CC7 – ECtAsIA dURAl Com mUltI-RAdICUlopAtIA lombAR:
UmA mAnIFEstAÇÃo RARA dA EspondIlItE AnqUIlosAntE
Joana Matos1, Joana Silva1, Miguel Guerra2, Jorge Moreira1, Sofia Toste1, Inês Táboas1
1. Medicina Física e de Reabilitação, Centro Hospitalar Entre Douro e Vouga, Aveiro, Portugal
2. Rheumatology Department, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal Introdução: As alterações neurológicas na Espondilite
Anquilosante (EA) são raras (2,1%), geralmente devi-das a subluxação atlanto-axial, deformidade/fratura ver-tebral ou canal lombar estreito.
As radiculopatias e o Síndrome de Cauda Equina (SCE) são ainda menos frequentes e surgem na EA de longa duração, associados a ectasia dural (dilatação di-verticular do saco dural, habitualmente lombossagrado, onde a pressão do LCR é maior). A relação entre radi-culopatias/SCE e ectasia dural na EA não está bem es-clarecida mas poder-se-à dever a inflamação crónica das meninges, e a uma interdependênica entre si e do processo inflamatório.
O tratamento é controverso e pouco eficaz, poden-do nos casos em que há progressão clínica e imagioló-gica, considerar-se a opção cirúrgica.
O objetivo deste trabalho é descrever um caso clíni-co de EA clíni-com ectasia dural e rever a literatura acerca do tema.
Caso Clínico: Homem, 62 anos, com EA
diagnostica-da há mais de 30 anos, controladiagnostica-da clínica e analitica-mente com etanercept e metrotexato 7,5mg/semana desde 2008.
Enviado à consulta de MFR por lombalgia e agrava-mento de alterações sensitivo-motoras dos membros inferiores e da marcha, com 4 anos de evolução.
À avaliação apresentava hipercifose dorsal; limitação da mobilidade do ráquis; diminuição da força muscu-lar (dorsiflexão dos dedos à direita, sobretudo D1); hi-postesia álgica (dermátomos L4-S1 bilateral); incapa-cidade para marcha em pontas à esquerda e calcanha-res bilateral; laségue negativo e ROT’s rotulianos
/aqui-Casos clínicos
acta reumatol port. 2018:43:143-185 (sup)
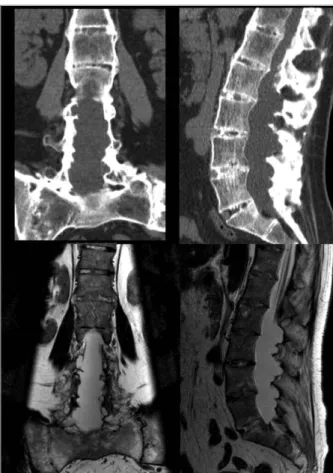
lianos diminuídos; sem alteração esfincterianas. Realizou EMG - alterações neurogénicas L5-S1 bila-teral; TC lombar - redução da altura discal, calcificações intra-discais e ligamentares e fusão dos arcos vertebrais posteriores; alargamento exuberante do canal raqui-diano central L2-S1, perda da diferenciação do con-teúdo intra-canalar e remodelação/erosão dos elemen-tos ósseos lombo-sagrados posteriores; redução da per-meabilidade dos canais de conjugação L4-S1 de pre-domínio direito, com conflito de espaço radicular foraminal; RM lombar - ectasia dural L3-S1 com múl-tiplos divertículos dorsais, condicionando moldagem e erosão óssea até aos buracos de conjugação, expansão do canal e conteúdo intracanalar com sinal de líquor,
FIGURA 1.Achados imagiológicos na tomografia axial computorizada lombar (em cima) e na ressonância magnética lombar (em baixo)
Case report: A twenty-year-old female presented in
the emergency department with rotational vertigo, tinittus, headeach and progressive hearing loss for 15 days and acute redness of eyes for 4 days. She also re-ferred pain on the right flanque with nausea for the last week. Redness of the eyes was associated with dimi -nished vision and photofobia, there was no discharge from the eyes, any restriction of movements or diplo -pia. Also, the patient had a progressive hearing loss for 15 days which was not associated with any ear dis-charge. There was no history of intake of any ototoxic drugs. There was no history suggestive of any other cranial nerve deficits/seizures/acute confusional state. A physical examination revealed circumciliary conges-tion of eyes with preserved visual acuity and bilateral severe sensorineural hearing loss. There was no nys-tagmus. There was no evidence of other cranial nerve or focal neurological deficit. Pain in the right abdomi-nal quadrant with no