de concentração conhecida, na sua forma
intacta. A eficiência foi estimada em 10
8transformantes por µg de DNA.
4.6- Construção da Biblioteca 16S rDNA
Para que fosse possível fazer o estudo filogenético das espécies encontradas nas
amostras de solo foi construída uma biblioteca de clones 16S rDNA de
Eubacterium
e
Archea
,
Para tal foram utilizados os pares de
primers
8F
(5´-AGAGTTTGATCCTGGCTCAG-3´) e 1391R (5´-GAGGGGCGGTGTGTRCA-(5´-AGAGTTTGATCCTGGCTCAG-3´) (Ouverney, não publicado) que amplificam
fragmentos de aproximadamente 1,3 Kb e COM1F (5´-CAGCAGCCGCGGTAATAC-3´,
posições 519 a 536) e COM2R (5´-CCCTCAATTCCTTTGAGTTT-3´, posições 907 a 926)
(Schwieger, 1998) amplificando fragmentos de 408 pb de
Eubacterium
. Para a amplificação de
rDNA
16S
de
Archea
foram
utilizados
os
primers
Arch341IF
(5´-
CCTA/ideoxil/GGGG/ideoxil/GCA/ideoxil/CAG-3´)
e
1492R
(5´-GGYTACCTTGTTACGATT-3´), amplificando fragmentos de 1,1 kb.
Na reação de PCR foram utilizados tampão de T
aq
10X (10 mM Tris-HCl pH 8.3; 5 mM
KCl, 1,5 mM MgCl
2), 20 µM de cada
primer
; 2,5 mM dNTP; 5 U enzima
Taq
polimerase e 10
ng de DNA de solo em uma reação de 50 µl de volume final.
A amplificação de rDNA 16S foi feita em termociclador Perkin Elmer através de uma
etapa de 95°C por 3 minutos de desnaturação inicial, 25 ciclos de 95°C por 1 minuto para
desnaturação do ciclo, 56°C por 1,5 minutos para anelamento dos primers, 72°C por 3 minutos
para extensão da fita de DNA. Após os 25 ciclos, 72°C por 30 minutos para extensão final e
mantido a 10°C até o momento de retirada do termociclador. Para os
primers
de
Archea
, a
amplificação de rDNA 16S foi feita em termociclador Perkin Elmer através de uma etapa de
95°C por 3 minutos de desnaturação inicial, 25 ciclos de 95°C por 1 minuto para desnaturação
do ciclo, 51°C por 1,5 minutos para anelamento dos
primers
, 72°C por 3 minutos para extensão
da fita de DNA. Após os 25 ciclos, 72°C por 30 minutos para extensão final da fita de DNA e
mantido a 10°C até o momento de retirada do termociclador.
Tanto para o conjunto de
primers
8F/1392R quanto para COM1F/COM2R, foram
de reações de PCR com gradientes de temperatura variando em 10°C da temperatura
recomendada pelo fabricante dos
primers.
Após estes testes, as amplificações continuaram apresentando um rastro de
amplificações não específicas que foram solucionadas baixando-se o número de ciclos da reação
de 30 para 25, o que minimiza a formação de
primer-dimer
, que vem a ser um produto de
anelamento entre dois
primers
que se complementam. Esta formação não só diminui a
concentração de
primers
na reação como também inicia a formação de produtos de PCR não
específicos (Hsu, 1999).
Os produtos de PCR foram corridos em gel de agarose 1% corado com brometo de
etídeo e os fragmentos de DNA amplificados com o tamanho previsto foram cortados do gel e
purificados com kit UltraCleanTM15 DNA Purification (MOBIO Laboratories Inc.). Ao final da
purificação, o rDNA foi ressuspendido em água milli-Q (Millipore).
Para a clonagem dos fragmentos amplificados de DNA de solo de cerrado
stricto sensu
,
foi utilizado vetor pGEM-T (Promega, Wisconsin, EUA), conforme as instruções do fabricante.
Para os fragmentos amplificados de solo de pastagem foi utilizado o vetor TOPO TA Cloning®
(Invitrogen). A solução foi dialisada em filtro de nitrocelulose 0,025 µm (Millipore - EUA) por
20 minutos e 10 µl da ligação fragmento 16S rDNA/vetor dialisado utilizada para transformação
por eletroporação em células competentes
E. coli
DH5
α
, e colocados em uma cuveta para
A seleção de transformantes foi realizada através de plaqueamento em meio S-gal/LB
Ágar Blend (Sigma) contendo ampicilina a 250 µg.mL
-1e incubação a 37°C por 16 horas. Foi
observada a formação de colônias pretas e brancas, sendo as colônias brancas as selecionadas
para a detecção de inserto. Foram selecionadas 400 colônias brancas das placas de clones de
cerrado
stricto sensu
e 100 colônias brancas das placas de pastagem. As colônias foram
crescidas em tubos Falcon contendo 5 ml de meio Lura-Bertani (LB) e ampicilina a 250 µg.mL
-1
durante 19 horas a 37°C a 240 rpm.
