First Edition: October 2012 Second Edition: December 2012
Front cover
By Cláudia S. F. Queiroga
ITQB-UNL/IBET Animal Cell Technology Unit Instituto de Tecnologia Química e Biológica/ Instituto de Biologia Experimental e Tecnológica Av. Da República EAN, 2780-157 Oeiras, Portugal Phone: +351 21 446 91 00; Fax:+351 21 442 11 61 http://tca.itqb.unl.pt
http://www.itqb.unl.pt http://www.ibet.pt
Copyright © 2012 by Cláudia S. F. Queiroga All Rights Reserved
From left to right: Prof. Carlos C. Romão, Dr. Roberto Motterlini, Prof. Cláudia N. Santos, Cláudia S.F. Queiroga, Dr. Helena L.A. Vieira, Prof. Sílvia V. Conde and Dr. Paula M. Alves
Supervisors
Dr. Helena L.A. Vieira, Head of the Biology of Cytoprotection Laboratory, Chronic Diseases Research Center (CEDOC), Faculdade de Ciências Médicas (FCM), Universidade Nova de Lisboa (UNL) (supervisor).
Dr. Paula Marques Alves, Principal Investigator and Head of the Animal Cell Technology Unit at Instituto de Tecnologia Química e Biológica (ITQB), UNL, and Executive Director of Instituto de Biologia Experimental e Tecnológica (IBET), Oeiras, Portugal (co-supervisor).
Jury
Dr. Roberto Motterlini, INSERM U955, Faculty of Medicine, Université Paris Est (Paris XII), France.
Professor Carlos B. Duarte, Head of Neuronal Cell Death and Neuroprotection Laboratory and Associate Professor, Faculdade de Ciências e Tecnologia, Universidade de Coimbra, Portugal.
Professor Sílvia V. Conde, Pharmacology Department, FCM-UNL, Portugal.
The present thesis dissertation is the result of more than four years of research at the Animal Cell Technology Unit of Instituto de Tecnologia Química e Biológica – Universidade Nova de Lisboa and Instituto de Biologia Experimental e Tecnológica (Oeiras, Portugal) and at the Biology of Cytoprotection Laboratory at Centro de Estudos de Doenças Crónicas – Faculdade de Ciências Médicas, Universidade Nova de Lisboa (Oeiras, Portugal), under the supervision of Dr. Helena L.A. Vieira and Dr. Paula M. Alves.
helped me during this thesis. I would also like to acknowledge the good conditions of the institutes where I worked: ITQB, IBET and CEDOC.
To my supervisor, Dr. Helena Vieira, it is a pleasure doing science with you. You taught me many different things without invading my space to grow. Also, in the personal side, there are some special people that we have the pleasure to find few times in our lives. In my life, you are one of them. Two feet give a better walk and the result of such good friendship is so many serious, scary and laughing stories, adventures, jokes, trips, cryings, scientific divagations almost philosophic, beers, orange hair, all the mental spirals. The Talking Cricket only exists because Pinnochio is in a search of courage, lealty and honesty. You don’t need to search it no more! To my co-supervisor, Dr. Paula Alves, for the excellent example of hard-working and for inspiring the others to do always more and better. Leading a team as Animal Cell Technology Unit can only be achieved by a true leader.
To all Animal Cell Technology Unit team, especially to the friends I was lucky to find. For sharing all the lab frustrations maintaining the sanity when no one else is seeing it! Sofia L. for the inspiration for the life and the romantic side, a little star of light and
energy to live in a roller coaster; Sofia A. for all the “nothing” conversations, text
messages, work, existencial doubts, cientific discussions, shared papers, parties, confessions, cryings and shakra calibrations; Tiago for all our moments as the morning coffees; Rita for all the sharings; Cristina, for all the warm reception after we
moved to CEDOC; Pedro, for all the “engenheirices”, “almocinhos”, “brigadeiros”, “bons bocados” e muppet music in the car; to the Italian committee, Marco and Zé, for all the laughing.
The CO-team, which started really small (nano-clusters): Raquel, Rita, Sara, for all the scientific discussions and for the good environment in the lab, so important for our brain.
Romão for allowing the access to carbon monoxide gas.
To the financial support provided by Fundação para a Ciência e Tecnologia (SFRH/BD/43387/2008).
To my FCUL friends, in particular Ana Maria and Pedro, some things never change. To all my ITQB/InTerQB friends. To Neuza for all the conversations that started as group works and presentations but evolved to conversations about personal life and personal decisions. Lia, Licas, Damas, Gonçalo, Rui, Bárbara, Margarida and all the others that I am not mentioning, our group is a great way to relax through laugh therapy.
Ao João e Sérgio pela inquestionável amizade, suportando o tempo, a distância, confusões, mudança de prioridades na vida e ajudando nas vicissitudes; Carlos e família pela intimidade criada, ajudando nos momentos complicados; Judas, Tony, Filipa, Cátia, Rita, Marta, Joana, Ana, Vítor e tantos outros; Daniela, Mário, Cuco, Susana, Magda, Tina, Guedes, Joi, Nuno por me darem a juventude em semanas e dias complicados e cansativos, são vocês a verdadeira fonte da minha jeunesse! Ao Sérgio, pela boa surpresa numa altura que nada fazia prever. Os desígnios de Deus são bons e inquestionáveis e acredito que Ele nos guarda muita coisa boa. A toda a minha extensa família, que me leva até à Lixa do Alvão por ser um
verdadeiro interruptor “off” para descansar, e “on” para as verdadeiras prioridades;
Perinatal complications are a serious clinical problem, in particular hypoxic-ischemic (HI) episodes, which are caused by birth asphyxia or uterine and fetal blood flow interruption. HI corresponds to 23% of all neonatal deaths worldwide, being one of the top 20 leading causes of burden of disease in all age groups (source: http://www.who.int/en/). Recent progress in neonatology has contributed to reductions in mortality rates. Nevertheless, there is still high neurological morbidity related to HI accounting for significant disability. Therefore, there is an urgent need for investigation in this field.
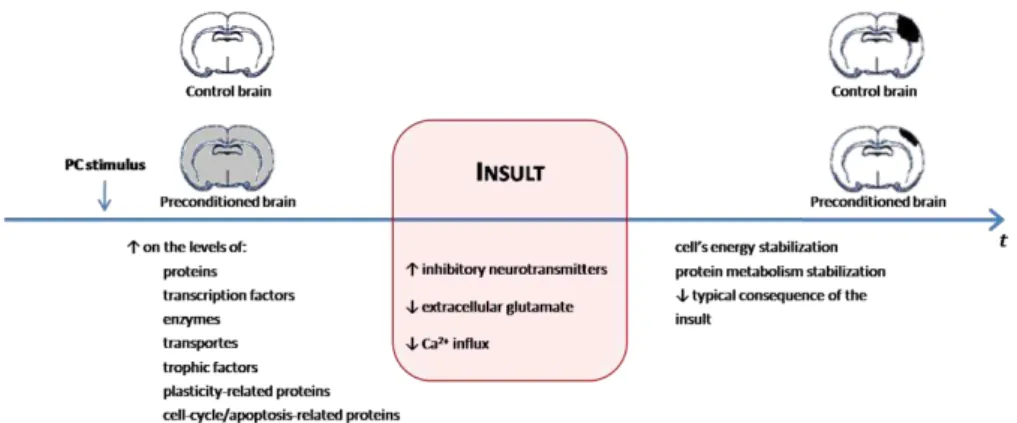
Preconditioning (PC) is defined as stimulation below the threshold of injury that activates endogenous protective mechanisms and prevents tissue and cellular damage. Because PC is a physiological process, the organism fully adapts to low doses of carbon monoxide (CO), an endogenously produced molecule that induces the process and confers protection in several systems.
The aim of this PhD project was to evaluate the role of CO in the pathological model of cerebral ischemia. Cellular mechanisms involved in CO-induced cytoprotection were also studied using different cerebral models.
Chapter I introduces some general concepts of the brain field, constitution and general functional pathways. Particular attention is given to the key role of mitochondria in the variety of cell death mechanisms occurring after cerebral HI episodes. Additionally, the advantages of PC induction as well as the potential clinical importance of CO are addressed.
beta-carotene, an antioxidant molecule, revealing the key signalling role of reactive
oxygen species (ROS) in CO’s biological functions.
