1
Clínica Universitária de Pediatria
Púrpura de Henoch-Schönlein: uma
entidade benigna? – Reflexão a propósito
de um caso com envolvimento renal
Joana Margarida Esteves Atabão
2
Clínica Universitária de Pediatria
Púrpura de Henoch-Schönlein: uma
entidade benigna? – Reflexão a propósito
de um caso com envolvimento renal
Joana Margarida Esteves Atabão
Orientado por:
António Eduardo Mendes Gil Lopes Figueiredo
3
Resumo
A púrpura de Henoch-Schönlein (PHS) é a vasculite mais frequente em idade pediátrica. É particularmente frequente em rapazes de raça caucasiana e durante os meses frios. Apesar da etiopatogénese ser desconhecida, sabe-se que as imunoglobulinas do tipo A1 (IgA1) têm um papel preponderante na doença, observando-se deposição da molécula nos vasos envolvidos.
Apresenta-se como púrpura e restante clínica variável, consoante os tecidos afetados – envolvendo mais frequentemente as articulações, trato gastrointestinal e rins. O diagnóstico da doença é clínico. Alguns exames complementares de diagnóstico podem ajudar no diagnóstico diferencial de outras vasculites.
A melhor estratégia terapêutica não é consensual. Em apresentações de menor gravidade, a terapêutica é de suporte; em apresentações mais severas - abdominal e renal - são utilizados corticoesteróides. É maioritariamente uma doença benigna, autolimitada, com remissão completa; contudo, quando há envolvimento renal - glomerulonefrite ou síndrome nefrótica - existe maior risco de desenvolvimento de doença renal crónica. É importante o follow-up nestas crianças.
É apresentado o caso clínico de uma criança de 10 anos do sexo masculino, que recorreu ao serviço de urgência por púrpura nos membros inferiores, com progressão para face, tronco e membros superiores. Nos membros inferiores evoluiu de forma exuberante, com formação de bolhas. Desenvolveu uma síndrome nefrótica e nefrítica e necessitou de um internamento prolongado para compensação clínica e otimização terapêutica. Dois dias após a alta, foi reinternado por recidiva da síndrome nefrótica, com edema acentuado na face e membros inferiores, no contexto de uma celulite no dorso dos pés secundária às lesões purpúrica. Foi iniciada antibioticoterapia endovenosa e desbridamento das lesões, tendo alta ao fim de dois dias de internamento.
Este caso permite refletir sobre a benignidade da doença uma vez que condicionou um internamento prolongado, necessidade de corticoterapia e acompanhamento em consulta de nefrologia pediátrica.
Palavras-chave: Púrpura de Henoch-Schönlein; biópsia renal; glomerulonefrite;
4
Abstract
Henoch-Schönlein Purpura (HSP) is the most frequent vasculitis in pediatric age occurring particularly in Caucasian males and during cold months. Though its pathology is not well known, type A1 immunoglobulins (IgA1) play an important role, with visible deposits on the blood vessels involved.
It is presented as purpura and other clinical features depending on affected tissues. It usually affects joints, the gastrointestinal system and kidneys. The diagnosis is based on clinical manifestations; further analysis and exams, as justified, may help in the differential diagnosis of other vasculitis.
There is not a consensus on the best therapeutic strategy. Usually, mild presentation is controlled with support therapy; for more serious presentations with abdominal pain and kidney involvement, corticosteroids are required. On most cases, it is a benign, self-limited disease with full remission; however, when there is kidney involvement, namely glomerulonephritis or nephrotic syndrome, the risk of progression to chronic kidney disease is higher. It is important to follow these children in a long term basis.
A clinical case is presented: a 10-year old boy goes to the emergency room (ER) with purpura in the lower limbs, which progressed to the face, body and upper limbs; skin blebs developed in the lower limbs. The child developed nephrotic and nephritic syndromes and a prolonged hospital-stay was required for clinical stabilization. Two days after discharge, he sought medical care due to worsening edema in the face and lower limbs, and inflammatory signs on skin lesions on the dorsum of both feet. Intravenous antibiotics and debriding of lesions were necessary; he was discharged after 2 days.
The clinical scenario, described above, allows for a reflection on the so-called benign nature of the disease considering the long hospital-stay, corticotherapy and long term follow-up required.
Key-words: Henoch-Schönlein purpura; kidney biopsy; glomerulonephritis; nephrotic
syndrome; corticotherapy
5 Índice Introdução ... 6 Fisiopatologia ... 6 Epidemiologia ... 6 Clínica ... 7
Meios Complementares de Diagnóstico ... 8
Tratamento ... 8 Prognóstico ... 9 Caso Clínico ... 10 Discussão ... 14 Conclusão ... 20 Agradecimentos ... 21 Bibliografia ... 22 Anexos ... 27
6
Introdução
A púrpura de Henoch-Schönlein (PHS), também denominada por vasculite IgA, é uma vasculite leucocitoclástica, não trombocitopénica, caracterizada por depósitos de IgA1 nos pequenos vasos[1][2]. É a vasculite mais frequente em idade pediátrica[3]. Afeta
principalmente pequenos vasos cutâneos, articulares, do trato gastrointestinal e rins[4].
Fisiopatologia
Estão reportadas várias associações entre antigénios, alimentos, fármacos, vacinas, infeções (principalmente do trato respiratório) e aparecimento de PHS [5][6], no entanto, a etiologia é desconhecida [7][8].
A maioria dos casos de PHS é precedida por infeções respiratórias da via aérea superior; os agentes mais comuns são Streptococcus (sobretudo do grupo B – beta hemolítico), Staphylococcus e parainfluenza. No entanto, há vários casos clínicos em que se verificam associações com outros agentes como vírus da hepatite B, adenovírus, VIH e Mycoplasma pneumoniae [2][9].
Pequenos estudos descrevem o aparecimento de PHS após vacinação, nomeadamente contra influenza, sarampo, Streptococcus pneumoniae e Neisseria meningitidis.
Também foram descritas associações com medicação, habitualmente antibióticos. Assim, é difícil diferenciar se o aparecimento tem como causa o fármaco ou a infeção subjacente [2][9].
É considerada uma doença imunomediada, em que as IgA1 têm um papel importante na patogénese[4].
Epidemiologia
Tem uma incidência que varia entre 10-20:100.000 por ano, em diferentes países, sugerindo que o ambiente e a genética têm alguma influência na patogénese da doença[10]. Também se notou maior incidência na raça caucasiana [11] bem como no sexo masculino com relação 1.2-1.5:1 [4] [12]. No entanto, nalguns estudos, não se verificou relação com o género [13].