Para confirmação da presença de inserto no vetor, foram realizadas reações de PCR de
10 clones de cada tipo de solo, utilizando-se
primers
SP6 e T7 para o vetor pGEM-T. Para o
vetor TOPO foram utilizados os
primers
T3 e T7, como recomendado pelos fabricantes. Para
isso foram realizadas extrações plasmidiais de 10 colônias previamente crescidas em 5 ml de
meio LB e 250 µg.mL
-1de ampicilina durante 19 horas a 37°C a 240 rpm, utilizando o kit
QIAprep
®Miniprep (Qiagen).
Após a confirmação da presença do inserto nos clones, 200 µl de cultura bacteriana de
cada inóculo foram misturados a glicerol para uma concentração final de 10% e colocados em
placas tipo Elisa para congelamento a -80C.
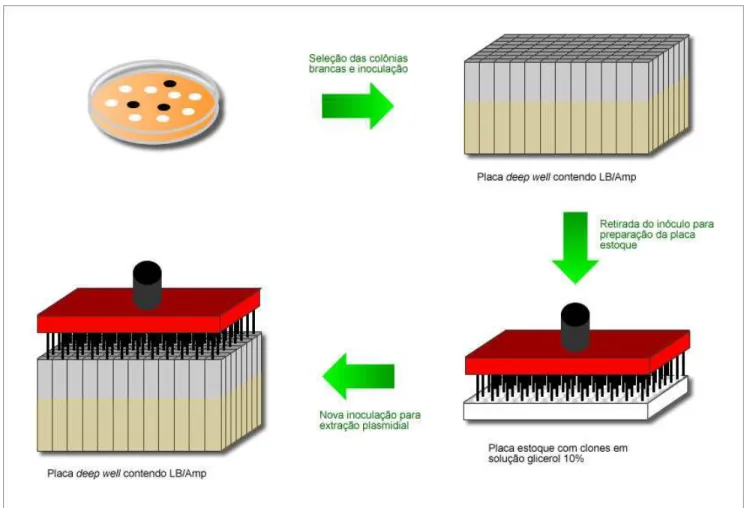
As colônias foram, mais tarde, descongeladas e inoculadas em uma micro-placa
Deep
Well
de 96 poços contendo 1 ml de meio LB e ampicilina 250 µg.mL
-1, utilizando-se um
Após o crescimento bacteriano, foi feita a extração de DNA plasmidial das culturas
(Birnboim
et al
,1979), segundo protocolo modificado, centrifugando-se as placas
Deep-Well
a
4000 rpm por 15 minutos e descartando o sobrenadante. O sedimento bacteriano foi
ressuspendido em 240 µl de solução I, centrifugado 10 minutos a 4000 rpm e descartado o
sobrenadante, secando a placa sobre papel absorvente. O sedimento bacteriano foi mais uma vez
ressuspendido em 70 ul de solução I adicionado 20mg de RNAse e incubado por 5 minutos a
temperatura ambiente. Foram transferidos 70 µl das suspensões de células para microplaca
fundo U e adicionado 70 µl de solução II e homogeneizada por inversão várias vezes. A placa
foi incubada por 10 minutos a temperatura ambiente e em seguida, adicionado 70 µl de solução
III gelada, invertendo várias vezes para homogeneização. A placa foi incubada por 10 minutos a
temperatura ambiente e em seguida, centrifugada por 20 minutos a 4.000 rpm e 120 µl do
sobrenadante foram transferidos para microplaca-filtro (Millipore) com a base fixada com
elásticos em uma microplaca fundo V. As placas fixadas foram centrifugadas por 6 minutos a
4.000 rpm. Os plasmídeos foram então precipitados adicionando-se 100 µl de isopropanol 100%
à placa fundo V. Após 20 minutos em temperatura ambiente, a placa foi centrifugada por 45
minutos a 4.000 rpm. O isopropanol foi retirado por inversão da placa. O precipitado foi lavado
adicionando-se 200 µl de etanol 70% e centrifugado por 15 minutos a 4.000 rpm. O
sobrenadante foi retirado e a placa foi centrifugada invertida por 30 segundos a 600 rpm para
remoção de traços de etanol. Para secagem do etanol, a placa foi incubada em estufa a 37°C por
30 minutos. Os plasmídeos foram, então, ressuspendidos em 50 ul de água milli-Q deixando-os
incubados a 4°C por 18 horas.
