Mara Vanessa Camacho Fernandes
Carcinoma da Tireóide
2011/2012
2
Mara Vanessa Camacho Fernandes
Carcinoma da Tireóide
Mestrado Integrado em Medicina
Área: Cirurgia Geral
Trabalho efetuado sob a Orientação de:
Professor Doutor António Taveira Gomes
Professor associado convidado da Faculdade de Medicina da Universidade do PortoMarço, 2012
5
Agradecimentos
Agradeço ao Professor Doutor António Taveira Gomes, orientador deste projeto de opção, pela disponibilidade, orientação, críticas e sugestões que muito contribuíram para o meu enriquecimento a nível pessoal e científico.
6
Lista de Abreviaturas
CT – Carcinoma da Tireóide
CMT – Carcinoma Medular da Tireóide
CDNMT – Carcinoma Diferenciado Não Medular da Tireóide CPT – Carcinoma Papilar da Tireóide
CFP – Carcinoma Folicular da Tireóide
MEN II – Neoplasias endócrinas múltiplas tipo II
CDNMFT – Carcinoma Diferenciado Não Medular Familiar da Tireóide PAF – Polipose Adenomatosa Familiar
SC – Síndrome de Cowden
HAD – Hereditariedade Autóssomica Cominante
CMv-CPT – Subtipo Cribiforme-Morular do Carcinoma Papilar da Tireóide AF – Adenoma Folicular
BMN – Bócio Multinodular
MNA – Múltiplos Nódulos Adenomatosos CC – Complexo de Carney
SW – Síndrome de Werner
7
Lista de Figuras
Figura 1: Frequência com que ocorre Carcinoma Diferenciado Não Medular da Tireóide no
8
Lista de Tabelas
Tabela 1:
Caraterísticas clínicas que podem sugerir a presença de patologia hereditária.Tabela 2: Carcinoma Diferenciado Não Medular da Tireóide associado a outras Síndromes
Tumorais Familiares (adaptado de Cameselle et al (28)).
Tabela 3: Carcinoma Diferenciado Não Medular Familiar da Tireóide sem associação a outas
Síndromes Familiares (adaptado de Nosé et al (2))
Tabela 4: Comparação entre caraterísticas clínicas nos doentes com Carcinoma Diferenciado
Não Medular Familiar da Tireóide em estudos publicados ao longo dos anos (adaptado de Robenshtok et al (3)).
9
Índice
Resumo ... 10 Abstract ... 11 Introdução ... 12 Objetivo ... 14 Métodos ... 15Carcinoma Diferenciado Não Medular Familiar da Tireóide ... 16
História Familiar ... 18
Caraterísticas Clínicas ... 19
Agressividade e Recorrência ... 20
Síndromes Familiares associados ... 21
A. Síndromes tumorais familiares com carcinoma diferenciado não medular da tireóide. ... 21
Polipose Adenomatosa Familiar ... 21
Síndrome de Cowden ... 22
Complexo de Carney ... 23
Síndrome de Werner ... 23
B. Carcinoma diferenciado não medular familiar da tireóide sem associação a outras síndromes ... 24
Carcinoma Papilar Familiar da Tireóide ... 24
Discussão ... 25
Conclusão ... 29
Referências bibliográficas... 31
10
Resumo
As neoplasias epiteliais malignas primárias da tireóide podem ser divididas em neoplasias com origem nas células foliculares, designadas de Carcinoma Diferenciado Não Medular da Tireóide e neoplasias com origem nas células C, denominadas Carcinoma Medular.
Em ambos os casos podem ocorrer em contexto esporádico ou familiar.
O Carcinoma Diferenciado Não Medular da Tireóide inclui dois subtipos principais: Carcinoma Papilar e Carcinoma Folicular.
O Carcinoma Medular Familiar pode ocorrer isoladamente ou no contexto de neoplasias endócrinas múltiplas tipo II. A alteração genética que predispõe a esta forma é a mutação do oncogene RET no cromossoma 10q11.2.
O Carcinoma Diferenciado Não Medular Familiar da Tireóide é raro. Por definição, a forma familiar engloba pelo menos dois familiares afetados pela patologia, na ausência de outra síndrome familiar. Os genes responsáveis pela doença ainda não foram totalmente identificados e o tipo de hereditariedade também não está esclarecido.
Neste trabalho abordámos o Carcinoma Diferenciado Não Medular Familiar da Tiróide como uma entidade clínica distinta. Foi realizada uma pesquisa bibliográfica numa base eletrónica de dados, PUBMED, e selecionados artigos sobre o tema.
O Carcinoma Diferenciado Não Medular Familiar da tiróide pode ser mais agressivo e apresentar pior prognóstico. Nos casos familiares as lesões são frequentemente multifocais, incidem em idade mais precoce, metastizam frequentemente para gânglios linfáticos e apresentam maior recorrência.
É importante o desenvolvimento de estudos genéticos que permitam identificar as alterações subjacentes. Na sua ausência, a identificação clínica de uma forma familiar de carcinoma da tiróide é a única forma de conseguir o tratamento atempado.
Palavras-chave: carcinoma não medular familiar da tiróide; carcinoma familiar da tiróide;
11
Abstract
Thyroid neoplasms corresponding to primary epithelial tumors can be divided into: tumors that originate in the follicular cells and tumors originating from C cells, named medullary thyroid carcinoma.
Although the majority is sporadic, familial forms have been described in recent years. Thyroid carcinomas derived from follicular cells are divided into two major subtypes: papillary carcinoma and follicular carcinoma, designated Nonmedullary Thyroid Carcinomas.
Medullary thyroid carcinoma may presents as pure familial syndrome or inherent in multiple endocrine neoplasia type II. These tumors are associated with a RET gene mutation on chromosome 10q11.2.
Familial nonmedullary thyroid carcinoma is rare. A familial case of nonmedullary thyroid cancer is defined as a patient with two or more first-degree relatives with thyroid cancer of follicular cell origin in the absence of another familial syndrome. The genetic inheritance remains unknown. In this paper we aim to characterize hereditary nonmedullary thyroid as a distinct clinical entity. We performed a review of the literature using an electronic bibliographic database(PUBMED) and selected papers on this topic.
Familial nonmedullary thyroid cancer can present a more aggressive phenotype of thyroid cancer and display poor prognosis. It is more often multifocal, occurs at a younger age, with a predisposition for lymph nodes metastasis, extrathyroidal invasion and has higher recurrence rate than its sporadic counterpart.
It is important to developing genetic studies and the clinician must also be aware of an underlying familial thyroid cancer in the presence of certain features in order to allow the correct treatment.
Keywords: familial nonmedullary thyroid carcinoma; familial thyroid carcinoma, papillary
12
Introdução
O carcinoma da tireóide (CT) é o sétimo cancro mais frequentemente diagnosticado nas mulheres (1). É notório o aumento da sua incidência, sendo que nos Estados Unidos são documentados cerca de 33 550 novos casos por ano (2,3). É mais frequente no sexo feminino e aparece, sobretudo, em idades mais jovens (2).
As neoplasias epiteliais primárias da tireóidepodem ser divididas em duas categorias principais:tumores com origem nas células foliculares (95% são deste tipo) e tumores com origem nas células parafoliculares, células C produtoras de calcitonina (cerca de 5% dos tumores) (2,4,5). Os tumores derivados das células C são denominados de Carcinoma Medular da Tireóide (CMT).
Os tumores malignos com origem nas células foliculares, designados de Carcinoma Diferenciado Não Medular da Tireóide (CDNMT), encontram-se divididos em 2 subtipos principais: Carcinoma Papilar da Tireóide (CPT) e Carcinoma Folicular da Tireóide (CFP) (1,2,6). O tipo histológico mais frequente é o papilar (80%), sendo o carcinoma folicular mais raro (15%) (2,4,7).
Quer o carcinoma diferenciado não medular da tireóide quer o carcinoma medular pode ocorrer em contexto esporádico ou familiar.