7
anos[14].
Relativamente a estações do ano, existem vários estudos que demonstram uma menor incidência no verão[4], relacionando a maior incidência de infeções nos meses mais frios
com o aparecimento de PHS [10][15].
Clínica
Atualmente, os critérios mais recentes da European League Against Rheumatism (EULAR), Paediatric Rheumatology European Society (PRES) e American College of Rheumatology (ACR) (Tabela 1) requerem a presença de púrpura palpável e um dos seguintes:
Artrite/artralgia; Dor abdominal difusa;
Envolvimento renal (hematúria e/ou proteinúria); Biópsia cutânea ou renal com depósitos de IgA [16].
Habitualmente os sintomas duram entre 4 e 6 semanas[6].
Podem existir outras manifestações mais raras, nomeadamente envolvimento urológico, tanto na fase aguda de doença como após recuperação, com obstrução do ureter ou ureterite. Também pode ocorrer envolvimento escrotal em 7-22% dos casos, sob a forma de epididimite ou orquite com complicações do cordão espermático.
Relativamente a envolvimento do sistema nervoso central (SNC), durante o curso da doença, podem verificar-se alterações do estado de consciência, convulsões, sinais neurológicos focais, disartria e alterações visuais. Nestes doentes, os métodos de imagem revelam lesões vasculares, hemorragia intracerebral, edema subcortical ou trombose do seio sagital posterior.
Existem também casos em que se verifica hemorragia pulmonar, pneumonite intersticial ou bloqueio auriculoventricular completo[12][17].
8
Meios Complementares de Diagnóstico
O diagnóstico é com base na clínica. Perante uma suspeita de diagnóstico de PHS, são recomendadas medição da pressão arterial e avaliação analítica com hemograma completo com plaquetas, creatinina sérica, análise sumária de urina e razão proteinúria/creatinúria ou proteinúria de 24h (Tabela 1), se se justificar [13][18].
Nos casos em que se detete proteinúria de valores nefróticos (≥50 mg/kg/dia ou 40 mg/m2/h) ou uma razão proteinúria/creatinúria>2, é importante identificar alterações laboratoriais típicas de síndrome nefrótica, nomeadamente albumina, colesterol total, triglicéridos, coagulação, imunoglobulinas e complemento [19][20].
Quando o diagnóstico não é claro, podem ser necessários exames mais invasivos, nomeadamente biópsia cutânea ou renal [13]. A biópsia renal é também recomendada em situações em que há significativo envolvimento renal – nefrite rapidamente progressiva ou proteinúria nefrótica ou persistente [3][9][19].
Nos casos em que há envolvimento abdominal, pode ser útil realizar ecografia abdominal, que permite diagnosticar complicações como invaginação [9].
Tabela 1: Investigação inicial em criança com púrpura e suspeita de PHS [13] Pressão arterial
Análise sumária de urina
Hemograma completo com plaquetas Creatinina sérica
Razão proteinúria/creatinúria
Tratamento
Os dados orientadores obtidos para tratamento de vasculite em idade pediátrica provêm de pequenos estudos ou de estudos em adultos, pelo que o tratamento varia de país para país [3].
Habitualmente, a doença é benigna e autolimitada, necessitando apenas de terapêutica de suporte em casos de apresentação ligeira [4][11], com analgésicos ou anti-inflamatórios não esteróides (AINE) para as dores articulares [12], adequada ingesta e hidratação [12][21]. No entanto, pode ser necessário tratamento mais agressivo com corticoterapia.
9
Consegue-se um bom controlo de dor com AINE em dores articulares e abdominais; no entanto, deve evitar-se este grupo de fármacos se houver envolvimento renal.
A utilização de corticoterapia está reservada para quadros mais agressivos, habitualmente com envolvimento gastrointestinal grave, com suspeita de invaginação intestinal, dor articular muito intensa [7][22] ou lesões cutâneas extensas; no entanto não previnem o aparecimento de doença renal[13][21][23][24].
Quando há alterações renais severas (proteinúria nefrótica, diminuição da filtração glomerular, ou proteinúria persistente) são necessários corticosteroides e agentes antihipertensores e antiproteinúricos, nomeadamente inibidores da enzima conversora de angiotensina (iECA) [3][25].
Manifestações unicamente cutâneas, sem envolvimento sistémico, não necessitam de tratamento específico. No entanto, se houver lesões bolhosas, existe boa resposta a tratamento com corticosteroides[17].
Em situações de lesão renal aguda ou complicações refratárias à corticoterapia, pode realizar-se plasmaferese ou fármacos imunossupressores (ciclofosfamida, azatioprina, ciclosporina) [3][26][11].
Prognóstico
A PHS tem um bom prognóstico, com remissão completa em 2 semanas em um terço dos doentes, em 4 semanas noutro terço e com recorrência dos sintomas nos primeiros 4 meses nos restantes casos.
A morbilidade precoce deve-se sobretudo ao envolvimento gastrointestinal, enquanto a morbilidade tardia se deve a envolvimento renal, com risco de progressão para doença renal crónica que pode demorar décadas a surgir [3].
Os fatores associados com a progressão da doença renal são a diminuição da função renal basal (50% de redução da taxa de filtração basal ou duplicação da creatinina), hematúria macroscópica, hipertensão arterial ou proteinúria (>1mg/dia) durante o follow up. Outros estudos acrescentam que o envolvimento renal per se é um fator de mau prognóstico, bem como anemia, hematúria na altura do diagnóstico, combinação de síndrome nefrótica e nefrítica, fibrose intersticial ou esclerose glomerular em biópsia renal. [8][27][28][29]
10
Caso Clínico
Apresenta-se o caso de uma criança de 10 anos, do sexo masculino, raça caucasiana que recorreu ao serviço de urgência por púrpura.
Tinha antecedentes familiares irrelevantes e antecedentes pessoais de asma, sendo acompanhado em consulta de Imunoalergologia; com alergia documentada a gramíneas, ervas daninhas, árvores e fungos, estando medicado com fluticasona + salmeterol (100 ug/50ug) diários, e salbutamol SOS, apresentando crises mensais; vacinas de acordo com o Programa Nacional de Vacinação; bom desenvolvimento estaturo-ponderal: peso 35,5 Kg (P50-P85) e altura 142,4 cm (P50).