A quantificação do DNA plasmidial foi feita em gel de agarose 1% corado com brometo
de etídeo, sendo utilizado o fago (lambda) de concentração conhecida como padrão de
comparação. Foi retirada uma alíquota para seqüenciamento e o restante foi armazenado a
-80°C.
As reações de seqüenciamento foram feitas por PCR utilizando-se o kit de
seqüenciamento DYEnamic ET Terminator Cycle Sequencing (Amersham Biosciences) para
seqüenciador automático ABI Prism 377 (Applied Biosystems), nas seguintes concentrações 0,3
µM de
primers
T7; 100 ng de DNA; 2 µl de
Sequencing reagent premix
(marcação fluorescente
para extensão. Os produtos das amplificações foram, em seguida, purificados, seguindo-se o
protocolo do kit de seqüenciamento.
4.7- Análises das Seqüências
As seqüências obtidas foram, inicialmente, analisadas em programa para determinação
de qualidade de eletroesferograma, desenvolvido pelo Departamento de Bioinformática da
Universidade
Católica
de
Brasília,
disponível
no
site
http://www.bioinformatica.ucb.br/electro.html
. Neste programa, é utilizado o algorítimo
PHRED (Ewing
et al
., 1998; Ewing & Green, 1998) que faz com que os dados brutos, que
foram gerados pelo seqüenciamento, sejam analisados e verificados na sua probabilidade de
erros, base a base. Neste caso, o valor de PHRED aceito foi maior que 20, em mais de 200 bases
de uma seqüência, o que significa dizer que uma base tem uma chance em cem de estar errada,
ou seja, esta base tem 99% de chance de estar correta. As seqüências que não apresentaram o
valor de PHRED maior que 20 em mais de 200 bases foram descartadas. Estes valores são os
mais utilizados e aceitos nas análises de seqüências provenientes de seqüenciamento automático
(Kaiser
et al
., 2003; Amos
et al
., 2000; Richterich, 1998). Além deste algorítmo, é executado
também o “
cross-match”
para a remoção das seqüências pertencentes aos vetores de clonagem.
Em seguida, as seqüências selecionadas foram comparadas ao banco de seqüências não
redundantes do National Center for Biotechnlogy Information (NCBI) usando-se BLASTn
(Altschul
et al
., 1990). Foram analisadas as similaridades das seqüências obtidas com aquelas
presentes no banco de dados. As seqüências que apresentaram identidade maior que 90% e
e-value igual a zero foram consideradas e nomeadas.
seqüências de dois parentes filogenéticamente distintos e ocorre quando um produto de
amplificação terminado prematuramente volta a se anelar a uma nova fita de DNA e é copiada
até o termino dos ciclos da PCR (Huber
et al
., 2004). A presença de quimeras pode levar a uma
falsa análise da biodiversidade presente na amostra ambiental por sugerirem a presença de
microrganismos não existentes (von Wintzingerode
et al
., 1997).
A detecção de quimeras foi feita utilizando-se o programa Bellerophon
(
http://foo.maths.uq.edu.au/~huber/bellerophon.pl
). Este programa é utilizado para a detecção
de seqüências quiméricas em conjuntos de múltiplas seqüências. Este programa foi
especialmente desenvolvido para a detecção de quimeras nas bibliotecas de amostras ambientais
geradas por PCR. Em seguida, as quimeras detectadas pelo programa Bellerophon foram
comparadas com aquelas obtidas pelo Check_Chimera (Maidak
et al
., 1996), para uma dupla
análise, como sugerido em von Wintzingerode
et al
. (1997), e retiradas.
4.8- Análise Filogenética
Para a análise filogenética primeiramente foi feita uma análise visando a identificação da
presença de seqüências redundantes na biblioteca seqüenciada comparando-se as seqüências
entre elas.
Em seguida foi feito o alinhamento múltiplo das seqüências utilizando-se o programa
T-Coffee (Notredame
et al
., 2000). Este programa permite que as seqüências sejam alinhadas, com
maior precisão, nas suas regiões mais conservadas, para que seja possível inferir se há relação
filogenética entre as mesmas. Para a inferência das árvores filogenéticas, foi utilizado o
programa Drawtree do pacote de programas PHYLIP.
mais provável que duas e, portanto, a árvore é formada de forma que requeira um menor número
de mudanças para explicar os dados no alinhamento (Hall, 2001; Mattioli, 2001).
Para testar a confiança da árvore filogenética gerada, foi utilizado o
bootstrap
(Felsenstein, 1985). Este método consiste em uma simples re-amostragem dos dados com
reposição pseudoaleatória destes. A cada re-amostragem, uma árvore réplica é construída e, ao
final, a confiabilidade das clades pode ser estimada (Mattioli, 2001). O
bootstrapping
das
seqüências foi realizado utilizando-se o programa
Seqboot
, também pertencente ao pacote de
programas Phylip, fazendo-se um total de 1000 repetições. A árvore foi visualizada
utilizando-se o programa ATV (Zmautilizando-sek & Eddy, 2001)
.