Quando ocorre em contexto de hereditariedade, o carcinoma medular da tireóide engloba cerca de 25% dos casos e pode ocorrer isoladamente ou como parte integrante de neoplasias endócrinas múltiplas tipo II (MEN- II). A alteração genética que predispõe à forma familiar do CMT é a mutação do oncogene RET no cromossoma 10q11.2. Pela sua elevada prevalência, a presença de história familiar deve ser pesquisada em todos os doentes com CMT
(7,8)
.
Uma vez que a genética dos tumores familiares derivados das células C é bem conhecida e a relação genótipo-fenótipo está bem estabelecida, a abordagem do CMT não foi incluída nos objetivos deste trabalho.
13 O CDNMT é a neoplasia endócrina mais frequente (2,7). No entanto, e em contraste com a história familiar nos doentes com CMT, a presença de hereditariedade nestes tumores só agora começou a ganhar relevo. A grande maioria ocorre de forma esporádica (7);no entanto, estima-se que aproximadamente 5% surja em contexto de história familiar (3,7,9). Com o aumento da incidência de CT, a existência de uma forma familiar tornou-se mais evidente. Estudos populacionais com mais de uma geração em estudo comprovam que o CDNMT constitui uma verdadeira síndrome hereditária (10).
14
Objetivo:
O objetivo deste trabalho centra-se em abordar a forma familiar do carcinoma diferenciado não medular da tireóide como uma entidade clínica distinta, procurando carateriza-la nomeadamente em recarateriza-lação à sua prevalência e incidência, caraterísticas clinico-histológicas, síndromes familiares que podem estar associados, meios de diagnóstico, agressividade, prognóstico e tratamento.
15
Métodos
Foi realizada uma pesquisa numa base bibliográfica eletrónica de dados, PUBMED, com o correspondente em português aos temas: carcinoma não medular familiar da tireóide e carcinoma familiar não-medular da tireóide, tendo sido dado preferência a artigos de revisão.
Os artigos identificados foram escolhidos pelos abstract’s, e nestes foram aplicados os seguintes critérios de inclusão/exclusão:
Critérios inclusão:
a) Artigos escritos em português ou inglês;
b) Artigos sobre Carcinoma Diferenciado Não Medular Familiar da Tireóide; c) Publicados desde Janeiro de 1997 até Dezembro de 2011;
d) Artigos com demonstração clara dos parâmetros descritos nos objetivos.
Critérios exclusão:
a) Artigos sem abstract disponível;
b) Artigos sobre Carcinoma Medular da Tireóide;
c) Artigos sobre Carcinoma Diferenciado Não Medular Familiar da Tireóide com mutações identificadas, excepto quando associado a outra síndrome familiar conhecida.
Dos resultados da pesquisa foi identificado um total de 105 artigos, dos quais 40 foram incluídos neste trabalho.
16
Carcinoma Diferenciado Não Medular Familiar da Tireóide
O carcinoma diferenciado não medular familiar da tireóide (CDNMFT) é raro, (11) contabilizando apenas 5% de todos os CT (3,7,9,11).No entanto, estudos recentes demonstram que a sua incidência tem vindo a aumentar devido ao seguimento médico mais cuidado e ao maior recurso a exames diagnósticos, permitindo a deteção precoce de pequenos nódulos que de outra forma poderiam passar despercebidos (12).
No estudo de Moses et al encontrou-se uma alta prevalência de CDNMFT entre os doentes: 8,8% entre todos os CT e 9,4% entre os casos de CPT (10).
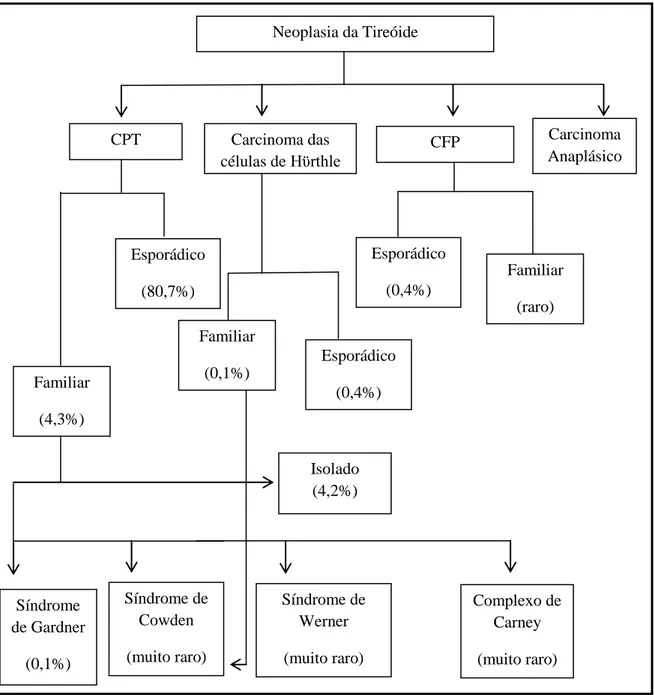
A figura 1, adaptada de Alsanea and Clark (13), pretende ilustrar a frequência relativa com que pode surgir CDNMT em contexto familiar e esporádico, segundo os dados do National
Cancer Database na Califórnia (Anexo 1).
Na última década o CDNMFT tem sido reconhecido como uma entidade clínica distinta. Inicialmente a forma familiar era apenas descrita como parte integrante de síndromes familiares como a Polipose Adenomatosa Familiar (PAF) ou a Síndrome de Cowden (SC). Foi descrita pela primeira vez, sem associação a outras síndromes, em 1955 por Robinson and Orr (14,15,16), em dois gémeos monozigóticos de 24 anos ambos com nódulos palpáveis na tireóide e forma histológica de CPT já metastizada aos gânglios linfáticos (2,7). Desde então, têm vindo a ser realizados uma série de estudos incluíndo doentes com CDNMT onde dois ou mais familiares são afetados por neoplasias da tireóide, demonstrando-se um risco 3 a 10 vezes superior de se encontrar CDNMT nos familiares de primeiro grau dos pacientes com neoplasia diferenciada da tireóide (9,14,17,18).
A definição de forma familiar de CDNMT é dada pela presença de dois ou mais familiares de primeiro grau com o diagnóstico de CDNMT, na ausência de outra síndrome familiar (14,19) ou exposição a fatores que predisponham a CT (3). Como a prevalência de CT na população em geral é elevada, alguns autores consideram que quando dois familiares são diagnosticados com CDNMT existe uma probabilidade de 53% de se tratar de uma forma
17 familiar e que quando são três elementos afetados, esta probabilidade aumenta para 99% (1,9,16). Assim, com apenas dois familiares afetados estaremos mais provavelmente na presença de um caso esporádico. A probabilidade de se tratar de uma forma hereditária de CDNMT aumenta com o número de familiares envolvidos (20).
O tipo de hereditariedade, contudo, ainda não está esclarecido. Baseando-se em estudos familiares com três ou mais indivíduos com diagnóstico de CDNMT, na transmissão entre indivíduos do sexo masculino, na percentagem de homens afetados quando comparado com a forma esporádica e na transmissão horizontal entre irmãos, crê-se tratar-se de hereditariedade autossómica dominante (HAD) com penetrância incompleta (1,3,11,21) e expressão variável
(14,22,23,24)
. No entanto, são necessários mais estudos genéticos para sedimentar esta afirmação. Visto que há poucas condições clínicas próprias, mesmo quando vários membros da família são afetados e por não existirem ainda testes genéticos específicos, a forma familiar não é facilmente distinguível da esporádica (25,26). Surge por vezes a dúvida se os casos de CDNMFT descritos têm realmente uma componente genética, ou ocorrem devido a fatores ambientais ou ainda se podem ser simplesmente atribuídos à elevada prevalência de CT na população em geral.
Como é sabido, os principais fatores de risco para desenvolver CT são o défice ou excesso de iodo (principalmente no desenvolvimento de CFT) e a exposição a agentes carcinogéneos como a radiação ionizante (sobretudo para desenvolver CPT) (1,6). Os indivíduos sujeitos a este tipo de radiação adquirem alterações que podem aumentar a predisposição para desenvolver CT, pelo que alguns autores defendem que doentes com história prévia de exposição a este agente não devem ser incluídos nos estudos de CDNMFT.