É observado no serviço de urgência pediátrica (SU) por exantema purpúrico nos membros inferiores com uma semana de evolução, mais exuberante na face posterior das pernas, também com envolvimento da região glútea e cotovelo esquerdo; apresentava ainda edema dos membros inferiores até ao joelho e dores articulares referidas à mão esquerda e pés. Negou febre, dor abdominal, perdas hemáticas nas fezes ou urina, ou outros sintomas. Cerca de uma semana antes do aparecimento do exantema, teve febre e odinofagia, acompanhadas de duas dejeções diarreicas sem sangue, muco ou pus. Foi medicado de forma sintomática com paracetamol e ibuprofeno, assumindo-se o diagnóstico de PHS.
Por agravamento franco das lesões cutâneas e dor articular que condicionava a marcha, recorreu de novo ao SU.
À observação, apresentava púrpura palpável e edema nos membros inferiores e exantema petequial nos membros superiores.
Analiticamente, Hb 14.7 g/dL, Leucócitos 10.9x109/L, Neutrófilos 5900, Eosinófilos
600, Linfócitos 3600, Monócitos 800. Plaquetas 405x109/L; Creatinina 0.51mg/dL,
Ureia 27mg/dL; TP 11.6 segundos, aPTT 23.9 segundos e PCR de 3 mg/dl. Análise sumária da urina pH 6, densidade 1.026, proteínas 200 mg/dL; glicose, corpos cetónicos, bilirrubina, urobilinogénio, hemoglobina, leucócitos, nitritos negativos.
Ficou internado para vigilância, repouso e controlo da dor com paracetamol endovenoso.
Durante o internamento, observou-se um agravamento das lesões cutâneas, com formação de bolhas nos membros inferiores e progressão da púrpura para o tronco, membros superiores e face (Figuras 1 e 2).
11
Ao terceiro dia de internamento (D3), foi medicado com prednisolona (1mg/Kg/dia) por manutenção das dores articulares ao nível dos tornozelos e mãos. As lesões cutâneas eram bastante exuberantes e pouco habituais numa PHS, pelo que foi pedida avaliação por Dermatologia. Realizada biópsia cutânea das lesões purpúricas que mostrou vasculite leucocitoclástica, compatível com o diagnóstico de Púrpura de Henoch-Schönlein (Figuras 3 e 4).
Do ponto de vista renal, observou-se a partir do 2º dia de internamento (D2) proteinúria de agravamento progressivo, com aumento para valores nefróticos em D9 de internamento (133 mg/m2/h); nesta altura, apresentava também microhematúria (1+) e pressão arterial no P95.
Adicionalmente, em D10, apresentou hipercolesterolémia (CT 231 mg/dL – máximo Figuras 1 e 2 - Lesões cutâneas bolhosas nos membros inferiores
12
em D25 295 mg/dL) com triglicéridos normais, hipoalbuminémia (2.79g/dL – mínimo 2g/dL em D19), diminuição das proteínas totais (5.83 mg/dL – mínimo 4.51 g/dL em D25).
Foi medicado com metilprednisolona (1g) em pulsos, seguida de prednisolona na dose de 2 mg/Kg/dia. Adicionalmente, foi medicado com enalapril 10 mg, AAS em dose antiagregante, cálcio, vitamina D e dieta hipossalina; foi vacinado com as vacinas antipneumocócica 13-valente e antigripal.
Apesar da otimização terapêutica, entre D12-D16 o doente desenvolveu oligúria (débito urinário mínimo de 0.6 mL/Kg/h), agravamento do edema e necessidade de restrição hídrica e furosemida.
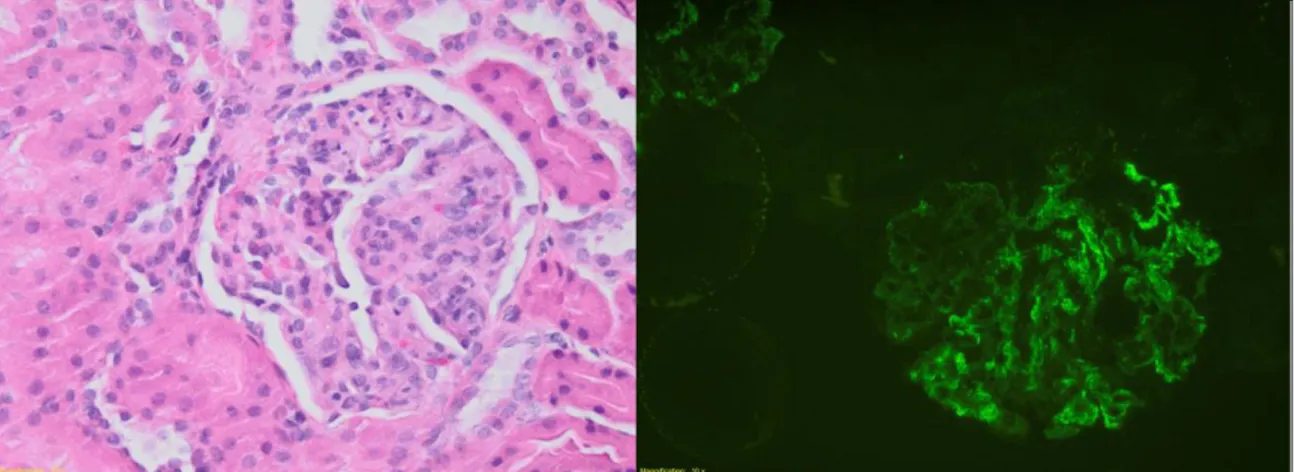
Por manutenção de proteinúria nefrótica, foi realizada biópsia renal guiada por ecografia em D14 que revelou glomerulonefrite proliferativa mesangiocapilar focal, compatível com o diagnóstico de nefropatia IgA, enquadrável no contexto clínico de púrpura de Henoch-Schönlein (Figuras 5 e 6).
Figura 5 – Histologia que revela glomerulonefrite proliferativa mesangiocapilar focal Figura 6 – Imunofluorescência com depósitos IgA
Em D17, apresentou razão proteinúria/creatinúria máxima de 9.5, com melhoria progressiva.
Ao longo do internamento verificaram-se valores de pressão arterial sistólica superiores ao P95 (PA máxima 140/72 mmHg em D19), controlados com o ajuste de enalapril. O doente nunca teve alterações dos valores de creatinina ou na coagulação.
Durante o internamento foram ainda realizados os seguintes exames complementares de diagnóstico, no contexto de protocolo do serviço de abordagem de uma síndrome nefrótica:
13
TASO negativo, Phadirect, e exame cultural do exsudado faríngeo sem alterações;
Serologias: EBV, CMV, Parvovírus, Adenovírus, Influenza A, Mycoplasma, Bartonella – IgM e IgG negativos;
Vírus nas fezes: adenovírus, enterovírus e rotavírus negativos;
Diminuição dos valores de IgG (545 mg/dL) e IgM (26 mg/dL); IgA e IgE normais;
IGRA negativo;
C3 normal (103 mg/dL) e C4 diminuído (19.6 mg/dL); ANCA-negativo, ANA, dsDNA negativos.