signs of peritoneal irritation was present. No other remarkable findings except for existence of low to moderate grade fever. She was diagno -sed with interstitial keratitis upon observation by Oph-thalmologist. Pure tone audiometry showed bilateral severe sensorineural hearing loss. Significant findings in laboratory investigations included raised V.S 70 mm and PCR 24 mg/dl; FA 700 U/L, GGT 300 U/L, AST 40 U/L, ALT 50 U/L, total bilirubin 5 mg/dl and direct bilirubin 3 mg/dl were also elevated, while rest of the routine metabolic profile was essentially within the nor-mal limits. Serum studies for ANCAs, ANAs and serolo-gies were negative. Abdominal CT Scan revealed mul-tiple liver abscesses. CE RMN showed “Enhancement of the VIII cranial pairs, as well enhancement of the membranous labyrinths of the inner ear, bilaterally, as-pects suggestive of neuritis and bilateral labyrinthitis”. Empiric antibiotic therapy with Meropnem and 1 mg/ /kg metilprednisolona were started. Blood cultures (4X) were negative, torax x-ray, echocardiogram, lumbar puncture and urinalysis were also negative for infec-tion. Inflammatory markers progressively normalized as well as red eyes and abscesses resolved. Neurossen-sorial deafness was not reversible and the patient was proposed for coclear implantation. Based upon the cli ni cal findings, Cougan Syndrome with systemic mani -festation was diagnosed. Hepatic vasculitis mimicking abcess were assumed to be a systemic manifestations of Cogan’s syndrome as septic screening including lumbar puncture were negative for infection.
Conclusion: The presence of interstitial
non-ulcera-tive keratitis in the absence of congenital syphilis, par-compatível com SCE e ectasia dural; raízes da cauda
equina junto à parede do saco dural, com indefinição dos limites.
Iniciou pregabalina e programa de reabilitação diri-gido à dor e aos défices.
Aos 2 meses apresentava alívio álgico mas agrava-mento progressivo da força, sobretudo flexão/abdução da anca, extensão do joelho, dorsiflexão/flexão plantar do tornozelo e extensão dos dedos bilateral; a marcha era instável, com padrão neuropático, trendelenburg bilateral e incapacidade para a marcha em pontas tam-bém à direita.
Manteve programa de reabilitação neurológica e foi referenciado para Ortopedia para eventual tratamento cirúrgico.
Conclusão: Apesar de infrequentes, as alterações
neu-rológicas na EA podem ser graves e devem ser cuida-dosamente investigadas com exame neurológico com-pleto e exames adequados. A ectasia dural e radiculo-patia/SCE constituem manifestações raras de EA e a melhor orientação destes doentes permanece incerta. O shunt lomboperitoneal é sugerido na literatura como a opção mais eficaz, a par de um programa de reabilita-ção individualizado como adjuvante no alívio álgico e reabilitação neurológica/funcional.
Este caso enfatiza a importância de uma boa comu-nicação entre as várias especialidades intervenientes (Reumatologia, Medicina Interna, Ortopedia, Neuro-cirurgia, MFR e MGF) a fim de otimizar a orientação destes doentes.
CC22 – CoGAns syndRomE: A RARE systEmIC vAsCUlItIs
João Pedro Freitas1, Flávio Campos Costa1, João Rovisco1
1. Serviço Reumatologia, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
Introduction: Cogan’s syndrome (CS) is a rare chronic inflammatory disorder, classically characterized by in-terstitial keratitis and sensorineural hearing loss. Inner ear disease might result in deafness. In some patients, it may also be accompanied by systemic vasculitis. Diagno sis of CS is often missed or delayed due to its ra -rity, the nonspecific clinical signs at onset, and the lack of a confirmatory diagnostic test. This syndrome is rare and very few cases have been reported since Cogan’s first characterization in 1945. The case of a 20-year--old female with the atypical form of the disease is pre-sented.