The CO effect in mitochondria is not limited to MMP prevention. Chapter III describes how CO improved cellular metabolism and reinforced oxidative phosphorylation by: (i) increasing ATP generation, (ii) augmenting O2 consumption
and (iii) decreasing the ratio between glucose consumption and lactate production. Moreover, CO increased cytochrome c oxidase (COX) specific activity and mitochondrial biogenesis. Silencing Bcl-2 expression by siRNA transfection reverted CO effects, showing that Bcl-2 cytoprotective property also involves cell metabolism modulation.
In Chapter IV a model of primary co-culture of neurons and astrocytes (2D) is used to explore the non-cell autonomous role of CO, namely in astrocyte-neuron communication. It is demonstrated that CO-pre-treated astrocytes were more efficient at reducing neuronal cell death than control astrocytes. CO stimulated purinergic signalling from astrocytes to neurons, by releasing adenosine and/or ATP, which activated neuronal A2A receptors and, in turn, transactivates TrkB receptors,
finally promoting neuronal survival.
After exploring CO biological functions in organelle and cellular models, a systemic approach, using animal models, is reported in Chapter V. In a neonatal brain HI rat model, CO was tested as a PC inducer for promoting cell death inhibition. In the brains of rat pups exposed to 250 ppm of CO before the cerebral HI induction, there was (i) apoptotic nuclei profiles reduction, (ii) caspase-3 activation decline, (iii) Bcl-2 expression increment and (iv) reduction on cytochrome c release from mitochondria.
Complicações perinatais são um problema clínico sério, em particular os episódios de hipóxia-isquémia (HI), causados por asfixia durante o nascimento ou por interrupção sanguínea no útero. HI corresponde a 23% do total mundial de mortes neonatais, sendo uma das 20 causas de morbilidade em todas os grupos etários (fonte: http://www.who.int/en/). Progressos recentes na neonatologia têm contribuído para uma redução na taxa de mortalidade. No entanto, existe uma elevada morbilidade neurológica relacionada com HI, aumentando a incapacidade mental e física. Assim, há uma necessidade crescente de investigação nesta área.
O pré-condicionamento (PC) é definido como um estímulo abaixo do limite de dano que activa os mecanismos endógeneos protectores prevenindo o dano tecidular e celular. Como o PC é um processo fisiológico, o organismo adapta-se totalmente a baixas concentrações de monóxido de carbono (CO), uma molécula produzido endogenamente que inicia o processo protector e a protecção em diferentes sistemas.
O objectivo deste projecto de doutoramento foi avaliar o papel do CO no modelo patológico de isquémia cerebral. Mecanismos celulares envolvidos na citoprotecção induzido pelo CO foram também estudados, usando diferentes modelos cerebrais.
O Capítulo I introduz alguns conceitos gerais no campo do cérebro, a sua constituição e vias de funcionamento geral. Foi dada uma atenção particular ao papel central da mitocôndria nos vários mecanismos de morte celular que ocorrem após episódios de HI cerebrais. Em adição, as vantagens da indução do PC, assim como a potencial relevância clínica do CO foram também contempladas.
mitocondriais (MMP) e a apoptose. Estes processos induzidos pelo CO são revertidos na presença do beta-caroteno, uma molécula antioxidante, revelando o papel fulcral das espécies reactivas de oxigénio (ROS) na sinalização das funções biológicas do CO.
O efeito do CO na mitocôndria não é limitado à prevenção da MMP. O capítulo III descreve como o CO melhora o metabolismo celular e reforça a fosforilação oxidative: (i) aumenta a producção de ATP, (ii) aumenta o consumo de O2 e (iii)
diminui o rácio entre o consumo de glucose e produção de lactato. Em adição, o CO aumenta a actividade específica da citocromo c oxidase (COX) e a biogénese mitocondrial. O silenciamento da expressão de Bcl-2 por transfecção com siRNA reverteu os efeitos do CO, mostrando que a propriedade citoprotectora da Bcl-2 também envolve modelação do metabolismo celular.
No Capítulo IV, um modelo 2D de culturas primárias de neurónios e astrócitos foi usado para explorar o papel celular não-autónomo do CO, especialmente na comunicação astrócito-neurónio. É demonstrado que os astrócitos pré-tratados com CO foram mais eficientes a reduzir a morte neuronal do que os astrócitos controlo. O CO estimula a sinalização purinérgica dos astrócitos para os neurónios, através da libertação de adenosina e/ou ATP, que activam os receptores neuronais A2A que, por
1. Queiroga C.S.F., Almeida A.S., Martel C., Brenner-Jan C., Alves P.M. and
Vieira H.L.A., “Glutathionylation of adenine nucleotide translocase induced by
carbon monoxide prevents mitochondrial membrane permeabilization and
apoptosis”, Journal of Biological Chemistry 2010 May 28;285(22):17077-88.
2. Almeida A.S., Queiroga C.S.F., Sousa M.F., Alves P.M. and Vieira H.L.A.,
“Carbon monoxide modulates apoptosis by reinforcing oxidative metabolism in
astrocytes: role of Bcl-2”, Journal of Biological Chemistry 2012 Feb 13; 287(14): 10761-10770.
3. Queiroga C.S.F, Tomasi S., Widoroe M., Alves P.M., Vercelli A. and Vieira
H.L.A., “Preconditioning triggered by carbon monoxide (CO) provides neuronal
Abbreviation Full form
2-VP 2-vinylpiridine
αβmeATP α,β-methyleneadenosine 5’-triphosphate lithium salt AGA 18α-glycyrrhetinic acid
AIF Apoptosis-Inducing Factor
ANT Adenine Nucleotide Translocase
AK Adenylate Kinase
Ap5A P1, P5-diadenosine-5’-pentaphosphate Apaf-1 Apoptotic Protease Activating Factor 1
ARL ARL67156
BBB Blood-Brain Barrier
Bcl-2 B-cell lymphoma-2
BCNU Carmustine
BCSFB Blood-Cerebrospinal Fluid Barrier BDNF Brain-Derived Neurotrophic Factor
BME Basal Medium Eagle’s
CCA Common Carotid Artery
CL Contralateral
CNS Central Nervous System
CO Carbon Monoxide
CORM’s Carbon Monoxide Releasing Molecules
COX Cytochrome c Oxidase
CsA Cyclosporine A
CSF Cerebrospinal Fluid
CTL Cytotoxic T Lymphocytes
Cyp D Cyclophilin D
DG Dentate Gyrus
DMEM Dulbecco’s Minimum Essential Medium DPCPX 8-cyclopentyl-1,3-dipropylxanthine DTNB 5, 5’-dithio-bis 2-nitrobenzoic acid
EA Ethacrynic Acid
EAA Excitatory Aminoacids
EPO Erythropoeitin
FBS Fetal Bovine Serum
GFAP Glial Fibrillary Acidic Protein
GSSG Oxidized Glutathione
GST Glutathione S-Transferase
HIF Hypoxia Inducing Factor
HIR Hypoxia-Ischemia and Reperfusion
HO Heme-Oxygenase
Hoe Hoechst 33342
IAP Inhibitor of Apoptotic Proteins ICAD Inhibitor of Caspase Activated DNAse
IL Ipsilateral
JAK Janus Kinase
MMP Mitochondrial Membrane Permeabilization NBTI S-(ρ-nitrobenzyl)-6-thioinosine
NDS Normal Donkey Serum
NO Nitric Oxide
NOS Nitric Oxidase Synthase
NSC Neural Stem Cells
ONOO- Peroxinitrite
PBS Phosphate Buffered Saline PARP-1 Poly (ADP-ribose) polymerase-1
PC Preconditioning
PI Propidium Iodide
PNS Peripheral Nervous System
PTP Permeability Transition Pore
ROS Reactive Oxygen Species
RT Room Temperature
RT-qPCR Real Time-quantitative Polymerase Chain Reaction
SCH SCH58261
SD Standard Deviation
sGC Soluble Guanilyl Cyclase
SOD Superoxidase Dismutase
STAT Signal Transducer and Activator of Transcription t-BHP tert-butyl hydroperoxide
TIAs Transient Ischaemic Attacks
TNB 5-thio-2-nitrobenzoate
TNF Tumour Necrosis Factor
tPA Tissue-Plasminogen Activator
Figure Page Legend Title
1.1. 4 Eschematic representation of the (A) human and (B) rat brain. 1.2. 5 Brain barriers.