Como intercorrências, no 15º dia de internamento teve agravamento da asma, com sibilos e fervores subcrepitantes bilaterais à auscultação pulmonar, com melhoria progressiva após terapêutica broncodilatadora e otimização da diurese.
Teve alta ao fim de 30 dias de internamento, clinicamente assintomático, com melhoria significativa das lesões vasculíticas, sem edema, com diurese adequada e sem proteinúria nefrótica. Analiticamente apresentava proteínas totais 4.5g/dL, proteinúria de 200 mg/dL e relação proteinúria/creatinúria 1.8.
Cinco dias depois, regressa ao SU por edema acentuado da face e membros inferiores, associado a um aumento ponderal de 2 Kg. Concomitantemente, apresentava no dorso dos pés duas lesões, com 4 e 2 cm de diâmetro, com placa necrótica, exsudado purulento e celulite circundante.
Analiticamente, verificava-se leucocitose (15100/uL), com 71% neutrófilos e 20,6% de linfócitos. PCR<0.29 mg/dL. Urina II: Hematúria ++, proteinúria de 300 mg/dL e relação proteinúria/creatinúria 3.8. O quadro clínico foi interpretado como uma recidiva da doença no contexto da infeção.
Foi internado para realização de antibioticoterapia endovenosa com flucloxacilina e desbridamento das lesões, após observação pela cirurgia pediátrica.
Teve alta 2 dias depois com parâmetros vitais estáveis, função renal preservada e a urina II revelava vestígios de proteínas e hemoglobina 1+.
14
Discussão
Descreve-se o caso de uma criança com PHS com envolvimento renal grave.
De acordo com o descrito na literatura, a doença teve início durante a primavera, sendo precedida numa semana por febre, odinofagia e diarreia. Não foi possível documentar agentes infeciosos e não existia história de fármacos ou vacinação recente; a sua idade não corresponde ao grupo etário mais frequentemente envolvido na PHS.
Relativamente à apresentação clínica, as principais manifestações são púrpura, artrite, manifestações gastrointestinais e envolvimento renal.
A púrpura é o elemento essencial para o diagnóstico. São lesões palpáveis com diâmetro de 2-10 mm, podendo existir também sufusões hemorrágicas. Os locais mais frequentes são as zonas de maior gravidade e pontos de maior pressão, podendo ocorrer também noutras localizações. Ocorre habitualmente de forma simétrica e pode ser acompanhada de edema subcutâneo no dorso das mãos, pés e região periorbitária [25][21]. Podem também verificar-se vesículas, bolhas e ulceração de equimoses. [17] No caso descrito observou-se púrpura na face posterior das pernas, nádegas e cotovelos, e ainda sufusões hemorrágicas nos membros inferiores. Durante o internamento, as lesões progrediram para o tronco, membros superiores e face. As lesões cutâneas evoluíram de forma exuberante, sob a forma de bolhas, manifestação pouco habitual. Mais tarde, evoluíram para necrose com infeção secundária, condicionando um reinternamento. Existe evidência na literatura que sugere que todas as suspeitas de diagnóstico de PHS, mesmo com quadro típico, devem ser confirmadas com biópsia cutânea, por ser pouco invasivo e permitir melhor follow up em doentes com PHS [29][13][30], enquanto outros
estudos sugerem a utilização apenas em casos atípicos[21]. Neste caso, as lesões cutâneas
apresentaram uma evolução pouco característica de PHS, o que motivou a realização de biópsia cutânea que mostrou vasculite leucocitoclástica, compatível com o diagnóstico de PHS. Habitualmente observa-se um infiltrado de células inflamatórias e deposição de IgA [21].
A artrite é a segunda manifestação mais frequente (75%). Caracteriza-se por ser oligoarticular, transitória, não migratória e não destrutiva, afetando, habitualmente os joelhos e tornozelos, sendo menos frequente nos membros superiores[31]. A dor é intensa, e pode impedir a marcha [7][13][11]. Esta manifestação pode preceder a púrpura em 25% dos casos[7]. Em conformidade com a literatura, o doente apresentou edema e dor articular intensa nos membros inferiores e nas mãos, com necessidade de
15
corticoterapia.
Em 50-75% dos casos, ocorrem sintomas GI, nomeadamente: dor abdominal tipo cólica, vómitos, diarreia ou hemorragia (habitualmente oculta). A apresentação gastrointestinal mais severa é a invaginação (1-5%)[31]. Os sintomas GI podem também preceder cerca de 1 semana o aparecimento da púrpura, mimetizando doenças inflamatórias intestinais. Apesar da elevada percentagem de manifestações gastrointestinais, o doente não apresentou qualquer alteração neste sistema.
Ocorre envolvimento renal em cerca de 40-50% dos doentes. A hematúria microscópica é o achado laboratorial mais frequente, sendo que 25% apresentam hematúria macroscópica. A proteinúria acompanha a hematúria em 60% dos casos. No caso descrito verificou-se hematúria microscópica e proteinúria.
O envolvimento renal é mais frequente e mais severo a partir dos 10 anos[32]. São fatores de risco para doença renal: sexo masculino, idade superior a 10 anos, dor articular, púrpura persistente – que se verificou no caso descrito; sintomas GI severos (dor, hemorragia ou angina intestinal), envolvimento neurológico, leucocitose >15.000/ul, trombocitose >500.000/uL, ASO elevado ou diminuição de C3, que não se verificou.[33]
No primeiro internamento, está descrito edema na apresentação; a partir de D2 com proteinúria de agravamento progressivo e aumento para valores nefróticos em D9 de internamento (133 mg/m2/h). Também tinha hipoalbuminémia (2 g/dL) e
hipercolesterolémia (CT 295 mg/dL), condicionados pela perda urinária de proteínas e síntese hepática compensatória, de outros elementos – nomeadamente lipoproteínas e fatores de coagulação. Apresentou diminuição dos níveis de IgG, por perda urinária de imunoglobulinas; alterações típicas de síndrome nefrótica. Casos com proteinúria nefrótica, como o apresentado, são menos frequentes e têm maior risco de complicações renais a longo prazo[13][34].
Apresentou ainda, ao longo do internamento, valores de pressão arterial sistólica acima do P95, controlados com enalapril, bem como períodos de oligúria. Estas alterações, juntamente com hematúria (mesmo sem elevação da creatinina), correspondem a uma síndrome nefrítica.