sedimentation rate was 21mm/h, and C-reactive pro-tein 0.76 mg/dl. Serological screening was negative. Tests for autoimmunity were positive to antinuclear an-tibodies, antibodies to double-stranded DNA (12.7IU/ /mL, reference value > 15U/mL), positive lupus anticoagulant and anticardiolipin antibodies (high-ti-tre IgG anticardiolipin and low-ti(high-ti-tre IgG anti-b2-gly-coprotein 1 antibodies). Complement was normal. Ce-rebrospinal fluid analysis was normal for glucose, pro-teins and cellular count, without oligoclonal bands. Septic screening in cerebrospinal fluid was negative. After methylprednisolone pulses, she initiated treat-ment with prednisolone 1 mg/kg/day. After 5 days of treatment, brain magnetic resonance imaging showed multiple bilateral cortical, subcortical and infratentorial T2 and fluid-attenuated inversion recovery high-in-tensity lesions in various stages of healing. These le-sions had contrast enhancement on T2* sequence. Ce-rebral angiography was normal. Clinical improvement was noticeable after 3 days of treatment: practically with out neurologic deficits, hemoglobin 10.3mg/dL and normalization of platelet and lymphocyte count. She re-mains stable, without new events until now. We esta-blished the diagnosis of SLE with central nervous sys-tem involvement based on 2012 Syssys-temic Lupus Col-laborating Clinics criteria and NPSLE-ACR definition.
Discussion: According to the available literature, a
small amount of patients with CLE can evolves to SLE, with rates ranging from 5 to 10%. (1) We described the case of a woman with CLE that presented with a full cli-nical picture of SLE after 7 years. This case emphasizes the importance of considering NPSLE as a possible diagnosis for onset neuropsychiatric symptoms.
REFEREnCEs:
1. Okon L, Rosenbach M, Krathen M, et al. Lenalidomide in treat-ment-refractory cutaneous lupus erythematosus: Efficacy and safety in a 52-week trial. J Am Acad Dermatol. 2014 Mar;70(3): 583-4.
2. Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and mana-gement of neuropsychiatric SLE manifestations. Nat Rev Rheu-matol. 2010 Jun;6(6):358-67.
3. Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 2014 Jun;10(6):338-47.
CC42 – ARtRItE psoRIÁsICA pmR-lIKE: novo FEnÓtIpo?
Tiago Costa1, 2, Teresa Pedrosa1, Inês Silva1, Sandra Falcao1, 2, Fernando Pimentel-Santos1, 2, Jaime C. Branco1, 2
1. Serviço de Reumatologia, Hospital de Egas Moniz, Lisboa, Portugal
ticularly, in conjunction with audiovestibular sym -ptoms, makes the diagnosis of Cogan’s. The atypical form of the disease is related to systemic manifesta-tions, being more aggressive and of worse prognosis. CC41 – CEntRAl nERvoUs systEm
vAsCUlItIs As pRImARy mAnIFEstAtIon oF systEmIC lUpUs ERythEmAtosUs In A pAtIEnt wIth CUtAnEoUs lUpUs ERythEmAtosUs
Luisa Brites1, Marília Rodrigues2, Mariana Luis1, Mariana Santiago1, Cátia Duarte1
1. Serviço Reumatologia, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
2. Centro Hospitalar de Leiria, Leiria, Portugal
Introduction: Cutaneous lupus erythematosus (CLE)
is an autoimmune and heterogeneous disease that can occur as a manifestation of systemic lupus erythema-tosus (SLE) or independently. The progression rate of CLE to SLE remains unknown but it is believed to be low1. Neuropsychiatric systemic lupus erythematosus syndromes (NPSLE) can occur in 12-95% of patients with SLE2. One third of all neuropsychiatric syndro-mes in patients with SLE occurs as the first manifesta-tion and is directly attributed to SLE but other causes needs to be excluded3.