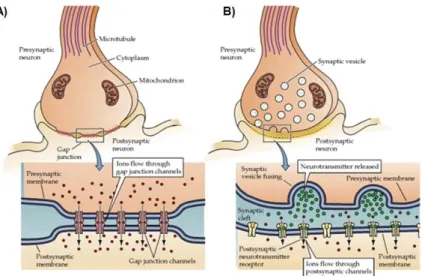
1.3. 6 Eschematic representation of a typical neuronal structure. 1.4. 7 Synapse types: (A) electrical and (B) chemical.
1.5. 8 Schematic representation of several brain cell populations. 1.6. 11 Schematic representation of a slice of the human brain. 1.7. 14 Apoptotic pathways.
1.8. 16 Regulated non-apoptotic cell death. 1.9. 22 Preconditioning and mitochondria.
1.10. 23 General preconditioning mechanism in the brain.
1.11. 24 Heme group metabolization by the enzyme heme-oxygenase (HO). 1.12. 31 Main questions and systems of this thesis.
2.1. 57
Carbon monoxide at 100 μM does not confer protection against astrocytic apoptosis but 50 μM of CO confers protection against apoptosis in delayed periods of time.
2.2. 58 Carbon monoxide confers protection against apoptosis. 2.3. 60 Relevance of ROS on carbon monoxide protective role.
2.4. 62
Carbon monoxide effect on the mitochondrial membrane depolarization, inner membrane permeabilisation, mitochondrial swelling and cytochrome c release.
2.5. 64 Influence of ROS on CO effect at mitochondrial level.
2.6. 65 Carbon monoxide effect on ADP/ATP translocase activity of ANT. 2.7. 66 Carbon monoxide effect on mitochondrial GSSG/GSH ratio. 2.8. 69 Role of ANT glutathionylation in MMP modulation.
2.9. 70 Effect of CO in ANT-GST interaction.
3.1. 94 Carbon monoxide confers protection against apoptosis. 3.2. 95 CO increases ATP generation in astrocytes.
3.3. 97 Effect of CO on ATP production and protection against cell death under glycolysis limiting conditions.
3.4. 99 Effect of CO on cytochrome c oxidase activity and mitochondria biogenesis.
3.5. 101 Role of CO in expression of Bcl-2 and Bcl-2-COX interaction. 3.6. 103 Role of Bcl-2 in CO-induced astrocytic metabolism modulation.
4.1. 123 Representation of experimental time-line.
4.2. 125 Effect of astroglial pre-treatment with carbon monoxide in neuronal apoptosis.
4.3. 126 CO influences ATP and adenosine content in co-culture supernatant.
4.4. 127 Adenosine and αβmeATP protect neurons against cell death. 4.5. 128 Effect of SCH58261 (SCH), suramin
and8-cyclopentyl-1,3-dipropylxanthine (DPCPX)in the CO mechanism.
4.6. 129 Effect of 18α-glycyrrhetinic acid (AGA), S-(ρ -nitrobenzyl)-6-thioinosine (NBTI) and ARL67156 (ARL) in the CO mechanism. 4.7. 130 Effect of K252a in the CO mechanism.
5.1. 149 Experimental groups and time-points schematic representation. 5.2. 153 Stereological measurement of apoptosis.
5.3. 155 Effect of carbon monoxide treatment on neuronal apoptosis. 5.4. 157 Carbon monoxide effect in hippocampus after perinatal
hypoxia-ischemia – apoptotic profiles.
5.5. 158 Carbon monoxide effect in hippocampus after perinatal hypoxia-ischemia – cleaved caspase 3 expression.
5.6. 160
Effect of carbon monoxide on apoptotic markers in hippocampal extracts after 6 and 24 h of HI – protein expression and sub-cellular localization.
Table Page Legend Title
1.1. 18 Mitochondrial Membrane Permeabilization (MMP) consequences in the cellular processes.
1.2. 20 Mitochondrial quality control system levels.
1.3. 21 Molecules intervenients on the preconditioning pathways. 1.4. 26 Protective actions of HO-1 and HO-2.
1.5. 27 Protective actions of CO. 1.6. 29 Protective actions of CORMs.
3.1. 96 Metabolic hallmarks
4.1. 122 List of compounds used to modulate CO effect.
Chapter Page Title
I 1 Introduction
II 41
Carbon Monoxide Direct Effect On Non-Synaptic Mitochondria: mitochondrial membrane permeabilization prevention
III 81 Carbon Monoxide Direct Effect On Non-Synaptic Mitochondria: mitochondrial metabolism reinforcement
IV 113 Carbon Monoxide Non Cell-Autonomous Role: purinergic signalling
V 139 Carbon Monoxide Effect On Perinatal Hypoxia-Ischemia: apoptosis prevention
I
INTRODUCTION
“A major part of brain function in decision-making is the testing of predictions against reality — in essence all people are 'scientists'”
CONTENTS
1. Brain ... 3 1.1. Neuronal Cells ... 5 1.2. Glial Cells... 7 1.3. Cell-to-cell communication ... 8 2. Hypoxia-Ischemia and Reperfusion ... 9 3. Cell Death ... 12 3.1. Apoptosis ... 12 3.2. Necrosis ... 15 3.3. Regulated non-apoptotic cell death ... 15 4. Mitochondria ... 17 4.1. Mitochondrial role in cell metabolism ... 17 4.2. Mitochondrial control of cell death ... 18 4.3. Mitochondrial quality control ... 19 5. Preconditioning ... 20 5.1. Preconditioning in the brain ... 22 6. Carbon Monoxide ... 23 6.1. Heme-oxygenase 1 and the central nervous system ... 25 6.2. Carbon monoxide and the central nervous system ... 27 6.3. Carbon monoxide as therapeutic molecule ... 28 7. Aims and Scope of the Thesis ... 29 8. References ... 32
C
hapter
I
1. BRAIN
The interest in the brain is very ancient, with many theories involving the function of this organ. In 1906, Camillo Golgi and Santiago Ramón y Cajal received the Nobel Prize in Physiology and Medicine for their work in the brain. This prize is a landmark of the beginning of a more systematic era in the field of Neurosciences.
The human brain is part of the central nervous system (CNS), together with the spinal cord, and is an organ of extremes: extreme importance and extreme complexity. It integrates and centralizes the information sensed all over the body and initiates the signaling for the organism to react to it. The sophisticated network allows a rapid response as a result of cooperation between several organs and muscles. Due to the major importance of the brain in the body, its function is preserved by (i) the thick bones of the skull, protecting against traumatic impacts (ii) the suspension in the cerebrospinal fluid (CSF), which acts as a shock absorber and (iii) biological barriers, as blood-brain barrier (BBB), decreasing the risk of infections (Purves et al. 2001). Despite all evolutionary strategies, the brain is still susceptible to several types of damage and disease. Stroke is the first cause for brain injury, constituting along with chronic neurodegeneratives diseases (as Parkinson, Alzheimer and Hungtinton Disease) and the psychiatric disorders an enormous threat with the increasing life expectancy, in particular because the nature of these pathologies is not well understood.
The cerebral areas are represented in Figure 1.1. In order to attribute functions to each brain region electrophysiological and behavioral studies can be performed. However, each area is responsible for more than a single function being its action the integrative communication between several areas. Despite all the knowledge already achieved, the exact correlation is still undisclosed.
Figure 1.1. Schematic representation of the (A) human and (B) rat brain. The organization of the areas in the brain vary according to the species.