Por outro lado, como não está ainda esclarecido se o CDNMFT ocorre apenas devido a herança genética ou se se trata de uma combinação entre fatores genéticos e ambientais, outros autores admitem que a exposição a baixas doses de radiação não deve excluir de todo o diagnóstico de CDNMFT (19).
Histologicamente, as formas esporádica e familiar também não são facilmente diferenciadas. Porém a forma familiar pode apresentar estroma trabecular com oxifília
18 caraterístico (19) e quando associada á PAF, o subtipo cribiforme-morular pode elucidar o seu diagnóstico (2,5,19,27).
Embora raro, o CDNMFT é uma entidade clínica distinta e as suas caraterísticas devem ser esclarecidas. Assim, o presente trabalho foi elaborado numa série de tópicos que focam os aspetos mais importantes referidos em diferentes estudos realizados sobre o CDNMT no contexto familiar.
História Familiar
Como já foi referido, não existem ainda testes genéticos que nos permitam distinguir a forma esporádica da familiar, mas existem caraterísticas que nos podem alertar para a presença de uma componente hereditária.
Quando os carcinomas são detetados em indivíduos do sexo masculino é mais provavelmente um caso familiar; no entanto, tal como acontece na forma esporádica, as mulheres são geralmente o grupo mais afectado (11,27).
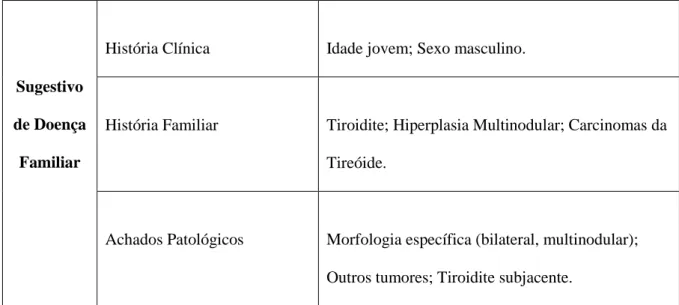
A tabela 1 pretende demonstrar, de forma resumida, algumas caraterísticas clinicopatológicas que podem traduzir hereditariedade (Anexo 2).
Embora não exista consenso entre estudos (23, 28), alguns demonstraram que o CDNMFT é frequentemente multifocal e é diagnosticado em idades mais jovens (25,26). Assim, segundo alguns autores, a presença de 3 ou mais familiares afetados (16,20,29), idade jovem aquando do diagnóstico, sexo masculino, tumores de grandes dimensões, bilaterais e multifocais, apontam para uma maior probabilidade de se tratar de um caso hereditário (2,6,7,24).
Como já foi referido, o risco familiar de um doente com CDNMT vir a apresentar doença é 5 a 10 vezes superior ao da população geral, e este aumenta quando o familiar é de primeiro grau, como por exemplo, um irmão ou ainda maior entre irmãs, sobretudo nos casos de CPT (1,9,19).
Quando comparamos a primeira com a segunda geração (pais-filhos), de doentes com CDNMFT verifica-se que a sua apresentação difere ligeiramente. A segunda geração é geralmente diagnosticada em idade mais precoce e os tumores têm um maior grau de
19 multifocalidade (11). Capezone et al (17) encontraram nos indivíduos da segunda geração maior número de indivíduos do sexo masculino afetados, tumores em estadios mais avançados e pior prognóstico.
Temos assim o chamado fenómeno de antecipação genética, onde a sintomatologia de uma doença genética se manifesta em idade mais precoce na geração seguinte e onde uma maior agressividade dos sintomas é também notória (11,17,30).
Resumindo, podem ser apontadas como principais caraterísticas para se suspeitar de neoplasia familiar da tireóide (31):
Idade jovem;
Incidência de géneros quase idêntica (embora com mais mulheres afetadas); História familiar de nódulos tiroideus ou de carcinoma da tireóide;
Tumores multifocais; Bilateralidade;
Existência de uma lesão percursora (como hiperplasia das células C no CMT);
Ao suspeitar-se de uma forma familiar, toda a informação que se possa recolher junto do doente é importante. Deve questionar-se sobre a história clínica completa, enfatizando sintomas locais e regionais (dor, disfagia, dispneia), exposição prévia a radiação, história familiar de patologia tiroideia benigna ou maligna e sobre história pessoal ou familiar conhecida de outra síndrome familiar que pode aparecer combinada com CDNMFT (19).
Caraterísticas Clínicas
Além da história familiar, não existem grandes diferenças entre as caraterísticas clínicas ou histológicas do CDNMT quando comparamos a forma esporádica com a familiar.
O CDNMFT está associado a uma maior incidência de condições benignas, como o adenoma folicular, o bócio multinodular (32) e a tiroidite de Hashimoto (19,26,30). Musholt et al (24) propuseram que o diagnóstico de bócio multinodular em 3 familiares de primeiro ou segundo
20 grau de um paciente com CPT constitui um critério primário para o diagnóstico de CDNMFT
(19)
. Está descrito que cerca de 36-57% dos doentes com CDNMFT tem história pregressa ou familiar de patologia benigna da tireóide (26,30).
A bilateralidade e multinodolaridade são critérios que também ocorrem com mais frequência nesta forma (1,16,21,33).
Um estudo retrospetivo a doentes com CDNMFT tratados entre 1980-1994 na Califórnia revelou que a incidência de multifocalidade era cerca de 90%, que a bilateralidade ocorria em 43% dos casos, que a metastização ao nível dos gânglios linfáticos e invasão local se verificava em 57% e que a taxa de recorrência era cerca de 50% (33).
Agressividade e Recorrência
A maioria dos estudos é ainda pouco clara no que toca a este assunto.
Estudos multicêntricos afirmam que a forma familiar é mais agressiva e tem maior taxa de recorrência pelo facto de aparecer em idades mais jovens e ser frequentemente multifocal, aconselhando por isso uma abordagem e tratamento mais dirigidos (19). O facto de metastizar frequentemente para os gânglios linfáticos e de invadir estruturas locais traduzem a sua maior agressividade (19). Num estudo caso-controlo retrospetivo multicêntrico (25), o CDNMFT é considerado como mais agressivo, com uma taxa de recorrência de 44% comparado com o grupo de controlo. O tempo livre de doença seria também mais curto.
Por outro lado, em estudos menores demonstra-se que esta forma não apresenta pior prognóstico quando comparada com a esporádica, pelo que a necessidade de vigilância não será necessário nos familiares de pacientes afetados (28).
Loh K-C (34) na sua revisão da literatura de 1955 a 1996 não encontrou evidências de se tratar de uma forma mais agressiva quando comparada com a forma esporádica. Por outro lado, Sippel et al (19) que reviram a literatura de 1996 a 2008 demonstraram maior agressividade e tempo livre de doença mais curto.
Quanto ao prognóstico as opiniões divergem igualmente. Estudos que demonstram o CDNMFT como mais agressivo também lhe atribuem pior prognóstico. A recorrência está
21 também associada a um prognóstico menos favorável. Cerca de 1/3 dos doentes com carcinoma da tireóide recorrente irão falecer desta doença (19).
Verifica-se ainda que nos estudos em que os doentes são divididos pelo número de familiares afetados as diferenças são mais evidentes: doentes com 3 ou mais familiares afetados apresentam pior prognóstico (16,25). Os fatores preditores de mau prognóstico são assim, o número de familiares com CDNMT e a presença de metástases locais e à distância (25,34,35).
Síndromes Familiares associados
O CDNMT engloba um grupo heterogéneo de doenças, podendo ser dividido em dois principais com base em características clinico-patológicas que foram já descritas previamente (idade jovem ao diagnóstico, a multicentricidade e a bilateralidade). O primeiro, A, é constituído por um conjunto de síndromes familiares onde o CDNMT aparece como uma componente minor, e não como tumor principal. O segundo grupo, B, é formado por um conjunto de tumores familiares, onde a neoplasia da tireóide, nomeadamente o CDNMT/CDNMFT, é predominante (2,4,5,7,8,29,31,36).