4.9- Construção da Biblioteca Metagenômica
Após a extração de DNA genômico como detalhado acima, o DNA que não foi utilizado
na construção da biblioteca 16S rDNA, foi então utilizado na construção da biblioteca
metagenômica.
O DNA genômico total foi digerido utilizando a enzima de restrição
Sau
3AI (Promega -
EUA). A enzima foi diluída em tampão de enzima para que sua eficiência fosse alterada, uma
vez que, com a enzima pura, o DNA era digerido em fragmentos de tamanhos excessivamente
pequenos. Para isso foi feito então a diluição da enzima
Sau
3AI em tampão XK (Promega
-EUA), que altera a eficiência da enzima para menos de 20%. Foi utilizado 1 µl de enzima para 9
µl de tampão XK. O mesmo tampão foi utilizado na reação.
Para a digestão foram utilizados 4 mg de DNA genômico, 4 µl de tampão XK 10X, 3 µl
de enzima de restrição
Sau
3AI diluída, e água milli-Q para o volume final de 40 µl. A reação foi
DNA foi visualizada em gel de agarose 0,8% corado com brometo de etídeo, juntamente com
marcador 1Kb plus ladder para identificação do tamanho dos fragmentos.
O DNA que se apresentava na faixa de 3 a 9 Kb de tamanho foi cortado do gel,
purificado com kit UltraCleanTM15 DNA Purification (MOBIO Laboratories Inc.) e
ressuspendido em 20 µl de água milli-Q. Em seguida o DNA foi tratado com enzima Klenow
(Promega) para a que os fragmentos ficassem com finais abruptos desta forma: 20 µl de DNA
digerido, 3 µl de tampão 10X, 1 µl de enzima Klenow (Promega), 2 µl de dNTPs; água milli-Q
para o volume final de 30 µl. A reação foi incubada a 37°C por 30 minutos e em seguida a
enzima foi inativada a 75°C por 10 minutos. O DNA tratado foi, então, precipitado utilizando-se
2,5 volumes de etanol 100% e 0,3 M de acetato de sódio. Incubado 30 minutos em gelo e
centrifugado por 20 minutos a 12.000 rpm. O precipitado foi lavado em etanol 70% e seco por 5
minutos no Speed-Vac e em seguida ressuspendido em 10 µl de água milli-Q. Esse DNA foi,
posteriormente, utilizado para clonagem em vetor de expressão.
O vetor de expressão de escolha para os primeiros testes de clonagem, na construção da
biblioteca metagenômica foi o vetor pBlueScript II KS (Stratagene). Para esta clonagem foi
necessário que o vetor fosse digerido com uma enzima de restrição que tornasse seus finais
abruptos. A enzima escolhida foi
Eco
RV (Promega). A reação foi feita utilizando-se 600 ng de
vetor pBlueScript II KS; 2 µl de tampão 10X; 2 µl de enzima de restrição
Eco
RV; água milli-Q
O plasmídeo digerido foi tratado com fosfatase alcalina de camarão (Promega) para
retirar os fosfatos da porção 5´. A reação de desfosforilação foi feita da seguinte forma: 10 µl de
pBlueScript II KS digerido por
Eco
RV; 2 µl de fosfatase alcalina; 3 µl de tampão 10X; água
para o volume final de 30 µl. A reação foi incubada por 1 hora a 37°C. Para a inativação da
enzima foi adicionado SDS para a concentração final de 1% e NaAc para a concentração final
de 0,1M e as amostras foram colocadas a 65°C por 20 minutos. Em seguida foram extraídas
duas vezes com fenol e uma vez com clorofórmio, adicionando um volume de fenol ou
clorofórmio á reação, misturando bem e centrifugando por 5 minutos a 12.000 rpm retirando-se
a fase aquosa, onde se encontra o DNA plasmidial. Este DNA foi então precipitado com 2,5
volumes de etanol 100% e 0,3 M de acetato de sódio. Incubado 30 minutos em gelo e
centrifugado por 20 minutos a 12.000 rpm. O precipitado foi lavado em etanol 70% e seco por 5
minutos no Speed-VAc e em seguida ressuspendido em 10 µl de água milli-Q. O DNA foi,
então, visualizado em gel de agarose 0,8% corado com brometo de etídeo para confirmação da
recuperação deste após o processo de purificação.
A ligação do DNA no vetor de expressão foi feita utilizando 2 µl de vetor; 14 µl de DNA
de solo digerido; 2 µl de tampão 10X da ligase; 1 µl de ATP; 1 µl de ligase. A reação foi
incubada a 16°C por 16 horas. Após o período de incubação, a ligação foi dialisada por 15
minutos.