There are two barriers, which are key players in the communication between its periphery and the brain (Figure 1.2.). The most described is the BBB (Figure 1.2. – A), which is constituted by the endothelial cells of the blood capillaries together with associated astrocytic end-feet processes and perivascular neurons. As cellular junctions are tight this physical barrier avoids the entrance of toxic factors into the brain and restricts the exchanges between blood and brain, maintaining its normal function. The second barrier is the blood-cerebrospinal fluid barrier (BCSFB, Figure 1.2. – B), constituted by the epithelial cells of the choroid plexus that separate the blood from the CSF (Siegel et al. 1999; Francis et al. 2003).
C
hapter
I
The cellular components in the brain comprise neuronal and glial cells.
Figure 1.2. Brain barriers.(A) The blood-brain barrier (BBB). (Adapted from (Francis et al. 2003)) (B) Blood—Cerebrospinal Fluid Barrier (BCSFB) (Adapted from (Siegel et al. 1999))
1.1. Neuronal Cells
Neurons are polarized cells, highly specialized in the transmission of information. They can be classified according to their function, location, the synthesized and released neurotransmitter, shape and number of extensions from the cell body. The basic structure of a neuron (Figure 1.3.) includes (i) dendrites that receive the information, (ii) the cell body, where the cellular processes occurs and (iii) the axon, which transmits the information to the following element in the network and is coated by myelin to facilitate the action potential conduction velocity. However, neurons are not standardized. The size of the axon together with the ramification and size of the dendrites are co-related with the brain region and function (Purves et al. 2001). Bridging information in the brain is a very specialized neuronal function, and, therefore neurons have lost other cellular abilities, namely presenting limited energy reservoirs and are not able to divide.
circuits and (v) survival (reviewed in Vieira et al. 2011). Nevertheless, neuronal renewal is limited, hence the advance on scientific knowledge on neurodegeneration is of extreme relevance.
Figure 1.3. Eschematic representation of a typical neuronal structure. Adapted from (Morris 1988). Typically, the dendrites receive the information, signaling it to the cell body and the signaling continues through the axon until the following element in the network.
Synapse is the process of interneuronal communication being described as signal transmission (electrical and chemical – Figure 1.4.) between two neurons, a pre-synaptic from which the signal is transmitted and the post-synaptic one that receives the information. The space between them is called synaptic cleft. Nevertheless, nowadays the definition of synapse also includes astrocytes – tripartite synapse (Mirko Santello 2012). These astroglial cells remove the excess of neurotransmitters and recycle some of these factors, avoiding the over-stimulation of neuronal cells, which assures their normal function.
C
hapter
I
amines, excitatory aminoacids, inhibitory aminoacids, purines, gases, lipids and polypeptides (Purves et al. 2001).
Figure 1.4. Synapse types:(A) electrical and (B) chemical. Adapted from (Purves et al. 2001).
1.2. Glial Cells
Astrocytes (from astro, star shaped, and cyte, cell, Figure 1.5.) are the main neuronal suppliers of extracellular matrix proteins, growth factors and recyclers of neurostransmitters in the CNS, establishing an important metabolic cooperation with neurons (there is 1.4 astrocytes per neuron in the human brain (Theodosis et al. 2008)). The extension of numerous processes form highly organized anatomical domains and with little overlap between adjacent cells, in close contact with synapses or blood vessels. The domains communicate through gap junctions, structuring a functional network.
The unique astrocytic phenotype, differential receptors and ion channels expression become these cells as central in the regulation of morphology, proliferation, differentiation, homeostasis and survival of different neuronal subpopulations (Matyash et al. 2010).
Whereas neurons communicate using electric or chemical transmission, astrocytes use gap junction channels that are regulated by extra- and intracellular signals allowing exchange of information (Giaume 2010).
Figure 1.5. Schematic representation of several brain cell populations.
1.3. Cell-to-cell communication
C
hapter
I
glutamate is released from neurons and rapidly removed from the synaptic space by astrocytes. Astrocytic glutamine synthase converts glutamate into glutamine, which is released to synaptic space and uptaken by the neurons. Secondly, astrocytes uptake glucose more efficiently than neurons and after its convertion into lactate, this substrate is released to the synaptic space to be used as an energy source by neurons (astrocyte-neuron lactate shuttle). In addition, astrocytic function also modulates extracellular ionic homeostasis, neurotransmission, glutathione metabolism and aminoacid recycling (Theodosis et al. 2008).
Astrocyte-neuron network is very complex (a single astrocyte enwraps multiple neurons and one neuron interacts with 4-8 astrocytes). The revealing of cell-to-cell communications can only add benefits, in particular in pathologic conditions. More and more astroglial cells arise with an important biological role in neuroinflammation and neuroprotection, namely in Alzheimer Disease, Amyotrophic Lateral Disease and Cerebral Ischemia. Alexander Disease is caused by a mutation in glial fibrillary acidic protein (GFAP) an astrocytic specific gene and the patients present symptoms also related to dysfunction on other brain cells (neurodegeneration and abnormal myelination) (Allaman et al. 2011).
2. HYPOXIA-ISCHEMIA AND REPERFUSION
tissue-type plasminogen activator (tPA) is the one approved protocol for the treatment of cerebral HIR. Still, there are limitations: the short time window and the risk of haemorrhage (Yepes et al. 2008; Yepes et al. 2009). Thus, there is a crucial and urgent need for novel therapies.
When blood flow is re-established (reperfusion), mitochondria can overproduce reactive oxygen species (ROS) exacerbating cell injury by oxidative stress, which can lead to cell death and/or to an increase on the BBB permeability. Likewise, the interruption of oxygen supply favours anaerobic metabolism and lactate production, leading to acidosis. Moreover, the Ca2+ intracellular concentration augments inducing damaging cellular effects: (i) interferes with oxidative phosphorylation, causing mitochondria failure and ATP production decrease; (ii) activates calpain, which degrades the cellular matrix and (iii) activates phospholipase A2, which acts on phospholipids, changing the membrane structure. Modifications in the membrane permeability induce the release of excitatory amino acids (EAA), overstimulating the excitatory receptors, which can induce glutamate excitotoxicity (Guo et al. 2011; Yenari et al. 2012).
C
hapter
I
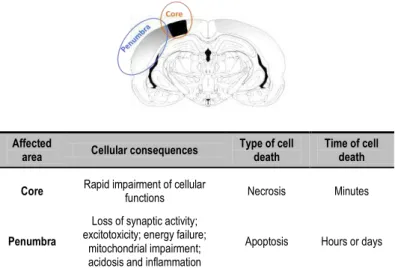
Figure 1.6. Schematic representation of a slice of the human brain. In an hypoxic-ischemic and reperfusion episode, one may consider the formation of distinct brain regions, core, where the more severe consequences occur and penumbra, where cell death is more controlled but with a broader time window.
In newborns cerebral HIR can be due to perinatal complications and is a common cause of damage in the immature brain (1-6 of every 1000 live term birth) (reviewed in Hossain 2008). In addition to the time of blood flow interruption, the lesion in perinatal brain depends on the developmental stage. Although there are still some aspects to unravel, it is accepted that the main molecular players, consequences and protective mechanisms differ between the mature and immature brain (Hossain 2008). The lesion affects determined areas and not the whole brain, however can cause cognitive, sensory and motor disabilities, which increases the importance on the finding of new treatments (Sanders et al. 2010). Hypothermia is an approved protocol for treatment of neonatal ischemia, however there is still an uncertainty on the involved cellular mechanisms.
Affected
area Cellular consequences
Type of cell death
Time of cell death
Core Rapid impairment of cellular
functions Necrosis Minutes
Penumbra
Loss of synaptic activity; excitotoxicity; energy failure;
mitochondrial impairment; acidosis and inflammation
It is accepted that, at least, three types of cell death can occur in HIR episodes: apoptosis, necrosis and/or autophagy, which can be interconnected by several proteins (Carloni et al. 2008). Due to the up-regulation of pro-apoptotic factors in early maturation ages, apoptosis has a major importance in developmental brain.
3. CELL DEATH
Nowadays, the definition of the different cell death types is under a strong scientific debate. One can assume three types of cell death: apoptosis, necrosis and regulated non-apoptotic cell death (autophagy, necroptosis and PARP-1 mediated cell death).