A. Síndromes Tumorais Familiares com Carcinoma Diferenciado Não
Medular da Tireóide
Neste grupo o CDNMT surge no contexto de outra síndrome familiar. Na base destas síndromes está provavelmente um gene responsável por aumentar o risco de desenvolver CDNMT. São exemplos: a Polipose Adenomatosa Familiar, a Síndrome de Cowden, o Complexo de Carney e a Síndrome de Werner, entre outros.
Polipose Adenomatosa Familiar
Trata-se de uma doença familiar causada por mutações germinativas no gene APC, cuja principal manifestação é o desenvolvimento de pólipos adenomatosos no cólon e reto.
22 Das manifestações extracólicas pode salientar-se o CPT que ocorre em cerca de 2-12% dos doentes com PAF (3,12). A baixa incidência de neoplasia da tireóide nestes doentes sugere que esta ocorre devido a suscetibilidade genética (12).
Histologicamente tem geralmente uma apresentação própria: variante cribiforme-morular do CPT (CMv-CPT). De entre os doentes com CPT na PAF, cerca de 90% apresentam este subtipo histológico (2), pelo que, em todos os doentes onde esta histologia está presente deve ser investigado se esta síndrome aparece subjacente. É frequentemente bilateral, atinge sobretudo jovens, é 10 vezes mais frequente em mulheres e apresenta prognóstico semelhante ao CPT clássico (12).
Algumas caraterísticas celulares e nucleares como núcleos claros, crescimento nuclear e
over-lapping que aparecem nas formas esporádicas, são muito raras nas formas familiares, o que
permite alertar para a possibilidade de se estar na presença de uma síndrome familiar (4,5,7,8,29). Devido á baixa incidência de CPT nos doentes com FAP, não estão padronizados exames de rotina, como ecografia ou TC cervical (12).
Síndrome de Cowden (SC)
Caraterizada pela presença de múltiplos hamartomas. É causada por um conjunto de mutações germinativas inativantes no gene supressor tumoral PTEN.
A patologia tiroideia maligna é a segunda manifestação mais frequente e afeta principalmente as células foliculares (12,37). O CFT é encontrado em 5 a 10% dos pacientes (37). É multicêntrico e crê-se que a lesão percursora seja o adenoma folicular (AF) tratando-se de um critério major para o diagnóstico de SC, enquanto a presença de bócio multinodular (BMN) ou de múltiplos nódulos adenomatosos (MNA) representam critérios minor. O CPT é mais raro mas também pode ocorrer (2).
Assim, a presença de doença tiroideia benigna multinodular e bilateral como AF ou BMN deve alertar o médico para a existência de uma possível síndrome subsequente como o SC e, tendo em conta que se trata de uma síndrome familiar, é importante o estudos dos respetivos familiares (2,4,8,29). A elevada incidência de patologia tiroideia nos doentes com SC determina
23 que estes devem ser submetidos a ecografia cervical de rotina, devendo ser ponderada tiroidectomia quando a ecografia ou a biópsia são inconclusivas (12).
Complexo de Carney (CC)
É uma doença autossómica dominante que se carateriza por hiperpigmentação cutânea e pela presença de várias neoplasias endócrinas como tumores da hipófise, tumores adrenais e neoplasias da tireóide. Geralmente apresenta-se com doença multinodular (MNA) ou AF e o CPT e CFT podem surgir em cerca de 15% dos doentes (2,7,8,29).
Uma vez que a presença de nódulos tiroideus é frequente, a ecografia de rotina pode ser importante na identificação precoce de CDNMT nestes doentes (12).
Síndrome de Werner (SW)
É uma doença rara, autossómica recessiva, que se inicia frequentemente na 3ª década, caraterizada por uma mutação no gene WRN. Estas mutações estão especificamente relacionadas com malignidade como melanomas, sarcomas e CDNMT (5).
A patologia tiroideia surge nos pacientes mais jovens (≈ 34 anos). Na população japonesa existe um risco 3 vezes superior de desenvolver CFT comparado com a população em geral. Nos doentes caucasianos encontramos apenas CPT (4,5,7).
A elevada prevalência de CT nesta síndrome predispõe a que o exame físico e ecografia de rotina estejam preconizados nestes doentes (12).
O CDNMT nos doentes com alguma destas síndromes familiares foi considerado como sendo mais agressivo em comparação com a forma esporádica. É importante que o clínico anteveja a possibilidade de que, na presença de CDNMT o doente possa apresentar outra síndrome subjacente ou vice-versa, uma vez que o CDNMT pode ser a apresentação inicial destas síndromes (12).
24
B. Carcinoma Diferenciado Não Medular Familiar da Tireóide sem associação
a outras síndromes
Este grupo inclui as famílias onde o CDNMT é o principal tumor, sem outras síndromes familiares associadas. Nele estão incluídos o CPT familiar (fCPT), CPT variante de células oxifílicas e o BMN familiar (37). Nestes, o tipo histológico mais frequente é também o CPT.
Carcinoma Papilar Familiar da Tireóide
Ao longo dos anos, tem sido descrito casos familiares nos doentes com CPT, sendo que este aparece frequentemente em pais-filhos, irmãos e em outros familiares de 1º grau (22). Ao analisar várias famílias com CPT, verifica-se que cerca de 3,5-10% dos doentes têm pelo menos um familiar de 1º grau com diagnóstico de CPT (37).
Um subtipo de fCPT é o microcarcinoma papilar. Nas famílias com CPT este subtipo é descrito como mais agressivo. Numa revisão da literatura, Harach et al verificaram que de um total de 33 famílias com microcarcinomas papilares, 15% apresentavam metástases regionais e 3% metástases à distância (37). É também duas vezes mais frequente nas neoplasias MEN-II em comparação com a população em geral (31).
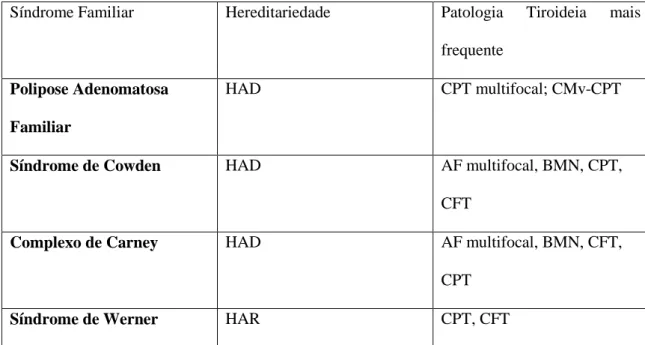
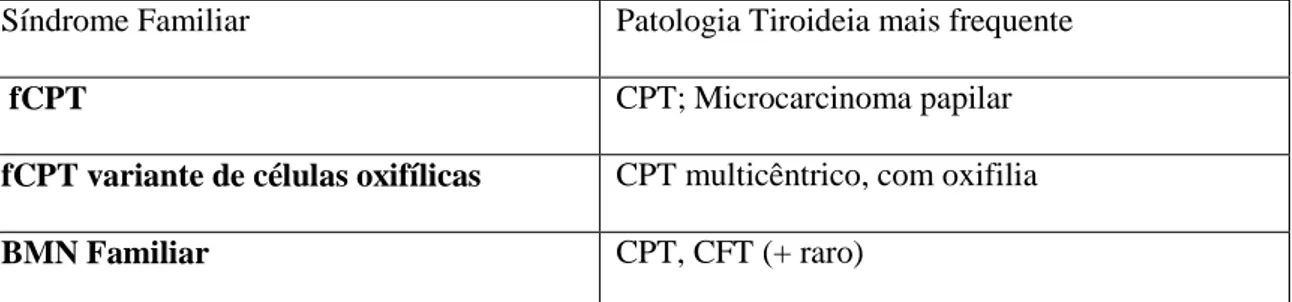
A Tabela 2 e 3 resumem as caraterísticas principais acabadas de referir, como o tipo de patologia tiroideia, de ambos os subgrupos A e B respectivamente (Anexo 3 e 4 respetivamente):
25
Discussão
Na revisão da literatura realizada pode verificar-se que as caraterísticas do CDNMFT não estão totalmente esclarecidas. Embora existam vários estudos sobre o tema há uma certa dificuldade em estudar a forma familiar do CDNMT, por um lado por ser relativamente rara, tornando-se difícil agrupar vários casos para um estudo, e por outro pela dificuldade em obter-se a história clínica dos respetivos familiares (38).