Foi realizada biópsia renal por apresentar proteinúria de valores nefróticos e sinais e sintomas de glomerulonefrite[13], que revelou glomerulonefrite proliferativa mesangiocapilar focal, compatível com o diagnóstico de nefropatia IgA, enquadrável no
16
contexto clínico de púrpura de Henoch-Schönlein. Doentes com PHS, apresentam habitualmente glomerulonefrite proliferativa que pode variar de focal segmentar até envolvimento extenso. Podem observar-se ao microscópio de fluorescência depósitos de IgA nas paredes dos vasos sanguíneos[26][21].
O doente apresentou concomitantemente síndrome nefrótica e glomerulonefrite, associação que ocorre em apenas 1% dos casos[35].
Relativamente aos exames complementares de diagnóstico, outros achados laboratoriais que podem sugerir PHS são o aumento sérico de IgA [6], aumentados em cerca de 50% dos doentes [25], que não se verificou no caso descrito. A ativação de complemento e da cascata inflamatória a nível renal tem um papel importante na lesão renal, condicionando hipocomplementémia transitória; neste caso, os valores de C3 eram normais (103 mg/dL – VR 75-107) e ligeiramente diminuídos de C4 (19.6 mg/dL – VR 21-38).
Diagnosticar uma vasculite pode ser difícil quando os sinais e sintomas são inespecíficos. É importante identificar clínica sugestiva das diferentes classes de vasculite. Existem subtipos de vasculite ANCA-positivo que têm também, como manifestação clínica, glomerulonefrite rapidamente progressiva, insuficiência renal aguda e, adicionalmente, sintomas respiratórios.
Assim, pode ser importante o diagnóstico diferencial com poliangeíte granulomatosa (PG) (antiga granulomatose de Wegener), poliangeíte microscópica (PM) e poliangeíte granulomatosa hipereosinofílica (PGH) (antiga síndrome de Churg-Strauss), sobretudo porque sem tratamento têm 100% de mortalidade, com sobrevivência média de apenas 5 meses. Habitualmente, são ANCA positivos.
É essencial biopsar órgãos afetados para estabelecer um diagnóstico. Habitualmente, o infiltrado tem poucos depósitos imunes, com granulomas na poliangeíte granulomatosa, e com hipereosinofilia na poliangeíte granulomatosa hipereosinofílica.
Características clínicas comuns a todas as vasculites ANCA-positivos são sintomas constitucionais em mais de 50% dos pacientes – febre, fadiga, anorexia, perda de peso – que precedem o envolvimento de órgão.
A poliangeíte granulomatosa afeta sobretudo o trato respiratório e rins. Cerca de 80% tem manifestações pulmonares: hemorragia pulmonar, nódulos, insuficiência
17
respiratória; 80% tem sintomas do trato respiratório superior: úlceras nasais, perfuração do septo nasal, epistaxis frequente, sinusite, mastoidite, estenose subglótica; clínica que não se verificou. Em 75% existe envolvimento renal: hematúria, proteinúria, glomerulonefrite ou insuficiência renal aguda, aspetos que se puderam observar.
Apesar do doente ter apresentado manifestações renais, não apresenta sinais/sintomas para completar os critérios necessários para o diagnóstico de poliangeíte granulomatosa (Quadro 3 - anexos).
Relativamente à poliangeíte microscópica, é uma vasculite de pequenos vasos não granulomatosa, cuja manifestação mais comum é glomerulonefrite necrotizante. A maioria das crianças tem sintomas constitucionais. Quase 100% das crianças tem envolvimento renal apresentando-se com hipertensão, hematúria ou proteinúria; perda de peso (70%), púrpura palpável em zonas dependentes da gravidade – mais nos membros inferiores (60%), neuropatia (60%) e febre (55%). Quando ocorre envolvimento pulmonar (cerca de 15-55%) as manifestações são severas com hemorragia alveolar.
Relativamente à poliangeíte granulomatosa hipereosinofílica, é uma vasculite de pequenos vasos que se apresenta habitualmente como uma asma de difícil controlo. A clínica mais frequente é asma (91%), infiltrados pulmonares (85%), sinusite (77%), púrpura (66%), doença renal (16%).
Não existem critérios para poliangeíte granulomatosa hipereosinofílica adaptados a idade pediátrica, no entanto, atendendo aos critérios em adultos, neste caso, não se cumprem critérios suficientes para o diagnóstico (Quadro 3 - anexos).
O doente tem uma asma de fácil controlo. Apesar de ter apresentado manifestações renais e pulmonares, as últimas deveram-se a uma agudização da asma, transitória, decorrente de edema pulmonar numa altura de menor débito urinário e não por manifestação pulmonar de vasculite.
A biópsia renal, também não é a favor do diagnóstico de vasculites ANCA-positivo
[11, 26, 36][37][38].
Não existem atualmente guidelines para o tratamento da doença. Esperavam-se orientações mais uniformes em 2015 com o projeto SHARE (Single Hub Access for pediatric Rheumatology in Europe), mas não foram criadas para PHS.
18
Como referido, a terapêutica é sintomática; a corticoterapia é utilizada em casos mais severos. Em doentes hospitalizados, podem ser utilizados pulsos de metilprednisolona (30 mg/kg máximo 1g/kg) ou prednisolona (1-2 mg/ kg/dia). A dose e duração do tratamento, ainda não estão bem definidos; sabe-se, no entanto, que duração curta ou diminuição rápida da dose pode precipitar o reaparecimento dos sintomas[11].
Outro aspeto a considerar, é a síndrome nefrótica apresentada pelo doente. Existem dados que mostram que os iECA diminuem o nível de proteinúria e reduzem a progressão da doença renal [3][12].
Crianças com síndrome nefrótica têm maior suscetibilidade a infeções por perderem imunoglobulinas pelo glomérulo e pela corticoterapia; têm risco de eventos tromboembólicos por aumento da produção hepática de fatores pro-trombóticos (fibrinogénio); perda urinária de fatores fibrinolíticos (antitrombina III, proteína C e S), aumento da produção de plaquetas (como reagente de fase aguda) e maior agregação plaquetária; apresentam hiperlipidémia por aumento da produção hepática de LDL.
Assim, é importante fazer a prevenção destas complicações com vacinação antipneumocócica 13 valente e da gripe, que foram administradas [21][39].