Case report: A 32 year-old woman with a 7-year
his-tory of CLE without any other manifestations and with a negative immunologic study) was admitted in the Emergency Department with paresthesias in the left side of her face with 1 week of evolution. Physical exa-mination revealed a left faciobrachial hemiparesis and a discrete anisocoria, without language deficits or other manifestations. Computed tomography scan showed bilateral cortical and subcortical frontal lesions of is-chemic nature, without contrast enhancement, sug-gestive of CNS vasculitis. She starts treatment with in-travenous methylprednisolone (1g daily – 3 days) and was admitted in Rheumatology department with a cli-nical suspicion of CNS vasculitis in SLE context. She denied any other complaints, history of recent infec-tions, thrombotic events or obstetric complicainfec-tions, al-cohol, tobacco, drug abuse or current medication. La-boratory investigations revealed autoimmune hemoly-tic anemia (hemoglobin 9mg/dL, a positive direct an-tiglobulin test, a decreased amount of haptoglobin, reticulocytosis and increased lactate dehydrogenase, low platelet and lymphocyte count. Activated partial thromboplastin time was prolonged. The erythrocyte
primeiro dedo da mão esquerda. Não se identificaram outros estigmas extra-articulares associados a Espon-diloartrites. O doente apresentava um sindesmófito grosseiro em ponte entre a 12ª vértebra dorsal e a 1ª vértebra lombar, sem sacroileíte radiográfica. Foi reobser vado em consulta de Dermatologia, assumin-do-se o diagnóstico de Psoríase cutânea. As queixas as-sociadas a entesite e dactilite cederam à terapêutica com anti-inflamantório não esteróide, sendo esta a sua te-rapêutica atual.
Conclusão: Trata-se de um doente com o diagnóstico
definitivo de AP cumprindo os critérios CASPAR, em que a forma de apresentação inicial com envolvimen-to rizomélico sugeriu o diagnóstico de PMR. O caso descrito procura alertar para a variável expressão feno-típica e a evolução insidiosa desta patologia, devendo o clínico estar atento para o amplo espectro de manifestações; algumas delas, em fase inicial, subtis. Segun -do o nosso conhecimento trata-se da primeira descri-ção de um caso clínico de AP com uma apresentadescri-ção PMR-like. Os autores propõem que o diagnóstico de AP seja considerado no diagnóstico diferencial de PMR. CC49 – doEnÇA dE KIKUChI-FUjImoto AssoCIAdA A doEnÇA mIstA do tECIdo ConjUntIvo: Um CAso dE RECIdIvA Bruno Miguel Fernandes1, Miguel Bernardes1, Helena Barroca2, Lúcia Costa1
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
2. Serviço de Anatomia Patológica, Centro Hospitalar de São João, Porto, Portugal
Introdução: A Doença de Kikuchi-Fujimoto (DKF) é
uma doença rara, normalmente auto-limitada e de evo-lução benigna, sendo ainda mais excecionais os casos com recidiva. Afeta mais frequentemente mulheres jo-vens e caracteriza-se pelo aparecimento de adenopa-tias cervicais dolorosas e sintomas constitucionais. A sua etiologia é desconhecida. Contudo, tem-se verifi-cado uma associação crescente a doenças auto-imunes do tecido conjuntivo, particularmente ao Lúpus Erite-matoso Sistémico. A histologia ganglionar é importan-te no diagnóstico diferencial com linfomas malignos.
Caso Clínico: Mulher, 35 anos, à qual foi
diagnostica-da DKF em 2006. Na altura, aos 24 anos, foi submeti-da a biópsia cirúrgica de uma adenopatia axilar direita dolorosa na sequência de um quadro clínico de febre e adenomegalias generalizadas, que resolveu em sema-nas. Em 2010, a doente foi referenciada à consulta de
2. CEDOC, NOVA Medical School. Faculdade de Ciências Médicas da Universidade NOVA de Lisboa, Lisboa, Portugal
Introdução: A Artrite Psoriásica (AP) é uma doença
reu mática inflamatória crónica, pertencente ao grupo das Espondiloartrites, cujo diagnóstico assenta nos cri-térios CASPAR (Classification Criteria for Psoriatic
Arth-ritis). Atualmente, são reconhecidos 5 subgrupos de
AP: oligoarticular assimétrica, poliarticular simétrica, envolvimento predominante das articulações interfa-lângicas distais, espondilite com ou sem sacro-ileíte e artrite mutilante. É importante realçar que o compro-misso articular é muito variável e a sobreposição de ma-nifestações entre os vários subgrupos frequente. En-contram-se descritos poucos casos de Espondiloartri-te indiferenciada do idoso que se apresenEspondiloartri-tem com sin-tomatologia sugestiva de Polimialgia Reumática (PMR). De acordo com informação disponível, nunca foi des-crito um caso de AP em que a forma de apresentação tenha sido um quadro de PMR.