A existência de hereditariedade é apoiada pelo facto de existir transmissão
vertical entre gerações, transmissão horizontal entre irmãos e uma percentagem elevada
de indivíduos do sexo masculino afetados
(17,18,19,34).Também a presença do fenómeno de
antecipação genética, isto é, na segunda geração o CDNMT se manifestar mais
precocemente e em estadio mais avançado
(31, 32), parece ser sugestivo da forma familiar. Existe alguma controvérsia e os diferentes estudos não obtêm resultados idênticos quanto a determinados tópicos como caraterísticas clínicas, agressividade e prognóstico do CDNMFT. Esta secção do trabalho tenta focar, resumidamente, as várias conclusões sobre as caraterísticas mais importantes.
No maior estudo feito a 6458 doentes (26), onde foram identificados 258 casos de CDNMFT, chegou-se à conclusão que a idade ao diagnóstico (49 anos) não era diferente da encontrada na forma esporádica. O tamanho entre formas também não apresentava diferenças. Os tumores multifocais eram mais frequentes nos doentes com CDNMFT (42%) e estes apresentavam frequentemente BMN (41% versus 29% na esporádica).
No que respeita à agressividade, por ser geralmente multifocal e estar associado a metastização aos gânglios linfáticos e invasão de estruturas locais, alguns autores consideram-no como uma forma mais agressiva e com maior taxa de recorrência comparada com os tumores esporádicos (14, 19, 26).
Contudo outros estudos contradizem esta opinião. O estudo de Robenshtok et al (3) demonstrou que o CDNMFT é geralmente diagnosticado mais cedo (≈ 5anos) e que para além
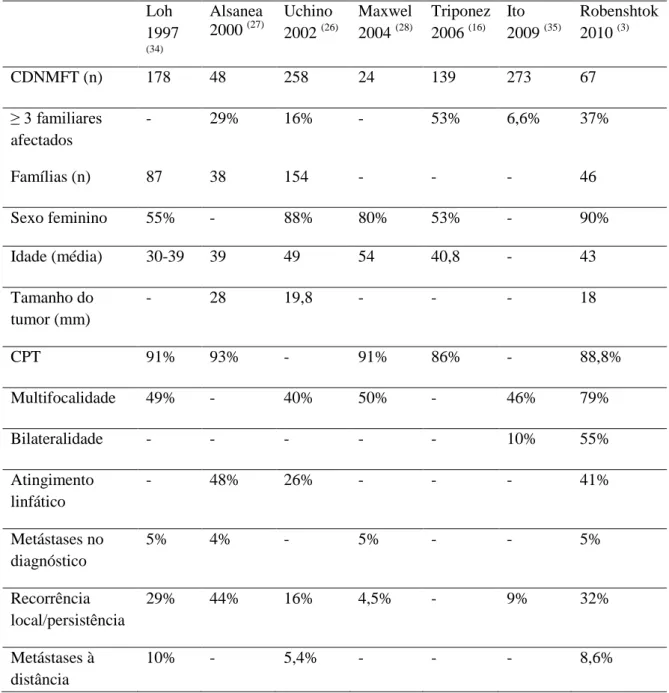
26 desta caraterística, a apresentação clínica, o prognóstico, tratamento a adoptar e o tempo livre de doença não diferem da forma esporádica. Este estudo fez a comparação entre diferentes estudos realizados ao longo dos anos que confrontavam vários parâmetros entre as duas formas. A tabela 4 (Anexo 5) procura resumir os resultados obtidos nestes estudos.
A meta-análise realizada em 1997 por Loh K-C (34) revelou que a forma familiar não seria mais agressiva em comparação com a esporádica. Um estudo caso-controlos (28) não encontrou diferenças entre as formas no que respeita à invasão, metastização à distância e tempo livre de doença. Uchino et al (26) demonstraram que a recorrência local era maior na forma familiar, mas em termos de sobrevivência e de ocorrência de metástases à distância não existiam diferenças evidentes.
A discrepância entre os resultados pode ser explicada pelo facto de na maioria
dos estudos serem incluídos doentes com apenas 2 familiares afetados, e como foi
referido, há probabilidade de se se tratar de um caso esporádico, o que pode traduzir nos
resultados um comportamento menos agressivo
(20). Uma outra razão prende-se com o facto de ainda não serem conhecidos os genes responsáveis pelo CDNMFT que poderiam explicar a existência de um defeito genético responsável por um fenótipo mais/menos agressivo (3).
Assim, verificamos que existe alguma concordância em certos aspetos: doentes com a forma familiar CDNMT são diagnosticados com idade ligeiramente inferior, apresentam frequentemente doença tiroideia benigna e frequentemente multifocal. Maior agressividade, pior prognóstico e maior taxa de recorrência podem, por vezes, aparecer associados a esta forma.
A importância de determinar as caraterísticas do CDNMFT e se este é realmente
mais agressivo quando comparado com a forma esporádica suscita algumas dúvidas.
Por exemplo, é necessário estudar todos os familiares de primeiro grau dos doentes com
CDNMT? Quem deve ser estudado? Com que idade se deve iniciar o rastreio? Como
deve ser feito o estudo? O exame físico é suficiente ou devem ser realizados exames
imagiológicos? Quem necessita ou beneficia de tratamento mais agressivo?
27
Quanto aos indivíduos que devem ser estudados, Sippel et al
(19)admitem que
todos os doentes, com pelo menos um familiar de primeiro grau com diagnóstico de
CDNMT, devem ser questionados sobre a sua história clínica e familiar (estruturar a
árvore genealógica da família pode ser importante para mais facilmente identificar e
quantificar familiares afetados)
(27).O exame físico completo também deve ser realizado.
A ecografia cervical deve ser utilizada para esclarecer eventuais casos duvidosos.
Nos doentes com CT multifocal diagnosticado ou BMN, mesmo sem familiares
afetados, deve ser realizada ecografia cervical, visto que estas condições clínicas estão
presentes com alguma frequência nos doentes com CDNMFT.
Na presença de 2 ou mais familiares com CDNMT a história clínica completa
deve ser sempre questionada e o exame físico e a ecografia cervical devem ser
realizados. Se forem negativos, é recomendado o follow-up anual destes doentes
(19).
Quando são detetados nódulos à palpação em adultos jovens ou indivíduos do
sexo masculino, estes devem ser avaliados com ecografia cervical e biópsia dos
respetivos nódulos
(27).Assim, não existindo nenhum teste genético para estudar a componente
hereditária do CDNMT, a ecografia cervical tem um papel importante no seu
diagnóstico
(19), permitindo identificar precocemente os casos familiares
(30). Através
deste exame uma elevada incidência de CPT assintomático foi detetada nos familiares
de doentes com CDNMFT, demonstrando a importância do rastreio ecográfico
(39).
Como se verificou, o CDNMFT não surge sempre na mesma idade, pelo que se
considera que o rastreio deve ser iniciado cerca de 5-10 anos mais cedo em relação à
idade de diagnóstico do primeiro familiar identificado
(19).
Quanto ao tratamento a ser utilizado, alguns autores admitem que os doentes
com história familiar positiva de CDNMT, pela elevada disseminação intraglandular e
28
metastização linfática, devem ser submetidos a uma terapia mais agressiva como
tiroidectomia total com dissecção cervical profiláctica
(19,20,33,40),diminuindo assim a
recorrência deste tumor. O estudo de Ito et al
(35)demonstrou que os doentes com
CDNMFT não submetidos a tiroidectomia total apresentavam maior recorrência na
tireóide remanescente, pelo que o tratamento recomendado é idêntico ao dos tumores
esporádicos.