Habitualmente, não está recomendado dar anticoagulação de forma profiláctica, a menos que haja antecedentes pessoais de evento tromboembólico. No entanto, o doente apresentava dor articular, que condicionou, durante algum tempo, imobilização no leito, fator de risco para tromboembolismo. Optou-se por iniciar ácido acetil salicílico.
Foi necessário administrar enalapril em doses crescentes para controlo da pressão arterial e dieta hipossalina.
A hiperlipidémia normaliza aquando a remissão da doença, pelo é dispensável a utilização de estatinas[19].
Outro aspeto a ter em conta é a hipocalcémia, quer pela perna urinária de vitamina D3, bem como pela osteopénia induzida pela corticoterapia. Assim, foi também iniciado colecalciferol[40]. No entanto, existe bibliografia que defende que só é necessária suplementação de vitamina D em população com carência da mesma, e que, apesar de melhorar os valores séricos, não existem diferenças na densidade mineral óssea entre doentes tratados e não tratados[41]. Também existe bibliografia que favorece a utilização de suplemento de vitamina D, por prevenir a diminuição e até aumentar a densidade minera óssea[42].
19
Relativamente ao prognóstico, a PHS é habitualmente autolimitada, benigna e com remissão completa dos sintomas em 4 semanas, ou menos, em 2/3 dos doentes. Cerca de 1/3 dos doentes têm recorrência da doença, que se apresenta com um quadro com clínica semelhante, mas mais leve que o quadro inaugural, nos primeiros 6 meses após remissão dos sintomas [3] [43] [44]. A recorrência é mais frequente em quadros iniciais mais severos [21]
Casos com glumerulonefrite associada a PHS podem ter recorrência da doença associada a infeções, apenas com envolvimento renal e sem outros sintomas[44]. Cinco dias após alta clínica, verificou-se recorrência da doença, associada a celulite - apresentando clinicamente edema acentuado da face e membros inferiores, associado a aumento ponderal de 2 Kg; e, analiticamente, para além do aumento dos parâmetros inflamatórios, aumento da relação proteinúria/creatinúria e hematúria.
A longo prazo, a principal complicação é evolução para doença crónica terminal, ocorrendo em cerca de 1-2% dos doentes com PHS [34][30][45][46]. No entanto, quando existe síndrome nefrótica, 20-40% dos casos evoluem para doença crónica terminal [14]
[46] [47] num período de 10-20 anos depois[47]. Assim, no caso descrito, há risco
aumentado de progressão para doença renal crónica terminal.
Os valores de proteinúria durante a fase aguda de doença não se correlacionam com pior prognóstico [46]. No entanto, ausência de proteinúria é um marcador de bom
prognóstico[14].
A idade de aparecimento da patologia é um fator prognóstico importante. Habitualmente, em idade superior a 10 anos o quadro clínico é mais severo e há maior progressão para doença renal crónica.
Se nunca houver alterações urinárias, é improvável que haja envolvimento renal a longo prazo; no entanto, quando existem alterações urinárias ou hipertensão arterial, como apresentado, é importante manter o seguimento em nefrologia pediátrica [21].
O principal objetivo do follow-up é detetar inflamação renal persistente que, se não for diagnosticada, pode provocar danos permanentes.
O período de seguimento não é consensual. No caso de PHS não complicada, recomenda-se 6-12 meses[14], com análises à urina e medição de pressão arterial, independentemente das manifestações clínicas apresentadas[12].
20
sintomas, como apresentado, devem ter um acompanhamento mais longo do que os restantes [32], até serem obtidos valores analíticos normais[25][35].
Existe literatura que afirma que, quando existe envolvimento renal, pode ser necessário seguimento até 5 anos [34].
É importante uma melhor definição dos doentes em risco para doença renal terminal, para definir qual a melhor estratégia terapêutica [47].
Atualmente, com 18 meses de seguimento, o doente mantém-se assintomático e sem alterações analíticas.
Conclusão
Este caso permite refletir acerca da benignidade da doença.
Neste caso, a PHS condicionou um internamento de cerca de 1 mês, absentismo escolar e necessidade de corticoterapia prolongada, com os seus efeitos adversos inerentes.
O envolvimento renal foi severo, com síndrome nefrótica e nefrítica.
Não existem sequelas na grande maioria, no entanto, existe risco de progressão para doença renal crónica, sobretudo se houver síndrome nefrótica.
Recomenda-se, em todos os doentes com envolvimento renal, o seguimento em consulta de nefrologia pediátrica por período não consensual.
Para além disto, podem ocorrer manifestações mais raras e graves, nomeadamente envolvimento do SNC, hemorragia pulmonar, alterações da condução cardíaca, perfuração intestinal.
Existe também uma incidência de hipertensão arterial e pré-eclâmpsia superior à da população normal (70% em comparação com 5-10%).
Existe pouco consenso em relação à melhor estratégia terapêutica e em relação ao follow up, sendo importante desenvolver projetos neste sentido.
Os factos acima descritos permitem questionar a benignidade da doença, e as implicações e custos em saúde associados nas formas graves de PHS.
21
Agradecimentos
A tese é um processo solitário de leitura, revisão e estudo. No entanto, tal não seria possível sem o apoio incansável do Dr. António Figueiredo, pediatra no hospital Fernando da Fonseca, que me ajudou nas várias etapas da mesma.
Quero também agradecer ao Dr. Nuno Martins, interno de 5º ano de especialidade de Medicina Interna no hospital Fernando da Fonseca, que me auxiliou com as melhores técnicas de pesquisa e formas de aceder a artigos.
Agradeço à Dra. Sandra Valente, pediatra no hospital de Santa Maria, que despertou o meu interesse por pediatria.
Agradeço ainda aos meus amigos e colegas que acompanharam este processo, bem como aos meus pais e família que me apoiaram e motivaram a fazer e ser sempre o melhor possível.
22
Bibliografia
1. Dalt, L. Da, Zerbinati, C., Strafella, M.S., Renna, S., Riceputi, L., Pietro, P. Di, et al. (2016) Henoch-Schönlein purpura and drug and vaccine use in childhood : a case-control study. Italian Journal of Pediatrics, 42, 1–5. http://dx.doi.org/10.1186/s13052-016-0267-2.
2. Hamdan, J.M. and Barqawi, M.A. (2008) Henoch-Schonlein purpura in children. Influence of age on the incidence of nephritis in children. Saudi Med J, 29, 549– 52.
3. Eleftheriou, D. and Brogan, P.A. (2016) Therapeutic advances in the treatment of vasculitis. Pediatric Rheumatology, 2016: 10.1186/s12969-016-0082-8. http://dx.doi.org/10.1186/s12969-016-0082-8.