Caso clínico: Apresenta-se o caso de um doente do
sexo masculino de 49 anos, referenciado à consulta de Reumatologia, por artralgias de ritmo inflamatório, com cerca de um ano de evolução, envolvendo os om-bros e as coxo-femorais acompanhadas de rigidez ma-tinal de longa duração, condicionando limitação im-portante nas atividades de vida diária. No exame obje-tivo destacava-se distrofia ungueal D1 e D2 da mão es-querda e D3 da mão direita; manobras sacroilíacas negativas, sem dactilite ou entesite. Analiticamente des-tacava-se velocidade de sedimentação 40mm/h, fator reumatóide, anticorpo anti-peptídeo citrulinado cícli-co (CCP), anticícli-corpos nucleares (ANAs), anti-corpos anti-antigenos nucleares extraíveis (ENAs), e Human leukocyte antigen (HLA-B27) negativos. Assu-miu-se o diagnóstico presuntivo de PMR, sendo medi-cado com prednisolona 10mg/dia e posteriormente adi-cionado metotrexato 15mg/semana por recidiva clíni-ca com o desmame da corticoterapia. Foi observado em consulta de Dermatologia tendo realizado o exame micológico das unhas que foi negativo; realizou, po-rém, terapêutica antifúngica empírica sem resolução da situação. Assumiu-se assim o diagnóstico de Pso-ríase ungueal. O desmame gradual da corticoterapia e metotrexato, com interrupção desta terapêutica, foi ini-ciado após 1 ano de remissão clínica. Três meses depois, o doente iniciou um quadro de dor ao nível das arti-culações condroesternais bilateralmente, fasciíte plan-tar direita, descamação palmo-planplan-tar e dactililte do
progressiva da dor e das dimensões da adenopatia axi-lar assim como normalização dos parâmetros analíticos em 8 semanas. Esta melhoria analítica permitiu pres-cindir da biópsia de medula óssea para exclusão duma eventual linfo-histiocitose hemofagocítica concomi-tante. Atualmente a doente encontra-se assintomática, sob prednisolona (7.5mg od), mantendo a hidroxiclo-roquina.
Conclusão: Os autores salientam a importância do
re-conhecimento da associação de DKF a outras doenças auto-imunes do foro reumatológico, podendo mesmo preceder o seu aparecimento. A singularidade deste caso clínico prende-se com a raridade da associação en-tre DKF e DMTC e com a recorrência tardia da DKF (11 anos), quando esta surge habitualmente num in-tervalo de alguns meses.
CC56 – ostEoARtRopAtIA hIpERtRÓFICA ApEnAs A pontA do ICEbERG
Flávio Campos Costa1, João Freitas1, Margarida Coutinho1, Armando Malcata1
1. Serviço Reumatologia, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
Introdução: As Síndromes Paraneoplásicas (SP)
afe-tam até 8% dos doentes com neoplasia, podendo pre-ceder o seu diagnóstico.
A Osteoartropatia Hipertrófica (OH) é uma síndro-me composta pela tríade de hipocratismo digital, pe-riostite dos ossos longos e artralgias e/ou artrite. Pode ser classificada como primária (hereditária ou idiopá-tica) ou secundária, apresentando uma prevalência de 4 a 32% nas neoplasias pulmonares primárias.
A apresentação clínica da síndrome varia de acordo com a rapidez de instalação da doença subjacente e res-petiva evolução. A artrite, quando presente, tende a en-volver cotovelos, punhos, metacarpofalângicas (MCF’s), joelhos e tibiotársicas (TT’s).