Em contraste com estes, outros estudos
(28,34)que chegaram a conclusão que a
agressividade não difere entre formas, assumem que não haverá necessidade de se
submeter os doentes a tratamentos radicais.
29
Conclusão
Da realização deste trabalho podemos concluir que a forma familiar do CDNMT é rara, contabilizando cerca de 5% dos tumores. O tipo de hereditariedade parece ser autossómico dominante.
A maior dificuldade em distinguir a forma esporádica da familiar prende-se com o facto de além de rara e praticamente indistinta histologicamente da forma esporádica, ainda não são conhecidos os genes responsáveis pela patologia (excepto em alguns casos de CPT variante de células oxifílicas).
Os critérios primários de definição propostos por Musholt et al (24) assentam na presença de pelo menos 2 familiares de primeiro grau afetados por CDNMT ou um doente com CPT com pelo menos 3 familiares de primeiro ou segundo grau com BMN. Como critérios secundários admite CT diagnosticado antes dos 35 anos, presença multifocalidade ou bilateralidade, grande crescimento tumoral, presença de metástases e patologia tiroideia em jovens. A predisposição hereditária para CPT é considerada se se cumprir qualquer um dos critérios primários, ou 1 critério primário e 3 secundários.
O CDNMT pode aparecer associado a outras síndromes familiares como a
Polipose Adenomatosa Familiar, a Síndrome de Cowden, o Complexo de Carney ou a
Síndrome de Werner pelo que, a história clínica e familiar completa deve ser sempre
questionada e o seu diagnóstico deve ser excluído nos doentes com patologia tiroideia.
Alguns estudos chegaram á conclusão que as principais caraterísticas clínicas do CDNMFT são a idade jovem ao diagnóstico, a multicentricidade e bilateralidade, a capacidade de invasão local, de atingimento de tecidos extra-tiroideus e de metastizar aos gânglios linfáticos, a elevada recorrência, a histologia específica e a patologia tiroideia benigna de base (como tiroidite, hiperplasia nodular e BMN). Baseando-se nestas características, bem como no diagnóstico em indivíduos do sexo masculino, o clínico deve estar alerta para a possibilidade de
30 se tratar de um caso hereditário. E, como há possibilidade de ser efectivamente mais agressivo, os doentes devem ser seguidos regularmente e a tiroidectomia total deve ser ponderada.
Para concluir, é importante o desenvolvimento de estudos genéticos que permitam identificar as alterações subjacentes. Na sua ausência, a identificação clínica de uma forma familiar de carcinoma da tiróide é a única forma de conseguir o tratamento atempado.
31
Referências
1.
Kebebew E. Hereditary non-medullary thyroid cancer. World J Surg 2008; 32:678-682.2.
Dotto J, Nosé V. Familial thyroid carcinoma: A diagnostic algorithm. AdvAnatPathol2008; 15(6):332-49.
3.
Robenshtok E, Tzvetov G, Grozinsky-Glasberg S, et al. Clinical characteristics and outcome of familial nonmedullary thyroid cancer: a retrospective controlled study. Thyroid 2011; 21(1):43-8.4.
Nosé V. Thyroid cancer of follicular cell origin in inherited tumor syndromes. AdvAnat Pathol 2010; 17(6):428-36.5.
Nosé V. Familial follicular cell tumors: classification and morphological characteristics. Endocrine Pathology 2010; 21:219-226.6.
Hemminki K, Eng C, Chen B. Familial risks for nonmedullary thyroid cancer. The Journal of Clinical Endocrinology & Metabolism 2005; 90(10):5747-5753.7.
Nosé V. Familial non-medullary thyroid carcinoma: An update. Endrocr Pathol 2008; 19:226-240.8.
Nosé V. Familial thyroid cancer: A review. Modern Pathology 2011; 24,S19-S33.9.
Santos R, Melo T, Assumpção L. Carcinoma diferenciado não medular familiar datireóide. Arq Bras Endocrinol Metab 2007; 51/5.
10.
Moses W, Weng J, Kebebew E. Prevalence, clinicophatologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid 2011; 21(4):367-71.11.
Hillenbrand A, Varhaug J-E, Brauckhoff M, et al. Familial nonmedullary thyroid carcinoma-clinical relevance and prognosis. A European multicenter study. Langenbecks Arch Surgery 2010; 395:851-858.12.
Richards ML. Familial syndromes associated with thyroid cancer in the era of personalized medicine. Thyroid 2010; 20(7):707-13.32
13.
Alsanea O, Clark OH. Familial thyroid cancer. Current Opinion in Oncology 2001;13:44-51.
14.
Mazeh H, Benavidez J, Poehls JL, et al. In patients with thyroid cancer of follicular cell origin, a family history of non-medullary thyroid cancer in one first degree relative is associated with more aggressive disease. Epub 2011 Dec 2.15.
Frich L, Glattre E, Akslen LA. Familial occurrence of nonmedullary thyroid cancer: A population-based study of 5673 first-degree relatives of thyroid cancer patients from Norway. Cancer Epidemiol Biomarkers Prev 2001; 10(2):113-7.16.
Triponez F, Wong M, Sturgeon C, et al. Does familial non-medullary thyroid cancer adversely affect survival?. World J Surg 2006; 30:787-793.17.
Capezzone M, Marchisotta S, Cantara S, et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocrine-Related Cancer 2008; 15 1075-1081.18.
Ito Y, Fukushima M, Yabuta T, et al. Prevalence and prognosis of familial follicular thyroid carcinoma. Endocrine Journal 2008; 55:847-852.19.
Sippel RS, Caron NR, Clark OH. An evidence-based approach to familial nonmedullary thyroid cancer: screening, clinical management, and follow-up. World J Surg 2007; 31:924-933.20.
Charkes ND. On the prevalence of familial nonmedullary thyroid cancer in multiply affected kindreds. Thyroid 2006; 16(2):181-6.21.
Fagin JA. Familial nonmedullary thyroid carcinoma-The case for genetic susceptibility. Journal of Clinical Endocrinology and Metabolism 1997; 82(2):342-4.22.
Pal T, Volg FD, Chappuis PO, et al. Increased risk for nonmedullary thyroid cancer in the first-degree relatives of prevalent cases of nonmedullary thyroid cancer: a hospital-based study. J EndocrinolMetab 2001; 86:5307-5312.33
23.
Leprat F, Bonichon F, Guyot M, et al. Familial non-medullary thyroid carcinoma: pathology review in 27 affected cases from 13 French families. Clin Endocrinol (Oxf) 1999, 50:589–594.24.
Musholt TJ, Musholt PB, Petrich T, et al. Familial papillary thyroid carcinoma:genetics, criteria for diagnosis, clinical features, and surgical treatment. World J Surg 2000; 24:1409-1417.25.
Alsanea O, Wada N, Ain K, et al. Is familial non-medullary thyroid carcinoma more agressive than sporadic thyroid cancer? A multicenter series. Surgery 2000:1043-105.26.
Uchino S, Noguchi S, Kawamoto H, et al. Familial nonmedullary thyroid carcinomacharacterized by multifocality and a high recurrence rate in a large study population. World J Surg 2002; 26:897-902.
27.
Alsanea O. Familial nonmedullary thyroid cancer. Current Treatment Options in Oncology 2000; 1:345-351.28.
Maxwell EL, Hall FT, Freeman JL. Familial non-medullary thyroid cancer: A matched-case control study. The Laryngoscope 2004; 2182-2186.29.
Cameselle-Teijeiro J. The pathologist’s role in familial nonmedullary thyroid tumors. Int J Surg Pathol 2010; 18:194S.30.
Prazeres H, Torres J, Soares P, et al. The familial counterparts of follicular cell-derived thyroid tumors. Int J Surg Pathol 2010; 18(4):233-242.31.
Livolsi VA, Baloch ZW. Familial thyroid carcinoma: the road less travelled in thyroid pathology. Diagnostic Histopathology 2009; 15:87-94.32.