4. Chen, O., Xb, Z., Ren, P., Yb, W., Rp, S. and De, W. (2013) Henoch Schonlein Purpura in children : clinical analysis of 120 cases. African Health Sciences, 13, 94–99.
5. Calvo-Río, V., Loricera, J., Mata, C., Martín, L., Ortiz-Sanjuán, F., Alvarez, L., et al. (2014) Henoch-Schönlein Purpura in Northern Spain Clinical Spectrum of the Disease in 417 Patients from a Single Center. Medicine, 93, 106–113.
6. Kraft, D.M., Mckee, D. and Scott, C. (1998) Henoch-Schönlein Purpura : A Review. American Family Physician, 58, 405–408.
7. Practice, C. and Lawee, D. (2008) Case Report Atypical clinical course of Henoch-Schönlein purpura. Can Fam Physician, 54, 1117–1120.
8. Woerner, A., Rudin, C., Bonetto, C., Santuccio, C., Ozen, S., Wise, R.P., et al. (2017) IgA vasculitis (Henoch–Schönlein): Case definition andguidelines for data collection, analysis, and presentation of immunisation safety data. Vaccine, 35, 1559–1566.
9. Trnka, P. (2013) Henoch-Schönlein purpura in children. Journal of Paediatrics and Child Health, 49, 995–1003.
10. Trapani, S., Micheli, A., Grisolia, F., Resti, M., Chiappini, E., Falcini, F., et al. (2005) Henoch Schonlein Purpura in Childhood : Epidemiological and Clinical
23
Analysis of 150 Cases Over a 5-year Period and Review of Literature. Seminars in Arthritis and Rheumatism, 35, 143–153.
11. Weiss, P. (2012) Vasculitis, Pediatric. Pediatr Clin North, 59, 407–423.
12. Bluman, J. and Goldman, R.D. (2014) Child Health Update Henoch-Schönlein purpura in children Limited benefit of corticosteroids. Canadian Family Physician,
60, 1007–1009.
13. Smith, G. (2008) Management of Henoch- Schönlein purpura. Paediatrics and Child Health, 18, 358–363.
14. Watson, L., Richardson, A.R.W., Holt, R.C.L., Jones, C.A. and Michael, W. (2012) Henoch Schonlein Purpura – A 5-Year Review and Proposed Pathway. PLOS ONE, 7.
15. Atkinson, S.R. and Barker, D.J.P. (1976) Seasonal distribution of Henoch-Schonlein purpura. British journal of preventive & social medicine, 90, 22–25.
16. Ozen, S., Pistorio, A., Iusan, S.M., Bakkaloglu, A., Herlin, T., Brik, R., et al. (2010) EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Annals of the Rheumatic Diseases, 69, 798–806. http://ard.bmj.com/cgi/doi/10.1136/ard.2009.116657.
17. Sag, E., Arici, Z.S. and Ozen, S. (2017) IgA vasculitis (Henoch–Schönlein purpura) in children. Expert Opinion on Orphan Drugs, 5, 405–410. http://dx.doi.org/10.1080/21678707.2017.1311783.
18. Correia, Manuela; Levy, António; Camilo, Cristina; Abecassis, Francisco; Vieira, Maria; Quintas, S. (2014) Protocolo de Urgência em Pediatria. 2014.
19. Pasini, A., Benetti, E., Conti, G., Ghio, L., Lepore, M., Massella, L., et al. (2017) The Italian Society for Pediatric Nephrology (SINePe) consensus document on the management of nephrotic syndrome in children: Part i - Diagnosis and treatment of the first episode and the first relapse. Italian Journal of Pediatrics, 43, 1–15.
20. Obiagwu, P.N., Aliyu, A. and Atanda, A.T. (2014) Nephrotic syndrome among children in Kano : A clinicopathological study. 17, 370–374.
24
21. Robert, M.K.M., Bonita, M.D.S.M., Joseph, S.G.M., Nina, F.S.M.P. and Richard, E.B.M. (2011) Nelson Textbook of Pediatrics 19th edition. 2011.
22. Lerkvaleekul, B., Treepongkaruna, S., Saisawat, P. and Thanachatchairattana, P. (2016) Henoch-Schönlein purpura from vasculitis to intestinal perforation : A case report and literature review. 22, 6089–6094.
23. Mrusek, S., Krüger, M., Greiner, P., Kleinschmidt, M., Brandis, M. and Ehl, S. (2004) Case report Henoch-Schönlein purpura. The Lancet, 363, 1116.
24. Graeff, N. De, Groot, N., Kamphuis, S., Avcin, T., Bader-meunier, B., Dolezalova, P., et al. SHARE – Workpackage 5 : Evidence Based Recommendations for Diagnosis and Treatment of Kawasaki Disease ( KD ) and Henoch Schonlein Purpura ( HSP ) Complete list of recommendations ( not yet published ) Continues on next page SHARE – Workpackage 5 : Eviden.
25. Of, P., There, P., The, P., The, P. and Gastrointestinal, P. (2007) Clinical update : Henoch-Schönlein purpura. The Lancet, 369, 25–27.
26. Janette, J.C. and Falk, R.J. (2017) Small-vessel vasculitis. The New England Journal of Medicine, 2017.
27. Audemard-Verger, A., Terrier, B., Dechartres, A., Chanal, J., Amoura, Z., Le Gouellec, N., et al. (2017) Characteristics and Management of IgA Vasculitis (Henoch-Schönlein) in Adults: Data From 260 Patients Included in a French Multicenter Retrospective Survey. Arthritis and Rheumatology, 69, 1862–1870.
28. Zaffanello, M. and Fanos, V. (2009) Treatment-based literature of Henoch-Schönlein purpura nephritis in childhood. Pediatric Nephrology, 24, 1901–1911.
29. Davin, J.C. and Weening, J.J. (2003) Diagnosis of Henoch-Schönlein purpura: Renal or skin biopsy? Pediatric Nephrology, 18, 1201–1203.
30. Guo, D. and Lam, J.M. (2016) Practice. CMAJ, 188, 5128.
31. Kawasaki, Y., Ono, A., Ohara, S., Suzuki, Y., Suyama, K., Suzuki, J., et al. (2013) HENOCH - SCHÖNLEIN PURPURA NEPHRITIS IN CHILDHOOD : PATHOGENESIS , PROGNOSTIC FACTORS AND TREATMENT. 59.
25
T., et al. (2010) Renal manifestations of Henoch-Schonlein purpura in a 6-month prospective study of 223 children. Archives of disease in childhood, 95, 877–882.