Da mesma forma que para a biblioteca 16S rDNA, a ligação foi transformada em células
competentes
E. coli
DH5
α
por eletroporação adicionando-se o 10 µl da ligação solo/vetor
1,8 KV) e após a eletroporação foi adicionado 1 ml de meio SOC na cuveta e misturado
gentilmente até que a solução células/meio estivessem bem homogêneos e transferidos para um
tubo eppendorf de 1,5 ml e incubados por uma hora a 37°C. A seleção de transformantes foi
realizada através de plaqueamento em meio S-gal/LB Ágar Blend (Sigma) contendo ampicilina
a 250 µg.mL
-1e incubação a 37°C por 16 horas. Foi observada a formação de colônias pretas e
brancas, sendo as colônias brancas as selecionadas para a detecção de inserto e crescidas em
placas
Deep-Well
com meio LB contendo ampicilina a 250 µg.mL
-1. Da mesma forma que na
biblioteca de 16S rDNA, foi colhido 200 µl de crescido de bactérias de cada inóculo e
adicionado glicerol para uma concentração final de 10% , colocados em placas tipo Elisa e
congeladas a -80C.
Foram feitas extrações plasmidiais de 10 clones para verificação da presença de
fragmento inserido nos vetores e determinação de seus tamanhos. Para isso foi utilizado kit
QIAprep
®Miniprep (Qiagen). Após a extração foi realizada uma reação de PCR para
confirmação da inserção dos fragmentos no plasmídeos. Para tal foram utilizados os
primers
T7
e M13R que possuem sítios de anelamento que flanqueiam o fragmento inserido. Na reação
foram utilizados 2 µl de tampão 10X de
Taq
polimerase; 2,5 mM de dNTPs; 10 µM de
primers
T7; 10 µM de
primers
M13R; 5U de
Taq
polimerase; 50 ng de DNA e água milli-Q para o
5- Resultados e Discussão
5.1- Extração de DNA
Em estudos moleculares, um dos pré-requisitos para que se possa analisar e obter
descrição da diversidade genética presente em uma amostra ambiental é o isolamento de DNA
com pureza suficiente para que se possa realizar análises moleculares e estimar-se a ocorrência
de diversidade genética (Liesack, 1992). Muitos métodos de extração de DNA de amostras
ambientais têm sido descritos. Entre eles, métodos de extração de DNA de solo, que se baseiam
em duas técnicas principais: a) extração de células de uma amostra de solo antes da lise celular
e, b) lise direta de células. O maior limitante do método de extração de células é o longo tempo
consumido e a limitação de se extrair somente algumas amostras por vez. No caso de extração
de DNA por lise direta das células, não é necessário isolar células, o que torna o método mais
rápido. Além disso, uma vez que a diversidade de microrganismos sujeito à lise é maior, este
protocolo para isolamento de DNA leva a uma melhor representação da diversidade microbiana
da amostra original. No entanto, a extração por lise direta de células aumenta a quantidade de
ácidos húmicos e outras substâncias orgânicas contaminantes presentes no extrato.
Ácidos húmicos têm propriedades físico-químicas similares aos dos ácidos nucléicos,
que fazem com que haja competição na absorção por colunas de purificação, tornando esse
processo mais laborioso. Além disso, esses contaminantes podem diminuir a eficiência de
hibridização DNA/DNA, inibir atividades de endonucleases de restrição e de enzimas
Taq
Em estudos sobre a otimização de extração de DNA de solo, Miller
et al
. (1999)
mostraram que maiores quantidades de DNA são obtidas por extração utilizando-se pó de vidro
(“
glass beads
”), ao invés de ciclos de congelamento e descongelamento e que a utilização de
fenol ou fenol/clorofórmio aumenta a quantidade de DNA extraído. Ainda no mesmo estudo foi
mostrado que a utilização de CTAB durante a extração diminui a quantidade de polissacarídeos
e ácidos húmicos contaminantes presentes na amostra. No entanto, nenhum dos métodos
utilizados remove completamente todos os contaminantes, e no que se refere à extração de altas
quantidades de DNA, alguns problemas ainda são encontrados. Por isso, a otimização dos
métodos de extração de DNA de amostras do solo é extremamente importante para se obter
DNA em qualidade e quantidade necessários para manipulação por técnicas moleculares.
A otimização do protocolo de purificação de DNA de solo baseado em Yeates
et al
.