Pitoia F, Cross G, Salvati ME, et al. Patients with familial non-medullary thyroid cancer have an outcome similar to that of patients with sporadic papillary thyroid tumors. Arq Bras Endocrinol Metabol 2011; 55(3):219-23.33.
Sturgeon C, Clark OH. Familial nonmedullary thyroid cancer. Thyroid 2005; 15 (6):588-593.34
34.
Loh K-C. Familial nonmedullary thyroid carcinoma: a meta-review of case series.Thyroid 1997; 7:107-113.
35.
Ito Y, Kakudo K, Hirokawa M, et al. Biological behavior and prognosis of familial papillary thyroid carcinoma. Surgery 2009; 145(1):100-5.36.
Malchoff CD, Malchoff DM. Familial nonmedullary thyroid carcinoma. Cancer Control 2006; 13(2):106-10.37.
Harach HR. Familial nonmedullary thyroid neoplasia. Endocrine Pathology 2001; 12 (2):97-112.38.
Galanti MR, Ekbom A, Grimelius L, Yuen J. Parental cancer and risk of papillary and follicular thyroid carcinoma. British Journal of Cancer 1997; 75(3):451-456.39.
Uchino S, Noguchi S, Yamashita H, et al. Detection of asymptomatic differentiated thyroid carcinoma by neck ultrasonographic screening for familial nonmedullary thyroid carcinoma. World J Surg 2004; 28(11):1099-1022.40.
Vriens MR, Suh I, Moses W, et al. Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid 2009; 19(12):1343-9.35
Anexos
Anexo 1:
Figura 1: Frequência com que ocorre Carcinoma Diferenciado Não Medular da Tireóide no
contexto de doença hereditária versus esporádica (adaptado de Alsanea et al (13)).
Abreviaturas: CPT: Carcinoma Papilar da Tireóide; CFP: Carcinoma Folicular da Tireóide.
Neoplasia da Tireóide CPT Carcinoma das células de Hϋrthle CFP Carcinoma Anaplásico Esporádico (80,7%) Familiar (4,3%) Isolado (4,2%) Complexo de Carney (muito raro) Síndrome de Werner (muito raro) Síndrome de Cowden (muito raro) Síndrome de Gardner (0,1%) Familiar (0,1%) Esporádico (0,4%) Esporádico (0,4%) Familiar (raro)
36
Anexo 2:
Tabela 1:
Caraterísticas clínicas que podem sugerir a presença de patologia hereditária.Sugestivo de Doença
Familiar
História Clínica Idade jovem; Sexo masculino.
História Familiar Tiroidite; Hiperplasia Multinodular; Carcinomas da Tireóide.
Achados Patológicos Morfologia específica (bilateral, multinodular); Outros tumores; Tiroidite subjacente.
37
Anexo 3:
Tabela 2: Carcinoma Diferenciado Não Medular da Tireóide associado a outras Síndromes
Tumorais Familiares (adaptado de Cameselle et al (28)).
Síndrome Familiar Hereditariedade Patologia Tiroideia mais frequente
Polipose Adenomatosa Familiar
HAD CPT multifocal; CMv-CPT
Síndrome de Cowden HAD AF multifocal, BMN, CPT, CFT
Complexo de Carney HAD AF multifocal, BMN, CFT, CPT
Síndrome de Werner HAR CPT, CFT
Abreviaturas: HAD: Hereditariedade Autossómica Dominante; CPT: Carcinoma Papilar da
Tireóide; CMv-CPT: Subtipo Cribiforme-Morular do Carcinoma Papilar da Tireóide; AF: Adenoma Folicular; BMN: Bócio Multinodular; CFT: Carcinoma Folicular da Tireóide; HAR: Hereditariedade Autossómica Recessiva.
38
Anexo 4:
Tabela 3: Carcinoma Diferenciado Não Medular Familiar da Tireóide sem associação a outas
Síndromes Familiares (adaptado de Nosé et al (2)).
Síndrome Familiar Patologia Tiroideia mais frequente
fCPT CPT; Microcarcinoma papilar
fCPT variante de células oxifílicas CPT multicêntrico, com oxifilia
BMN Familiar CPT, CFT (+ raro)
Abreviaturas: fCPT: Carcinoma Papilar Familiar da Tireóide; CPT: Carcinoma Papilar da
39
Anexo 5:
Tabela 4: Comparação entre caraterísticas clínicas nos doentes com Carcinoma Diferenciado
Não Medular Familiar da Tireóide em estudos publicados ao longo dos anos (adaptado de Robenshtok et al (3)). Loh 1997 (34) Alsanea 2000 (27) Uchino 2002 (26) Maxwel 2004 (28) Triponez 2006 (16) Ito 2009 (35) Robenshtok 2010 (3) CDNMFT (n) 178 48 258 24 139 273 67 ≥ 3 familiares afectados - 29% 16% - 53% 6,6% 37% Famílias (n) 87 38 154 - - - 46 Sexo feminino 55% - 88% 80% 53% - 90% Idade (média) 30-39 39 49 54 40,8 - 43 Tamanho do tumor (mm) - 28 19,8 - - - 18 CPT 91% 93% - 91% 86% - 88,8% Multifocalidade 49% - 40% 50% - 46% 79% Bilateralidade - - - 10% 55% Atingimento linfático - 48% 26% - - - 41% Metástases no diagnóstico 5% 4% - 5% - - 5% Recorrência local/persistência 29% 44% 16% 4,5% - 9% 32% Metástases à distância 10% - 5,4% - - - 8,6%
Abreviaturas: CDNMFT: Carcinoma Diferenciado Não Medular Familiar da Tireóide; CPT:
40 Anexo 6:
Normas de publicação segundo a revista Arquivos de Medicina
O texto deve ser limitado a 5000 palavras, excluindo referências e tabelas, e apresentar um máximo de 5 tabelas e/ou figuras (total). As revisões quantitativas devem ser organizadas em introdução, métodos, resultados e discussão/conclusão. As revisões devem apresentar resumos não estruturados em português e em inglês, com um máximo de 250 palavras cada, devendo ser estruturados no caso das revisões quantitativas.
Formatação dos Manuscritos
A formatação dos artigos submetidos para publicação nos ARQUIVOS DE MEDICINA deve seguir os “Uniform Requirements for Manuscripts Submitted to Biomedical Journals”. Todo o manuscrito, incluindo referências, tabelas e legendas de figuras, deve ser redigido a dois espaços, com letra a 11 pontos, e justificado à esquerda. Aconselha-se a utilização das letras
Times, Times New Roman, Courier, Helvetica, Arial, e Symbol para caracteres especiais.
Devem ser numeradas todas as páginas, incluindo a página do título.
Devem ser apresentadas margens com 2,5 cm em todo o manuscrito. Devem ser inseridas quebras de página entre cada secção. Não devem ser inseridos cabeçalhos nem rodapés.
Deve ser evitada a utilização não técnica de termos estatísticos como aleatório, normal, significativo, correlação e amostra. Apenas será efectuada a reprodução de citações, tabelas ou ilustrações de fontes sujeitas a direitos de autor com citação completa da fonte
Unidades de medida
Devem ser utilizadas as unidades de medida do Sistema Internacional (SI), mas os editores podem solicitar a apresentação de outras unidades não pertencentes ao SI.
41
Nomes de medicamentos
Deve ser utilizada a Designação Comum Internacional (DCI) de fármacos em vez de nomes comerciais de medicamentos. Quando forem utilizadas marcas registadas na investigação, pode ser mencionado o nome do medicamento e o nome do laboratório entre parêntesis.
Abreviaturas
Devem ser evitados acrónimos e abreviaturas, especialmente no título e nos resumos. Quando for necessária a sua utilização devem ser definidos na primeira vez que são mencionados no texto e também nos resumos e em cada tabela e figura, excepto no caso das unidades de medida.