33. Chan, H., Tang, Y., Lv, X., Zhang, G., Wang, M., Yang, H., et al. (2016) Risk Factors Associated with Renal Involvement in Childhood Henoch-Scho ¨ nlein Purpura : A Meta-Analysis. PLOS ONE, 11.
34. Narchi, H. (2005) Risk of long term renal impairment and duration of follow up recommended for Henoch-Scho ¨nlein purpura with normal or minimal urinary findings: a systematic review. Archives of disease in childhood, 90, 916–920.
35. Zaffanelo, M. (2011) Henoch-Schönlein Purpura Nephritis in Childhood. An update on glumerulopathies - Clinical and treatment aspects, 2011.
36. Eleftheriou, D., Batu, E.D., Ozen, S. and Brogan, P.A. (2015) Vasculitis in children. Nephrology Dialysis Transplantation, 30, i94–i103. https://doi.org/10.1016/j.paed.2017.10.009.
37. Calatroni, M., Oliva, E., Gianfreda, D., Gregorini, G., Allinovi, M., Ramirez, G.A., et al. (2017) ANCA-associated vasculitis in childhood : recent advances. 2017: 10.1186/s13052-017-0364-x.
38. Wang, H., Sun, L. and Tan, W. (2015) Clinical Features of Children with Pulmonary Microscopic Polyangiitis : Report of 9 Cases. 2015: 10.1371/journal.pone.0124352.
39. Kodner, C. (2009) Nephrotic syndrome in adults: Diagnosis and management. American Family Physician, 80, 1185–1189.
40. A Goldenstein, D., Haldimann, B., Shermann, D., Anthony, W.N.A. and Massry, S. (1981) Vitamin D Metabolites and Calcium Metabolism in Patients with Nephrotic Syndrome and Normal Renal Function. The Journal of Clinical Endocrinology & Metabolism, 52, 116–121.
41. Banerjee, S., Basu, S., Sen, A. and Sengupta, J. (2017) The effect of vitamin D and calcium supplementation in pediatric steroid-sensitive nephrotic syndrome. Pediatric Nephrology, 32, 2063–2070.
26
osteoprotection in children with new-onset nephrotic syndrome treated with steroids: A prospective, randomized, controlled, interventional study. Pediatric Nephrology, 29, 1025–1032.
43. Loricera, J., Palmou-fontana, N., Armesto, S. and Blanco, R. (2016) Relapses in patients with Henoch – Schönlein purpura. Medicine, 95, e4217.
44. Lau, K.K., Suzuki, H., Novak, J. and Wyatt, R.J. (2010) Pathogenesis of Henoch-Schönlein purpura nephritis. Pediatric nephrology, 25, 19–26.
45. Coakley, J.C. and Chambers, T.L. (1975) Should we follow up children with Henoch-Schonlein syndrome ? Archives of disease in childhood, 54, 903–904.
46. Feng, D., Huang, W., Hao, S., Niu, X., Wang, P., Wu, Y., et al. (2017) A single-center analysis of Henoch- Schonlein purpura nephritis with nephrotic proteinuria in children. Pediatric Rheumatology, 15, 1–8.
47. Davin, J.C. (2011) Henoch-Schönlein purpura nephritis: Pathophysiology, treatment, and future strategy. Clinical Journal of the American Society of Nephrology, 6, 679–689.
48. Watts, R. (2015) Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrology Dialysis Transplantation, 30, 14-22
27
Anexos
Quadro 1 – Critérios European League Against Rheumatism (EULAR), Paediatric
Rheumatology European Society (PREs) e American College of Rheumatology (ACR)
Critérios Glossário
Púrpura (obrigatório) e pelo menos um dos seguintes:
Púrpura (habitualmente palpável) ou petéquias, com predomínio nos membros inferiores
*sem trombocitopénia
1 – Dor abdominal Dor difusa tipo cólica de início súbito. Pode ocorrer invaginação ou hemorragia 2 - Histopatologia Vasculite leucocitoclástica com
depósitos IgA ou glomerulonefrite proliferativa com depósitos IgA
3 – Artrite ou artralgia Artrite de início súbito (edema, dor, limitação funcional)
Artralgia de início súbito (dor)
4 – Envolvimento renal Proteinúria >0.3g/24h ou rácio urinário albumina/creatina >30 mmol/mg em amostra urinária
Hematúria ou >5 eritrócitos/campo alta resolução ou cilindros eritrocitários no sedimento urinário ou >2+ em tiras de teste
Quadro 2 – Glossário[34][32]
Hematúria Hemoglobina positiva em análise sumária de urina ou
Observação de >5 glóbulos vermelhos por campo em microscopia
Proteinúria Proteínas positivas em análise sumária de urina ou
Proteinúria 4-40 mg/m2/h ou >200 mg/l,
ou
Albuminúria >30 mg/l
Síndrome nefrótica Proteinúria > 40 mg/m2/h ou > 50 mg/kg/24 horas Hipoalbuminémia
Com/sem:
Edema
Síndrome nefrítica Hematúria associada a pelo menos 1:
Aumento da ureia e da creatinina Hipertensão
28
Quadro 3: Critérios de diagnóstico de vasculite ANCA-positivo
Poliangeíte microscópica – Definições Chapel Hill revistas em 2012[48]:
Vasculite necrotizante com depósitos imunes escassos ou ausentes que afeta pequenos vasos.
Glumerulonefrite necrotizante é muito comum. Capilarite pulmonar é frequente. Sem inflamação granulomatosa.
Poliangeíte granulomatosa hipereosinofílica (adultos) - Diagnóstico requer 4 dos seguintes critérios:[11]
História de asma História de alergia
Eosinofilia periférica > 10% Mono ou polineuropatia
Infiltrados pulmonares migratórios
Dor nos seios perinasais ou opacificações em radiografia Biópsia com eosinófilos extravasculares
Poliangeíte granulomatosa - pelo menos 3 dos critérios:[37] Critério Glossário
Histopatologia Inflamação com granulomas na parede arterial
Envolvimento vias aéreas superiores Rinorreia purulenta crónica ou epistáxis/granulomas nasais
Perfuração do septo nasal ou nariz em sela
Inflamação seios crónica ou recorrente Envolvimento
laringo-traqueo-brônquico
Estenose ou envolvimento subglótico, traqueal ou brônquico
Envovimento pulmonar Nódulos, cavitações ou infiltrados fixos em radiografia tórax ou TC
ANCA Positivo por imunofluorescência ou ELISA
Envolvimento renal Proteinúria >0.3 g/24h ou >30 mmol/mg de relação albuminúria:creatinúria em urina ocasional