(1998), foi feita acrescentando-se uma etapa de precipitação do DNA com PEG 8000, que pode
ser usado ao invés da precipitação com etanol ou isopropanol, pois, a precipitação com etanol
favorece a precipitação de ácidos húmicos e diminui a quantidade de DNA recuperado
(Porteous
et al
., 1994; Xia
et al
., 1995). Portanto, baseado na literatura e em testes empíricos,
foi escolhido fazer uma precipitação inicial com isopropanol para remoção dos contaminantes
seguida da precipitação com PEG 8000. Mesmo com diminuição da quantidade final de DNA
extraído, a pureza mostrou-se mais satisfatória e a quantidade de DNA recuperado ainda foi
grande, como foi avaliado através da análise em espectofotômetro, PCR e digestão com enzimas
de restrição.
com clorofórmio: álcool isoamílico na proporção 24:1. Para finalizar o processo de purificação,
utilizou-se purificação com mini-colunas, utilizando-se o kit UltraCleanTM15 DNA Purification
(MOBIO Laboratories Inc.).
Vários autores têm escolhido fazer a purificação utilizando-se gel de eletroforese
seguido da utilização de mini-colunas ou gel de eletroforese e um micro-concentrador para que
o DNA estivesse puro o suficiente para a realização de PCR e digestão com enzimas de restrição
(Zhou
et al
., 1996; Boivin-Jahns
et al
., 1996). Mas, durante os nossos testes, notamos que esses
passos de purificação em gel de eletroforese fazem com que haja grande perda de DNA.
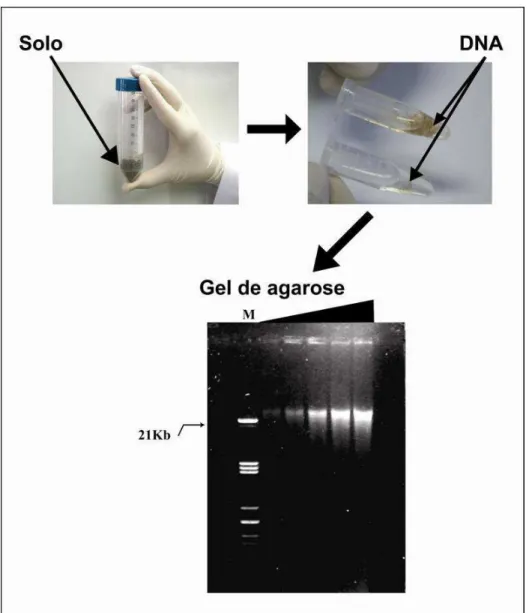
Figura 7: Resultado da extração de DNA. 1° foto: solo a ser extraído, 2° foto: DNA precipitado com isopropanol e 3° foto: gel de agarose mostrando a quantidade e qualidade do DNA extraído. (Poço 1: Marcador 1kb plus ladder; poço 2, 3, 4, 5, 6:; 1 µl, 2 µl, 3 µl, 4 µl e 6 µl de DNA de solo, respectivamente). Foto tirada no laboratório de Biotecnologia da UCB.
5.2- Construção da Biblioteca 16S rDNA
Para o conjunto de
primers
8F/1391R foram obtidas bandas de amplificação de
aproximadamente 1,3 Kb e para o conjunto de
primers
COM1F/COM2R foram obtidas bandas
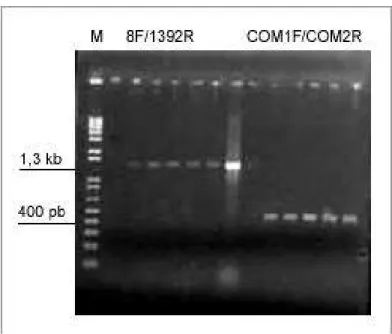
reações os controles negativos não foram amplificados. Um gel característico é mostrado nas
figuras 8 e 9.
Figura 8: Gel contendo os produtos de amplificação do DNA de solo nativo utilizando os dois conjuntos de primers, após purificação. Poço 1(M): marcador 1 kb plus ladder; poço 3: controle negativo; poços 4 a 6 : amplificação com primers 8F/1392R; poço 8: controle negativo; poços 09 a 11: amplificação com primers COM1F/COM2R .
Da clonagem destes fragmentos em vetor de clonagem pGEM-T foram obtidas 200
colônias contendo fragmento amplificado de solo de cerrado
stricto
sensu
com cada conjunto de
primers
de
Eubacterium
(8F/1391R e COM1F/COM2R) e 200 colônias contendo fragmento
amplificado de solo de cerrado
stricto
sensu
com
primers
de
Archea
, totalizando 600 colônias.
Para o solo de pastagem não foi possível obter a mesma quantidade de colônias, por
limitação do vetor de clonagem TOPO, que não apresentou a mesma eficiência com fragmentos
maiores que 1 kb, gerando somente clones de fragmentos de 400 kb, originados da amplificação
com o conjunto de
primers
COM1F/COM2R. Para estes fragmentos, foram colhidas 100
colônias para análise.