3.1. Apoptosis
Apoptosis is a programmed and energy-dependent form of cell death. The term apoptosis (from Greek, apo – from, off, without, -ptosis – falling) was first described in 1842, but only published in 1972 by Kerr, Wyllie and Currie (Kerr et al. 1972). Sidney Bremer, Robert Horvitz and John E. Sulston received the Nobel Prize in 2002 for their work in apoptosis.
During this tidy regulated process there are several morphological modifications: cell shrinkage, chromatin condensation (pyknosis), DNA and nuclei fragmentation (karyorhexis). Apoptotic bodies are formed (budding), which are packed organelles with the integrity preserved, are engulfed by macrophages, parenchymal or neoplastic cells and degraded within phagolysosomes. Thus, it is a way of removing non-needed, harmed, old or mutated cells without interfering with the remaining ones, with no inflammatory response or damage perpetuation. For the phagocytic cells to recognize the cell fragments, the apoptotic bodies expose phosphatidylserine in the outer leaflet of the plasma membrane – “eat me” signal (Tsujimoto et al. 2006; Elmore 2007; Galluzzi et al. 2007; Wu et al. 2007).
C
hapter
I
great importance to avoid serious defects. The excess of apoptosis can cause atrophy, elimination of healthy cells or difficulty in wound healing. On the other hand, the absence of apoptosis leads to tumour formation, autoimmune, inflammatory and viral diseases (Elmore 2007).
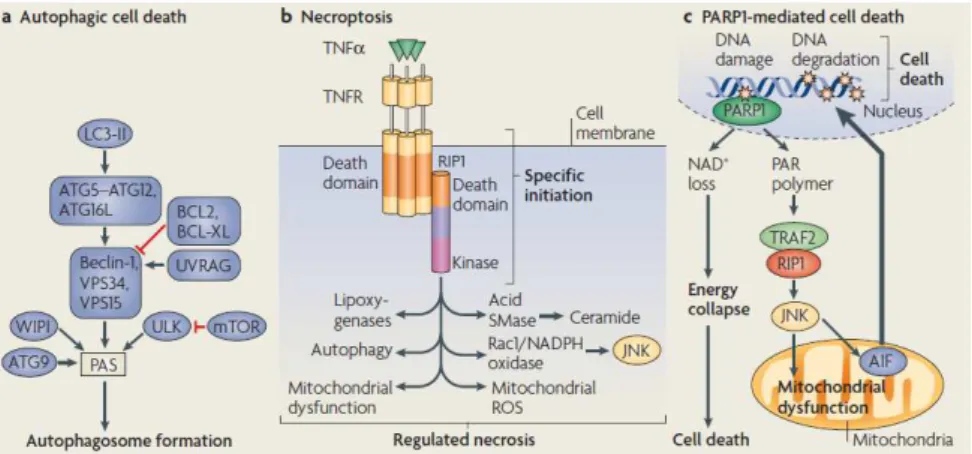
One can distinguish three apoptotic pathways (Figure 1.7.): intrinsic, extrinsic and perforin/granzyme pathways, which differ in the initiator stimuli, however converging in the execution phase (Elmore 2007).
Figure 1.7. Apoptotic pathways. Adapted from (Elmore 2007).
The already described pathways (Figure 1.7.) converge in caspase-3 activation, initiating the execution phase of apoptosis. The executioner caspases (3, 6 and 7) can be activated by any initiator caspase (caspase 2, 4, 8, 9, 10 or 12). Caspase 1, 5 and 11 are considered to be inflammatory ones (Ow et al. 2008).
Caspase-3 has different downstream targets, namely ICAD (Inhibitor of Caspase Activated DNAse) whose cleavage releases its ligand CAD that, after the translocation to the nucleus, degradates chromosomal DNA and causes chromatin condensation. Furthermore, caspase-3 assumes a key role in apoptosis since it is also involved in the cytomorphological changes and in the disintegration of the cell into the apoptotic bodies (Elmore 2007).
C
hapter
I
3.2. Necrosis
Necrosis (from Greek, “dead”) involves plasmatic membrane disruption with the leakage of cytoplasmatic content leading to inflammation and enlarging the lesion (Morse et al. 2009). It is associated with a bioenergetic catastrophe resulting from ATP depletion, with calpains activation, resulting from infection, toxins or trauma (Rami et al. 2008). Calpains are cytosolic proteases Ca2+-dependents required for normal cell function. In models of acute neurodegeneration such as ischemia, traumatic brain injury or epilepsy, calpains are pathologically activated, cleaving neural proteins (Vosler et al. 2008).
3.3. Regulated non-apoptotic cell death
Regulated non-apoptotic cell death types (Figure 1.8.) includes autophagy or type II, necroptosis and PARP1-mediated necrotic cell death (Degterev et al. 2008).
production. However, autophagy can allow apoptosis and delay necrosis by providing energy substrates to the cell or through molecular interconnections with apoptosis (Ginet et al. 2009). Despite the fact that autophagy role and intensity is not well classified, it is clear that it is a crucial process in homeostasis and in response to injury.
Necroptosis is defined as having similar initiator stimuli as apoptosis, however with necrotic morphological features (Fig. 1.8. b). Thirdly, Poly (ADP-ribose) polymerase-1 (PARP1) is a nuclear enzyme responsible for maintaining genome stability. Conversely, the excess of PARP1 activity can initiate caspase-independent cell death pathways. One is driven for a rapid depletion of cytosolic NAD+, which will lead to an energetic catastrophe, culminating in necrotic death. Secondly and associated with acute neuronal injury, in excitatory neuronal cells, PARP1 translocates to the cytosol, and conducts AIF translocation to the nucleus, mediating cell death.
Figure 1.8. Regulated non-apoptotic cell death. Adapted from (Ow et al. 2008)
C
hapter
I
insult (as cerebral HIR) the occurring mechanisms are variable, according to each cellular state. Thus, a combined or adjustable therapy might provide maximal benefit.
4. MITOCHONDRIA
4.1. Mitochondrial role in cell metabolism
Mitochondria are dynamic organelles that have a key cellular role both in metabolism (energy production) and in cell death (MMP).
Cellular energy is mostly produce in the inner mitochondrial membrane by oxidative phosphorylation (80-90% of ATP). This is a very productive metabolic pathway (26 of 30 ATP molecules per 1 glucose molecule) based on electron transfer along an electron transport chain, from Complex I to Complex IV, through oxidation-reduction reactions.
Glycolysis produces metabolites to be used in the tricarboxylic acid cycle, producing compounds to supply the first electronic transporters in the oxidative phosphorylation: NADH and succinate to complex I (NADH ubiquinone reductase) and complex II (succinate dehydrogenase), respectively. The generated electrons move to complex III (ubiquinol-cytochrome c oxidoreductase), and finally to complex IV (cytochrome c oxidase - COX), via cytochrome c, in which O2 is the final electronic acceptor, leading to H2O formation. While electrons are transported from one transporter to the next, H+ ions are pumped to the intermembranar mitochondrial space; leading to the formation of a gradient, maintaining a high transmembranar potential (ΔΨm). ATP is synthesized in the final enzymatic complex, ATP synthase, by driving back protons to the mitochondrial matrix (Nelson et al. 2004). In response to injury, ΔΨm can decrease, the gradient of protons is unbalanced, disturbing ATP production, ion homeostasis and protein transport into mitochondria, which has an impact on cellular functions (Tait et al. 2008).
specie, H2O2. ROS formation can augment in stress conditions, activating others signaling pathways, which can be damaging or protective, according to ROS concentration (Guo et al. 2011). In ischemia-reperfusion episodes and, for preserving ΔΨm, ATP synthase can revert its activity and hydrolises ATP; this is another source of ROS overproduction (Christophe et al. 2006).
4.2. Mitochondrial control of cell death
Mitochondria are key organelles in the cell and their damage can lead to cell death by two means: (i) the release of various pro-apoptotic proteins that are confined to the intermembranar space, after MMP and (ii) due to disruption of the respiratory chain (affecting energy cellular state associated pathways) (Kroemer et al. 2007; Galluzzi et al. 2009). (Table 1.1.)
Table 1.1. Mitochondrial Membrane Permeabilization (MMP) consequences in the cellular processes.