Reumatologia por poliartralgias simétricas, de ritmo in-flamatório, envolvendo os punhos e as articulações MTFs e IFPs das mãos, com 6 meses de evolução, e fe-nómeno de Raynaud. Do estudo analítico, salientavam--se: positividade para ANAs (>1/1000), anticorpos anti--RNP em título elevado (97U/mL), leucopenia (2820/mm3) e neutropenia (920/mm3). O diagnóstico de doença mista do tecido conjuntivo (DMTC) foi es-tabelecido e a doente foi medicada com hidroxicloro-quina (400mg od) e prednisolona (5mg od), consta-tando-se uma boa resposta clínica. Em 2017, a doente apresentou-se, novamente, com uma adenopatia axilar esquerda dolorosa e febre (inicialmente picos semanais e, posteriormente, diários). O estudo analítico revelou: elevação das proteínas de fase aguda [VS (67mm) e PCR (54,4mg/L)]; leucopenia (2290/mm3), neutropenia (1390/mm3) e trombocitopenia (131000/mm3); eleva-ção das transaminases [AST (42U/L) e ALT (67U/L)] e da ferritina (808 ng/mL); normotrigliceridemia e nor-mofibrinogenemia. As serologias para exclusão de in-feção vírica aguda foram negativas, nomeadamente VEB, VCM, parvovírus B19, VIH, VHC, VHB, vírus her-pes simplex (1 e 2) e vírus herher-pes tipo 6, 7 e 8. O es-tudo imunológico recapitulou a positividade para os ANAs (1/320), a negatividade para os anticorpos anti-DNA nativo e a normocomplementemia (C3c e C4). A ecografia axilar mostrou adenopatias axilares à esquerda e a mais volumosa (36x31mm) foi biopsiada por agulha fina. O exame citológico revelou detritos apoptóticos, população linfóide polimórfica, numero-sos macrófagos com corpos tingíveis e histiócitos (por vezes binucleados ou com formas nucleares em cres-cente), na ausência de polimorfonucleares. No estudo imunofenotípico, não se observaram populações B mo-noclonais nem linfócitos T com expressão fenotípica aberrante, corroborando o diagnóstico de DKF. A cor-ticoterapia oral foi aumentada para 15mg de predniso-lona diárias, ocorrendo resolução da febre, diminuição FIGURA 1.Evolução do caso clínico ao longo do tempo. Dgx – diagnóstico
Aug. 20(8):1386-1387.
3. Nguyen S, Hojjati M. Review of current therapies for seconda-ry hypertrophic pulmonaseconda-ry osteoarthropathy. ClinRheumatol. 2011 Jan. 30(1):7-13.
4. King MM, Nelson DA. Hypertrophic osteoarthropathy effective-ly treated with zoledronic acid. Clin Lung Cancer. 2008;9(3):179. 5. Yao Q, Altman RD, Brahn E. Periostitis and hypertrophic pul-monary osteoarthropathy: report of 2 cases and review of the li-terature. Semin Arthritis Rheum. 2009;38(6):458.
CC60 – toCIlIzUmAb: UmA AltERnAtIvA tERApÊUtICA pARA A vAsCUlItE pRImÁRIA do snC?
Bruno Miguel Fernandes1, Miguel Bernardes1, Carina Reis2, Lúcia Costa1, Elsa Azevedo3, Pedro Abreu3
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
2. Serviço de Neurorradiologia, Centro Hospitalar de São João, Porto, Portugal
3. Serviço de Neurologia, Centro Hospitalar de São João, Porto, Portugal
Introdução: A Vasculite Primária do Sistema Nervoso
Central (VPSNC) é uma vasculite rara que afeta exclu-sivamente os vasos do SNC. A etiologia é desconhecida e a clínica é inespecífica: cefaleias e disfunção cogni -tiva são os achados mais comuns. Dado o carácter in-vasivo das biópsias do SNC, a angiografia e a angio-res-sonância magnética (angio-RM) têm particular relevo para o diagnóstico. A exclusão de vasculite sistémica é fundamental.
Caso Clínico: Mulher, atualmente com 51 anos que,
no início de 2016, se apresentou com um quadro tran-sitório de hipostesia da hemiface direita e parésia do membro inferior direito, apesar da TAC cerebral reve-lar já isquemia frontoparietal esquerda. A angio-RM ce-rebral, além da área isquémica, mostrou múltiplas es-tenoses das artérias cerebrais médias (ACMs) e da ar-téria cerebral posterior (ACP) esquerda, não permitin-do excluir vasculite. Do estupermitin-do etiológico permitin-do AVC realçava-se apenas um perfil hipertensivo. O estudo do líquido cefalorraquidiano (LCR) revelou hiperprotei-norráquia (0.89 g/L), 4 linfócitos/L, ligeira hipoglicor-ráquia (49% da glicemia), negatividade para agentes infeciosos e para bandas oligoclonais e IgG ligeiramente elevada (6.87 mg/dL). Para exclusão de vasculite sisté-mica, foi efetuado estudo imunológico que revelou nor-mocomplementemia (C3c e C4) e negatividade para ANCAs, ANAs, anti-dsDNA e imunocomplexos circu-lantes. Não apresentava assimetrias tensionais e de pul-sos entre os membros nem estigmas muco-cutâneos de Em situações de OH secundária a remoção da
neo-plasia subjacente ou o tratamento de outras causas se-cundárias resulta, na maioria dos casos, em regressão das manifestações clínicas. Contudo, a utilização de analgésicos simples ou AINE’s está preconizada para alívio álgico, sendo que, em casos refratários, têm sido utilizados bifosfonatos devido à sua potencial ativida-de anti-inflamatória.