Foram construídas, então, 4 bibliotecas, sendo 2 bibliotecas de
Eubacterium
(
primers
8F/1391R e COM1F/COM2R) e uma biblioteca de
Archea
de solos de cerrado
stricto sensu
e
uma biblioteca de
Eubacterium
(
primers
COM1F/COM2R) de solo de pastagem.
Para confirmação da inserção dos fragmentos no vetor de clonagem, foram realizadas
reações de PCR utilizando-se
primers
para a região flanqueadora, como descrito anteriormente,
confirmando a presença dos fragmentos nos tamanhos desejados. Um gel característico é
mostrado na figura 10.
5.3- Análise das Seqüências
Na análise das 373 seqüências feitas pelo programa de Bioinformática da Universidade
Católica de Brasília, 42 seqüências foram descartadas por não apresentarem boa qualidade de
seqüenciamento.
Após análise das seqüências para a detecção de quimeras foram descartadas 12
seqüências quiméricas num total de 319 seqüências analisadas. As seqüências obtidas foram
comparadas entre si e nenhuma seqüência repetida foi encontrada.
5.4- Análise de Diversidade Microbiana
As seqüências amplificadas por PCR diretamente de DNA de solo apresentaram uma
vasta diversidade de grupos filogenéticos bacterianos. Isto foi concluído a partir do fato de que
dentre as 319 seqüências analisadas não foram encontradas seqüências redundantes.
Foram criadas bibliotecas de cerrado
stricto sensu
utilizando-se
primers
tanto para
Eubactérias
(COM1F/COM2R e 8F/1392R), como para
Archea
(Arch341IF/1492R). Para solos
de pastagem foi construída uma biblioteca de
Eubactéria
utilizando-se os
primers
COM1F/COM2R.
Em todas as bibliotecas de cerrado
stricto sensu
criadas, o maior número de seqüências
identificadas foi de microrganismos não-cultiváveis ou não-conhecidos. Eles representaram
29% das seqüências de
Eubactérias
de cerrado
stricto sensu
(figuras 11 e 13)
e 46% das
seqüências em amplificações utilizando-se
primers
para
Archea
(figura 14). Na biblioteca de
Eubactérias
de pastagem, este número foi menor (21%) (figura 12). Esta distribuição mostrou
Através da análise da diversidade microbiana presente nos dois tipos de solo (cerrado
stricto sensu
e pastagem), com um mesmo conjunto de
primers
(COM1F/COM2R), observou-se
que, nas duas bibliotecas, as seqüências pertencentes ao grupo dos A
ctinomicetos
foi muito
abundante, representando cerca de 20 a 30% das seqüências rDNA 16S amplificado por PCR
encontradas.
Outra divisão representativa nestes dois tipos de solo foi de
Acidobactérias
, que
representaram cerca de 10 a 20% das seqüências, uma divisão largamente encontrada em
análises de amostras ambientais marinhas, terrestres, animais, aerossóis e fontes termais. Este
grupo é um exemplo de um grande grupo de microrganismos que somente se tornou conhecido
após a utilização de análises de seqüências de rDNA (Barns
et al
., 1999). As
Acidobactérias
foram primeiramente identificadas na análise das seqüências de solo uma floresta nos Estados
Unidos, quando o grupo de Kuske
et al
. (1997) identificaram que, das seqüências encontradas,
um grande número destas se agrupavam como organismos desconhecidos, onde o único
membro conhecido era o
Acidobacterium capsulatum,
uma bactéria cultivável presente no solo.
A partir daí, muitas das seqüências até então consideradas desconhecidas, foram também
agrupadas dentro deste grande grupo.
Outro grande grupo isolado, tanto em solo de cerrado
stricto sensu
, quanto em pastagem,
foi o de
Proteobactérias
, sendo a subdivisão mais abundante a de
alfa-proteobactérias,
22% em
cerrado
stricto sensu
e 17% em solo de pastagem. Estas seqüências ocorreram com a mesma
freqüência que em outros estudos de solo. Em estudos de análises de comunidades bacterianas
em solos de manejado para pastagem na Grã-Bretanha, McCaig
et al
. (1999) observou que a
maioria de suas seqüências 16S rDNA eram provenientes de
alfa-proteobactéria
(22 a 53%). No
número de
alfa-proteobacterias
ainda menor (4%). Estas diferenças podem ter ocorrido por
vários fatores, entre eles o conjunto de
primers
escolhidos, método de extração do DNA do solo,
a região e época de colheita de solo e número de clones testados.
Entre as seqüências de
alfa-proteobactérias
, em solos de cerrado
stricto sensu
, foram
identificadas quatro seqüências de
Bradyrhizobium sp.
, bactérias fixadoras de nitrogênio. Entre
as seqüências de
beta-proteobactérias
, uma seqüência de
Bulkholderia sp.
foi identificada.