The mitochondrial membrane permeability is regulated via the interaction with pro or anti-apoptotic proteins from Bcl-2 (B-cell lymphoma-2) family. Bcl-2 family members contain Bcl-2 homology domains and can be divided in three classes: apoptosis inhibitors, as Bcl-2 or Bcl-xL, apoptosis inducers, as Bax and Bak, and BH3-only proteins that regulate anti-apoptotic bcl-2 proteins to promote apoptosis, as Bad or Bik (Tait and Green 2008; Youle et al. 2008). Bcl-2 proteins are localized in the endoplasmic reticulum, nucleus and in the outer membrane of mitochondria (Chen et al. 2009; Galluzzi et al. 2009).
The exact mechanism for MMP is still under the focus of intense investigation, with three potential models. One model is based on oligomerization of the
pro-Cell Death-related Cell Metabolism-related ROS overproduction
Release of numerous pro-apoptotic factor (which will activate several proteases and nucleases)
Mitochondrial transmembrane potential dissipation
C
hapter
I
apoptotic protein Bax; in response to death stimulus, translocates from the cytosol into the outer mitochondrial membrane, where there is pore formation and membrane permeabilization. A second model is based on VDAC (voltage dependent anion channel), which is a physiological pore in the outer mitochondrial membrane allowing the passage of solutes up to 5 kDa. Whenever VDAC interacts with Bax, the opening of the pore enlarges with the leakage of factors superior than 5 kDa (such as cytochrome c). Finally, the third model involves the permeabilization of the outer and inner membranes, through the dynamic interaction between VDAC, cyclophilin D and adenine nucleotide translocase (ANT). The complex of proteins is denominated permeability transition pore (PTP) and allows the release of solutes up to 1500 Da. PTP model appears to be the more relevant in ischemia-reperfusion injury (Vieira et al. 2000; Belzacq et al. 2003; Mattson et al. 2003; Hausenloy et al. 2009).
As discussed previously, MMP is considered as the point of no return, since it is the responsible for the activation of caspase dependent and independent apoptotic pathways (section 3).
4.3. Mitochondrial quality control
Table 1.2. Mitochondrial quality control system levels. Based in (Rugarli and Langer 2012).
5. PRECONDITIONING
In 1943, Noble was the first to propose that short periods of global hypoxia can protect the entire mammalian organism and preserve brain energy metabolism during longer periods of hypoxia. However, was only in 1964 that Janoff (Janoff 1964), introduced the term ‘preconditioning’.
The preconditioning (PC) phenomenon has been described as an insult that does not cause damage but induces a cellular protective state (tolerance) against a subsequent and more severe challenge (Kirino 2002; Alkan 2009). It can be an event of early response (tolerance induced in minutes or hours), or late response within days with de novo protein synthesis. PC induction involves (i) a stimuli, as ischemic insult or chemical agent; (ii) the stimuli recognition by the cell, through neurotransmitters, receptors, ion channels and redox-sensitive enzymes; (iii) the transduction of the recognized stimuli with signalling molecules such as ROS, adenosine, caspases and/or transcription factors and (iv) the integration of the previous steps to effect the signal. The effectors are ubiquitous including variations on enzymatic activities or gene transcription (Gidday 2006; Stenzel-Poore et al. 2007; Dirnagl et al. 2008) (Table 1.3). Because the protection results from an endogenous boost, the knowledge of these cellular pathways is of extreme importance for the design of new therapies to induce or reinforce cellular defences.
Intraorganellar Quality Control System
Organellar Quality Control System Within mitochondria a combination of
chaperones and proteases to assure: (i) the correct folding of newly imported proteins;
(ii) protection of the existing proteins from stress;
(iii) degradation of irreversible damaged proteins
Equilibrium between mitochondrial fission and fusion to maintain mitochondrial population:
(i) stress conditions (nutrient deprivation, oxidative stress, impairment of cytosolic protein synthesis) – mitochondrial hyperfusion
C
hapter
I
Table 1.3. Molecules intervenients on the preconditioning pathways. Sensors, the molecules that recognize the PC stimulus; transducers, that signal the induction of a protective state to effectors, in order for the cell accomplish the preconditioning level. Adapted from (Dirnagl et al. 2003; Gidday 2006).
Sensors Transducers Effectors
Neurotransmitters Cytokins Toll-like receptors Ion channels
Redox-sensitive enzymes
Depending on the PC stimulus
ROS MAPKs Adenosine NF-kB PkB/Akt Caspases Transcription factors
Genes transcription Transient or protracted During minutes, hours or days
Another interesting aspect of PC is cross-tolerance. One PC inducer can promote tolerance to another different type of damage. Moreover, the tolerance initiation in one organ has demonstrated to cause protection in a distinct organ (Kirino 2002).
Additionally, patients suffering angina before a myocardial infarction have smaller infarcts and longer survival – transient ischaemic attacks (TIAs, (Dirnagl et al. 2003)). Furthermore, there are already several clinical trials to test the efficacy of PC strategies. One of the most described is erythropoietin (EPO), already used in patients with anemia. EPO is a kidney-derived glycoprotein hormone that acts as (i) erythroid progenitor cell proliferation, (ii) cytoprotective, (iii) a gene target of HIF (hypoxia inducible factor), which is activated in the vast majority of stress episodes and (iv) PC induction increases EPO amounts. The reported protective cascade includes the action of Janus Kinase 2 (JAK 2), Akt (or protein kinase B) and STAT (signal transducer and activator of transcription) (Gidday 2006).
It is also described in heart that transient interruptions in blood flow during early reperfusion (postconditioning), reduced ischemic injury to levels similar to that achieved with PC (Vinten-Johansen et al. 2005) with promising results in clinical trials (Gerczuk et al. 2011).
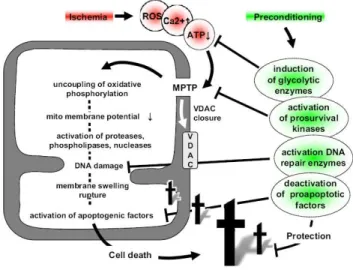
mitochondrial energy metabolism, which all together, increase ATP levels (Galeffi et al. 2000; Dave et al. 2001; Zhang et al. 2003); (ii) in heart, PC cytoprotection includes the inhibition of MMP (Javadov et al. 2000); (iii) PC-induced phophorylation of mitochondrial proteins regulates MMP and apoptotic proteins (Maurer et al. 2006; Zhao et al. 2006; Nishihara et al. 2007); and (iv) PC activates mitochondrial DNA repair (Chen et al. 2003; Li et al. 2006). In summary, mitochondria play a key role in PC regulation and is a possible therapeutic target.
Figure 1.9. Preconditioning and mitochondria. Adapted from (Dirnagl and Meisel 2008).
5.1. Preconditioning in the brain
Neuronal cells present higher energy demand than glial cells, since they conduct excitatibility events. Nevertheless, they present lower energy reservoirs, which position this cellular population as more dependent on mitochondria and on astrocytes (Galluzzi et al. 2009). Thus, neurons are particularly susceptible to HIR; however there are limited therapeutic options to this severe condition, being PC-induction a potential therapy.
C
hapter
I
hypoxic preconditioning during the prenatal period is effective for reducing hypoxic-ischaemic injury after birth (Gidday 2006). Although many aspects need more scientific attention, the cerebral factors can be resumed in Figure 1. 10.
Figure 1.10. General mechanisms of preconditioning in the brain. Adapted from (Dirnagl et al. 2003; Gidday 2006).
There are increasing efforts to translate PC into clinical solutions, because it takes advantage of endogenous cytoprotective mechanisms. For applying PC-based strategies it is necessary at least, partially predictable pathological situations of cerebral ischemia, such as pre-surgery period and in the case of perinatal complications leading to asfixia.
Clinical trials are already approved, most of them based in ischemic PC (clinicaltrials.gov). As examples, “The neuroprotective effect of remote ischemic preconditioning on ischemic cerebral vascular disease” (NCT01321749) and “Remote ischemic preconditioning in subarachnoid hemorrhage (RIPC-SAH)” (NCT01158508).