Página do título
Na primeira página do manuscrito deve constar: 1) O título (conciso e descritivo);
2) Um título abreviado (com um máximo de 40 caracteres, incluindo espaços);
3) Os nomes dos autores, incluindo o primeiro nome (não incluir graus académicos ou títulos honoríficos);
4) A filiação institucional de cada autor no momento em que o trabalho foi realizado;
5) O nome e contactos do autor que deverá receber a correspondência, incluindo endereço, telefone, fax e e-mail;
6) Os agradecimentos, incluindo fontes de financiamento, bolsas de estudo e colaboradores que não cumpram critérios para autoria;
7) Contagem de palavras separadamente para cada um dos resumos e para o texto principal (não incluindo referências, tabelas ou figuras).
42
Autoria
Como referido nos “Uniform Requirements for Manuscripts Submitted to Biomedical
Journals”, a autoria requer uma contribuição substancial para:
1) Concepção e desenho do estudo, ou obtenção dos dados, ou análise e interpretação dos dados; 2) Redacção do manuscrito ou revisão crítica do seu conteúdo intelectual;
3) Aprovação final da versão submetida para publicação.
A obtenção de financiamento, a recolha de dados ou a supervisão geral do grupo de trabalho, por si só, não justificam autoria. É necessário especificar na carta de apresentação o contributo de cada autor para o trabalho. Esta informação será publicada.
Agradecimentos
Devem ser mencionados na secção de agradecimentos os colaboradores que contribuíram substancialmente para o trabalho mas que não cumpram os critérios para autoria, especificando o seu contributo, bem como as fontes de financiamento, incluindo bolsas de estudo.
Resumos
Os resumos de artigos de investigação original, publicações breves, revisões quantitativas e séries de casos devem ser estruturados (introdução, métodos, resultados e conclusões) e apresentar conteúdo semelhante ao do manuscrito.
Os resumos de manuscritos não estruturados (revisões não quantitativas e casos clínicos) também não devem ser estruturados. Nos resumos não devem ser utilizadas referências e as abreviaturas devem ser limitadas ao mínimo.
Palavras-chave
Devem ser indicadas até seis palavras-chave, em portugês e em inglês, nas páginas dos resumos, preferencialmente em concordância com o Medical Subject Headings (MeSH)
43 utilizado no Index Medicus. Nos manuscritos que não apresentam resumos as palavras-chave devem ser apresentadas no final do manuscrito.
Introdução e Objetivo
Deve mencionar os objetivos do trabalho e a justificação para a sua realização. Nesta secção apenas devem ser efectuadas as referênciasindispensáveis para justificar os objetivos do estudo.
Métodos
Nesta secção devem descrever-se: 1) A amostra em estudo;
2) A localização do estudo no tempo e no espaço; 3) Os métodos de recolha de dados;
4) Análise dos dados.
As considerações éticas devem ser efectuadas no final desta secção.
Análise dos dados
Os métodos estatísticos devem ser descritos com o detalhe suficiente para que possa ser possível reproduzir os resultados apresentados. Sempre que possível deve ser quantificada a imprecisão das estimativas apresentadas, designadamente através da apresentação de intervalos de confiança. Deve evitar-se uma utilização excessiva de testes de hipóteses, com o uso de valores de p, que não fornecem informação quantitativa importante. Deve ser mencionado o
software utilizado na análise dos dados.
Considerações éticas e consentimento informado
Os autores devem assegurar que todas as investigações envolvendo seres humanos foram aprovadas por comissões de ética das instituições em que a investigação tenha sido desenvolvida, de acordo com a Declaração de Helsínquia da Associação Médica Mundial
44 (www.wma.net). Na secção de métodos do manuscrito deve ser mencionada esta aprovação e a obtenção de consentimento informado, quando aplicável.
Resultados
Os resultados devem ser apresentados, no texto, tabelas e figuras, seguindo uma sequência lógica. Não deve ser fornecida informação em duplicado no texto e nas tabelas ou figuras, bastando descrever as principais observações referidas nas tabelas ou figuras. Independentemente da limitação do número de figuras propostos para cada tipo de artigo, só devem ser apresentados gráficos quando da sua utilização resultarem claros benefícios para a compreensão dos resultados.
Apresentação de dados numéricos
A precisão numérica utilizada na apresentação dos resultados não deve ser superior à permitida pelos instrumentos de avaliação. Para variáveis quantitativas as medidas apresentadas não deverão ter mais do que uma casa decimal do que os dados brutos. As proporções devem ser apresentadas com apenas uma casa decimal e no caso de amostras pequenas não devem ser apresentadas casas decimais. Os valores de estatísticas teste, como t ou χ2, e os coeficientes de correlação devem ser apresentados com um máximo de duas casas decimais. Os valores de p devem ser apresentados com um ou dois algarismos significativos e nunca na forma de p=NS, p<0,05 ou p>0,05, na medida em a informação contida no valor de P pode ser importante. Nos casos em que o valor de p é muito pequeno (inferior a 0,0001), pode apresentar-se como p<0,0001.
Tabelas e figuras
As tabelas devem surgir após as referências. As figuras devem surgir após as tabelas. Devem ser mencionadas no texto todas as tabelas e figuras, numeradas (numeração árabe separadamente para tabelas e figuras) de acordo com a ordem em que são discutidas no texto. Cada tabela ou figura deve ser acompanhada de um título e notas explicativas (ex. definições de
45 abreviaturas) de modo a serem compreendidas e interpretadas sem recurso ao texto do manuscrito.
Cada tabela ou figura deve ser apresentada em páginas separadas, juntamente com o título e as notas explicativas. Nas tabelas devem ser utilizadas apenas linhas horizontais. As figuras, incluindo gráficos, mapas, ilustrações, fotografias ou outros materiais devem ser criadas em computador ou produzidas profissionalmente. As figuras devem incluir legendas. Os símbolos, setas ou letras devem contrastar com o fundo de fotografias ou ilustrações. A dimensão das figuras é habitualmente reduzida à largura de uma coluna, pelo que as figuras e o texto que as acompanha devem ser facilmente legíveis após redução. As figuras, criadas em computador ou convertidas em formato eletrónico após digitalização devem ser inseridas no ficheiro do manuscrito. Os gráficos não deverão ter cores. As legendas, símbolos, setas ou letras devem ser inseridas no ficheiro da imagem das fotografias ou ilustrações.
Discussão/Conclusão
Na discussão não deve ser repetida detalhadamente a informação fornecida na secção dos resultados, mas devem ser discutidas as limitações do estudo, a relação dos resultados obtidos com o observado noutras investigações e devem ser evidenciados os aspectos inovadores do estudo e as conclusões que deles resultam. É importante que as conclusões estejam de acordo com os objectivos do estudo, mas devem ser evitadas afirmações e conclusões que não sejam completamente apoiadas pelos resultados da investigação em causa.
Referências
As referências devem ser listadas após o texto principal, numeradas consecutivamente de acordo com a ordem da sua citação. Os números das referências devem ser apresentados entre parêntesis. Não deve ser utilizado software para numeração automática das referências. Pode ser encontrada nos “Uniform Requirements for Manuscripts Submitted to Biomedical
46
Anexos
Material muito extenso para a publicação com o manuscrito, designadamente tabelas muito extensas ou instrumentos de recolha de dados, poderá ser solicitado aos autores para que seja fornecido a pedido dos interessados.”
Conflitos de interesse
Os autores de qualquer manuscrito submetido devem revelar no momento da submissão a existência de conflitos de interesse ou declarar a sua inexistência. Essa informação será mantida confidencial durante a revisão do manuscrito pelos avaliadores externos e não influenciará a decisão editorial mas será publicada se o artigo for aceite.
Autorizações
Antes de submeter um manuscrito aos ARQUIVOS DE MEDICINA os autores devem ter em sua posse os seguintes documentos que poderão ser solicitados pelo corpo editorial: - Consentimento informado de cada participante;
- Consentimento informado de cada indivíduo presente em fotografias, mesmo quando forem efectuadas tentativas de ocultar a respectiva identidade;
- Transferência de direitos de autor de imagens ou ilustrações; - Autorização para utilização de material previamente publicado;