Clones desta bactéria com atividade antimicrobiana foram identificados em solo e zonas de
inibição de crescimento foram observadas quando estes clones eram cultivados em placas
contendo outras bactérias, fungos e leveduras (Cain
et al
., 2000).
Além da subdivisão
alfa e
beta-proteobactérias
, alguns exemplares da subdivisão
gama
e
delta
também foram
identificados. Entre as seqüências de
delta-proteobactérias
encontradas destaca-se uma
seqüência de
Byssophaga cruenta
, uma mixobacteria que degrada celulose (Reichenbach
et al
,
2004).
Em estudos de ambientes aquáticos, Beja
et al
. (2000) identificaram 16S rDNA de uma
bactéria pertencente ao grupo de
gama-proteobactérias
. A seqüência flanqueadora do DNA
revelou um gene similar à bacteriorodopsina, cujo produto é um foto-receptor encontrado, até
então, exclusivamente em
Archea
, tem se mostrado ser abundante entre
proteobactérias
(Handelsman, 2004).
Outro grande grupo encontrado em solo de cerrado
stricto sensu
foi o de
Verrucomicrobia
(6%) (figura 13). Este grupo da mesma forma que os demais tem
freqüentemente sido isolado em amostras de solo de diferentes áreas (Dunbar
et al
., 1999;
Sessitsch
et al
., 2001). Apesar deste grupo não ter sido isolado em solo de pastagem em nosso
específicos (O'Farrell & Janssen, 1999). Isto pode ter sido causado pelo tamanho da amostragem
ou pela diferença nos
primers
utilizados.
Utilizando-se o
primer
para amplificação de
Archea
Arch341IF e o
primer
universal
1492R em amplificações de DNA de solo de cerrado
stricto sensu
, foi observado o isolamento
de somente um exemplar do reino
Crenarchaeota
, pertencente ao domínio
Archea
(figura 14).
Isto pode ter ocorrido pelo fato de que, predominatemente, microrganismos
Archea
têm sido
isolados de ambientes extremos ou de nichos ecológicos muito especializados. Em estudos de
ambientes marinhos, microrganismos
Archea
podem constituir até 34% da população
procariota, mas o solo pode ser considerado um ambiente inóspito para este grupo de
microrganismo (Amann
et al
., 1995; DeLong
et al
., 1994). Ainda assim, alguns microrganismos
extremófilos puderam ser isolados no solo de cerrado
stricto sensu
, entre eles,
Thermobrachium
sp
., um termófilo, alcalino tolerante e anaeróbio obrigatório que havia sido isolado
anteriormente em ambientes geotermais, como fontes termais e habitats com elevadas
temperaturas (Engle
et al
., 1996). Sua característica mais interessante é o fato de serem
microrganismos proteolíticos. Todos os membros cultiváveis do reino
Crenarchaeota
são
termófilos. O outro reino de
Archea,
Euryarchaeota
, é composto de halófitos extremos,
heterótrofos termofílicos e metanogênios (Bintrim
et al
., 1997). Deste reino, nenhuma seqüência
Cerrado sensu stri ctu (COM1F/COM2R)
Acidobacteria 12%
Actinomicetos 17%
Alpha_proteobacterium 22%
Beta_proteobacteria 2% Chloroflexi
3% Delta_proteobacterium
3%
Planctomicetos 3%
Verrucomicrobia 2%
Proteobacterium 3% Gamma_proteobacteria
2%
Fibrobacteria 3%
bactérias desconhecidas ou não cultiváveis
28%
Cerrado Pastagem (COM1F/COM2R)
Acidobacteria 19%
Beta_proteobacterium 8%
cloroflexi 1% Delta_proteobacterium
1%
Planctomicetos 4%
gamma_proteobactéria 1%
Fibrobacteria 1%
bactérias desconhecidas ou não cultiváveis
21%
Alpha_proteobacterium 17%
Actinomicetos 29%
Cerrado sensu strictu (8F/1392R)
Acidobacteria 26%
Chloroflexi 3% Delta_proteobacterium
1% bactérias desconhecidas ou não
cultiváveis 29%
Gamma_proteobacterium 1%
planctomicetos 1%
Verrucomicrobia 6%
Beta_proteobactéria 1%
Alpha_proteobacterium 19%
Actinomicetos 13%
Archea cerrado sensu strictu
Crenarchaeote 1%
Verrucomicrobia 3%
Gram-positive_bacteria 1%
Proteobacterium 3%
bactérias desconhecidas ou não cultiváveis
46%
Firmicutes 10% Spirochete
4%
Gamma_proteobacterium 3%
Alpha_proteobacterium 7%
Delta_proteobacterium 3%
Beta_ proteobacterium 3%
Cloroflexi 4%
Actinomicetos 4%
Acidobacteria 8%