6. CARBON MONOXIDE
in cells (via cytochrome c oxidase inhibition) (Ahlstrom et al. 2009). Claude Bernard first published an accurate description of the physiology of carbon monoxide poisoning (Bernard 1857).
This gas is formed by heme-oxygenase (HO) activity after heme degradation, along with free iron and biliverdin (a strong antioxidant molecule), rapidly converted into bilirrubin, by biliverdin reductase (Figure 1.11.).
Figure 1.11. Heme group metabolization by the enzyme heme-oxygenase (HO),giving rise to free iron, biliverdin (rapidly converted to bilirubin) and carbon monoxide.
C
hapter
I
Despite the biological functions associated to CO in vivo and the existence of several proteins capable of binding CO in vitro; it is still a matter of discussion the actual physiological targets of CO.
6.1. Heme-oxygenase 1 and the central nervous system
Table 1.4. Protective actions of HO-1 and HO-2.
HO isoform Protective action Model Reference
HO-1
Prevents apoptosis in transplanted organs Transplanted heart Akamatsu et al. 2004
Reduces neuroinflammation Autoimmune
encephalomyelitis Chora et al. 2007
Prevents damage in cerebral ischemia Brain Atkins et al. 1999; Wang et al. 2011
Anti-inflammatory Gastrointestinal Vascular
Durante 2011; Zhu et al. 2011
Prevents development of ischemia-reperfusion injury
Prevents graft rejection
Liver Wang et al. 2011
Attenuates hypertension
Lower blood pressure in established hypertension Vascular Hosick et al. 2012
HO-2 HO-2 deficiency increases brain swelling and inflammation after intracellular hemorrhage Brain Wang et al. 2008
C
hapter
I
6.2. Carbon monoxide and the central nervous system
Carbon monoxide beneficial role has been described in the literature, namely the proliferative in vascular smooth muscle cells (Stanford et al. 2003); anti-inflammatory in lung (Ryter et al. 2007) and heart (Otterbein 2002); protective against organ rejection (Musameh et al. 2007) and HIR injury in various models (Chatterjee 2007; Nakao et al. 2008). Some examples of CO anti-apoptotic effect are reported in endothelial cells (Brouard et al. 2000), pulmonary cells (Otterbein et al. 1999) and muscle (Harder et al. 2008). For review (Ryter et al. 2007) and Table 1.5. However, the protective effect in the brain environment has been poorly described. We previously described that in primary cultures of neuronal cells exogenous CO prevents apoptosis by inducing a PC state (Vieira et al. 2008). Still, CO pathways remain elusive and further studies must be carried to improve the possibility to use this molecule as therapeutic agent.
Table 1.5. Protective actions of CO.
Protective action Model Reference
Prevents apoptosis
Endothelial cells Rat mitochondria
Brouard et al. 2000 Piantadosi et al. 2006 Reduces inflammatory responses and endoplasmatic
reticulum stress Metabolic disorders Joe et al. 2001
Reduces neuroinflammation Brain Chora et al. 2007
Energy maintenance Ischemic myocardium Ahlstrom et al. 2009
Reduces brain injury after transient middle cerebral
artery occlusion Brain Zeynalov et al. 2009
Preventing cerebral injury resulting from cardiac bypass procedures using deep hypothermic circulatory arrest
6.3. Carbon monoxide as therapeutic molecule
CO administered in combination with others strategies may extend the therapeutic window. In particular, CO-induced PC would enhance endogenous properties with clinical benefits, is not invasive and would not require very specialized equipment. Others inhalation therapies have been introduced in clinics (Robinson et al. 2009), for reducing toxicity resulting from metabolization of administered drugs, since drug extraction occurs by exhalation. The therapeutic application of nitric oxide (NO) as vasodilator in several disease models and also in injured lungs of premature and newborn babies is now widely accepted (Bloch et al. 2007). NO is chemically similar to CO; however, unlike CO, NO reacts rapidly with molecular oxygen, and produces peroxynitrite (ONOO-), which is highly reactive. Likewise, noble gases have also been studied for medical applications, especially xenon. Recently, Ryang and co-workers (Ryang et al. 2011) described the efficacy of argon in protecting rat brains in a model of transient middle cerebral artery occlusion. Taken all together, potential CO-inhaled based therapy has the added value of integrating two critical advantages: it is chemically inert compared to NO and is an endogenous molecule compared to noble gases.
C
hapter
I
At present there are two clinical trials phase II on CO gas inhalation-based therapy: for treating patients with intestinal paralysis after colon surgery, for prevention of post-operative ileus (NCT01050712) and for the improvement of tolerability in patients receiving kidney transplants (NCT00531856).
Table 1.6. Protective actions of CORMs.
Protective action Model Reference
Alleviate vascular and immune-related dysfunctions Motterlini et al. 2002
Protects against HI and oxidative stress Cardiac cells (CORM-3)
Clark et al. 2003 Stein et al. 2012
Late PC induction Myocardial infarction
(CORM-3) Stein et al. 2005
Reduces inflammation Microglia
(CORM-3) Bani-Hani et al. 2006
Protects against seizure-induced neonatal vascular injury
Brain
(CORM A1) Zimmermann et al. 2007 Protects against myocardial infarction in
hyperglycaemic rats
Heart
(CORM-3) Filippo et al. 2011 Regulation on mitochondrial respiration Heart
(CORM-3) Lo Iacono et al. 2011
7. AIMS AND SCOPE OF THE THESIS
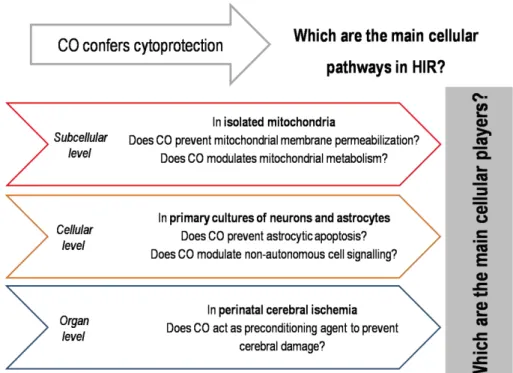
This PhD thesis aims at clarifying the cellular pathways involved in the carbon monoxide (CO) capacity to confer cytoprotection against apoptosis following hypoxia-ischemia and reperfusion (HIR) in the brain. Three different model systems were used to address this objective:
mitochondrial membrane permeabilization (chapter II), mitochondrial metabolism (chapter III) and its upstream events were studied;
(ii) primary monocultures of astrocytes and primary co-cultures of neurons and astrocytes (in vitro) – CO protective action in primary cultures of neurons was already demonstrated (Vieira et al. 2008), thus CO capability to revert cell death was tested in primary cultures of astrocytes (chapter II). Furthermore, approaching with a more physiological model – co-cultures – the relevance of carbon monoxide pre-treatment to cell-to-cell communication and its downstream events were investigated (chapter IV);
(iii) perinatal model of hypoxia-ischemia (HI) in rats (in vivo) – complementing the cellular and subcellular role of CO in vitro, the in vivo performance in order to validate CO as neuroprotector and its influence in anti-apoptotic pathways was pursued. The model used was a perinatal model of hypoxia-ischemia in rat pups (chapter V).
C
hapter
I
8. REFERENCES
Ahlstrom, K., B. Biber, et al. (2009). "Metabolic responses in ischemic myocardium after inhalation of carbon monoxide."Acta Anaesthesiol Scand 53(8): 1036-42.
Akamatsu, Y., M. Haga, et al. (2004). "Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury."Faseb J 18(6): 771-2.
Alkan, T. (2009). "Neuroproctective effects of ischemic tolerance (preconditioning) and postconditioning."Turk Neurosurg 19(4): 406-12.
Allaman, I., M. Belanger, et al. (2011). "Astrocyte-neuron metabolic relationships: for better and for worse."Trends Neurosci 34(2): 76-87.
Atkins, C. M. and J. D. Sweatt (1999). "Reactive oxygen species mediate activity-dependent neuron-glia signaling in output fibers of the hippocampus."J Neurosci 19(17): 7241-8.
Babu, A. N., S. S. Damle, et al. (2007). "Hemoglobin-based oxygen carrier induces hepatic heme oxygenase 1 expression in Kupffer cells."Surgery 142(2): 289-94.
Bani-Hani, M. G., D. Greenstein, et al. (2006). "Modulation of thrombin-induced neuroinflammation in BV-2 microglia by carbon monoxide-releasing molecule 3." J Pharmacol Exp Ther 318(3): 1315-22.
Belzacq, A. S., H. L. Vieira, et al. (2003). "Bcl-2 and Bax modulate adenine nucleotide translocase activity."Cancer Res 63(2): 541-6.
Bernard, C. (1857). "Lecons sur les Effets des Substaces Toxiques et Medicamenteuses." Paris: J-B Bailliere et Fils.
Bloch, K. D., F. Ichinose, et al. (2007). "Inhaled NO as a therapeutic agent."Cardiovasc Res 75(2): 339-48.
Boczkowski, J., J. J. Poderoso, et al. (2006). "CO-metal interaction: Vital signaling from a lethal gas."Trends Biochem Sci 31(11): 614-21.
Brouard, S., L. E. Otterbein, et al. (2000). "Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis."J Exp Med 192(7): 1015-26.
Buchan, A. M. and J. Kennedy (2007). "Strategies for therapy in acute ischemic stroke."Nat Clin Pract Neurol 3(1): 2-3.
C
hapter
I
Carloni, S., G. Buonocore, et al. (2008). "Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury."Neurobiol Dis 32(3): 329-39.
Chatterjee, P. K. (2007). "Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: a comprehensive review." Naunyn Schmiedebergs Arch Pharmacol 376(1-2): 1-43.
Chen, D., M. Minami, et al. (2003). "Upregulation of mitochondrial base-excision repair capability within rat brain after brief ischemia."J Cereb Blood Flow Metab 23(1): 88-98.
Chen, Z. X., R. Velaithan, et al. (2009). "mitoEnergetics and cancer cell fate." Biochim Biophys Acta 1787(5): 462-7.
Chora, A. A., P. Fontoura, et al. (2007). "Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation."J Clin Invest 117(2): 438-47.
Christophe, M. and S. Nicolas (2006). "Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion."Curr Pharm Des 12(6): 739-57.
Clark, J. E., P. Naughton, et al. (2003). "Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule."Circ Res 93(2): e2-8.
Cooper, C. E. and G. C. Brown (2008). "The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance."J Bioenerg Biomembr 40(5): 533-9.
Cosgrove, K. P., C. M. Mazure, et al. (2007). "Evolving knowledge of sex differences in brain structure, function, and chemistry."Biol Psychiatry 62(8): 847-55.
Dave, K. R., I. Saul, et al. (2001). "Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus."J Cereb Blood Flow Metab 21(12): 1401-10.
Degterev, A. and J. Yuan (2008). "Expansion and evolution of cell death programmes." Nat Rev Mol Cell Biol 9(5): 378-90.
Dirnagl, U. and A. Meisel (2008). "Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning?"Neuropharmacology 55(3): 334-44.
Dirnagl, U., R. P. Simon, et al. (2003). "Ischemic tolerance and endogenous neuroprotection." Trends Neurosci 26(5): 248-54.
Dore, S. (2002). "Decreased activity of the antioxidant heme oxygenase enzyme: implications in ischemia and in Alzheimer's disease."Free Radic Biol Med 32(12): 1276-82.
Elmore, S. (2007). "Apoptosis: a review of programmed cell death."Toxicol Pathol 35(4): 495-516.
Filippo, C. D., M. Perretti, et al. (2011). "Acute myocardial infarction in streptozotocin-induced hyperglycaemic rats: protection by a carbon monoxide-releasing molecule (CORM-3)." Naunyn Schmiedebergs Arch Pharmacol 385(2): 137-44.
Francis, K., J. Beek, et al. (2003). "The Blood-brain Barrier (BBB)."Expert Rev Mol Med 5: 1-19.
Galeffi, F., S. Sinnar, et al. (2000). "Diazepam promotes ATP recovery and prevents cytochrome c release in hippocampal slices after in vitro ischemia."J Neurochem 75(3): 1242-9.
Galluzzi, L., K. Blomgren, et al. (2009). "Mitochondrial membrane permeabilization in neuronal injury."Nat Rev Neurosci 10(7): 481-94.
Galluzzi, L., M. C. Maiuri, et al. (2007). "Cell death modalities: classification and pathophysiological implications."Cell Death Differ 14(7): 1237-43.
Gerczuk, P. Z. and R. A. Kloner (2011). "Protecting the heart from ischemia: an update on ischemic and pharmacologic conditioning."Hosp Pract (Minneap) 39(3): 35-43.
Giaume, C. (2010). "Astroglial Wiring is Adding Complexity to Neuroglial Networking."Front Neuroenergetics 2.
Gidday, J. M. (2006). "Cerebral preconditioning and ischaemic tolerance."Nat Rev Neurosci 7(6): 437-48.
Ginet, V., J. Puyal, et al. (2009). "Enhancement of Autophagic Flux after Neonatal Cerebral Hypoxia-Ischemia and Its Region-Specific Relationship to Apoptotic Mechanisms."Am J Pathol.
Grochot-Przeczek, A., J. Dulak, et al. (2012). "Haem oxygenase-1: non-canonical roles in physiology and pathology."Clin Sci (Lond) 122(3): 93-103.
Guo, M. F., J. Z. Yu, et al. (2011). "Mechanisms related to neuron injury and death in cerebral hypoxic ischaemia."Folia Neuropathol 49(2): 78-87.
Harder, Y., M. Amon, et al. (2008). "Ischemia-Induced Up-Regulation of Heme Oxygenase-1 Protects from Apoptotic Cell Death and Tissue Necrosis."J Surg Res.
Hausenloy, D. J., S. B. Ong, et al. (2009). "The mitochondrial permeability transition pore as a target for preconditioning and postconditioning."Basic Res Cardiol 104(2): 189-202.
C
hapter
I
Hossain, M. A. (2008). "Hypoxic-ischemic injury in neonatal brain: involvement of a novel neuronal molecule in neuronal cell death and potential target for neuroprotection."Int J Dev Neurosci 26(1): 93-101.
Hotchkiss, R. S., A. Strasser, et al. (2009). "Cell death."N Engl J Med 361(16): 1570-83.
Janoff, A. (1964). "Alterations in Lysosomes (Intracellular Enzymes) During Shock; Effects of Preconditioning (Tolerance) and Protective Drugs."Int Anesthesiol Clin 2: 251-69.
Javadov, S. A., K. H. Lim, et al. (2000). "Protection of hearts from reperfusion injury by propofol is associated with inhibition of the mitochondrial permeability transition." Cardiovasc Res 45(2): 360-9.
Joe, Y., M. Zheng, et al. (2001). "The role of carbon monoxide in metabolic disease."Ann N Y Acad Sci 1229: 156-61.
Kerr, J. F., A. H. Wyllie, et al. (1972). "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics."Br J Cancer 26(4): 239-57.
Kirino, T. (2002). "Ischemic tolerance."J Cereb Blood Flow Metab 22(11): 1283-96.
Koch, S., R. L. Sacco, et al. (2012). "Preconditioning the Brain: Moving on to the Next Frontier of Neurotherapeutics."Stroke.
Kroemer, G., L. Galluzzi, et al. (2007). "Mitochondrial membrane permeabilization in cell death."Physiol Rev 87(1): 99-163.
Li, W., Y. Luo, et al. (2006). "Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway."J Cereb Blood Flow Metab 26(2): 181-98.
Lo Iacono, L., J. Boczkowski, et al. (2011). "A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species."Free Radic Biol Med 50(11): 1556-64.
Locksley, R. M., N. Killeen, et al. (2001). "The TNF and TNF receptor superfamilies: integrating mammalian biology."Cell 104(4): 487-501.
Mahan, V. L., D. Zurakowski, et al. (2012). "Inhaled carbon monoxide provides cerebral cytoprotection in pigs."PLoS One 7(8): e41982.
Mattson, M. P. and G. Kroemer (2003). "Mitochondria in cell death: novel targets for neuroprotection and cardioprotection."Trends Mol Med 9(5): 196-205.