Os autores apresentam um caso clínico de Osteoar-tropatia Hipertrófica secundária.
Caso Clínico: Homem de 69 anos, com quadro de
po-liartralgias de ritmo misto, envolvendo ombros, coto-velos, MCF’s, interfalângicas proximais e distais, coxo-femorais, joelhos e TT’s bilateralmente, com 1 ano de evolução.
Antecedentes pessoais de tabagismo (40UMA) e de adenocarcinoma primitivo do pulmão diagnosticado há 4 meses.
Ao exame reumatológico, destacavam-se altera-ções compatíveis com tendinopatia da coifa dos rota-dores (bilateralmente), osteoartrose polifocal (mãos e joelhos) palpação dolorosa dos punhos e TT’s e pre-sença de acentuado hipocratismo digital das mãos e dos pés.
Do estudo complementar realizado destacou-se ele-vação de marcadores de fase aguda (VS: 32mm/h; PCR: 7,42mg/dL) e anemia normocítica. A ecografia articu-lar relevou sinovite moderada (sem sinal de power
dop-pler) de ambos os punhos e TT’s. A cintigrafia óssea
(so-licitada para despiste de lesões ósseas secundárias), re-velou acentuada captação de radiofármaco ao longo do periósteo dos ossos longos, compatível com periostite, em contexto de osteoartropatia hipertrófica. O doente foi medicado com AINE e encontra-se atualmente sob quimioterapia neoadjuvante.
Conclusão: A OH representa um dilema clínico,
sen-do o seu diagnóstico relativamente simples, mas a sua abordagem difícil. Esta dificuldade é devida ao desco-nhecimento do mecanismo etiopatogénico, variadas opções terapêuticas e à resposta individual ao trata-mento. A abordagem e prognóstico da OH dependem da doença subjacente e, alguns estudos, demostraram que esta entidade regride com o tratamento do tumor subjacente.
REFERÊnCIAs
1. Pereira J, Eugénio G, Calteras S, Santos R, Carvalho A. More than just a case of polymyalgia rheumatica. EJCRIM 2016;3: 2. Martínez-Lavín M, Matucci-Cerinic M, Jajic I, Pineda C.
Hy-pertrophic osteoarthropathy: consensos on its definition, clas-sification, assessment and diagnostic criteria. J Rheumatol. 1993
2. Rheumatology, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal
Introdution: Eosinophilic Granulomatosis with
Po-lyangiitis (EGPA) is a rare multisystemic disorder, cha-racterized by necrotizing vasculitis affecting small to medium sized vessels, associated with asthma and eo-sinophilia. Cardiac involvement is the most important predictor of mortality and it seems to be more frequent in anti–neutrophil cytoplasmic antibodies (ANCA) ne-gative patients.
Case description: A 65-year-old woman, with a past
medical history of allergic rhinosinusitis since the age of 27 and a recent diagnosis of asthma, treated with montelukast, fluticasone/formoterol and hydroxyzine, was admitted in the emergency department presenting with a lancinating neuropathic pain and asymmetrical lower limb edema more pronounced on left side with 2 days of duration. Fifteen days prior she developed multiple purpuric lesions and dysesthesias of the upper and lower limbs. On admission to the rheuma-tology department, she was apyretic and hemodyna-mically stable. Cardiopulmonary auscultation revealed rhythmic beats, without murmurs, diffused wheezing and inspiratory basal crackles. Some petechial and pur-puric lesions were evident on the palmar and dorsal side of the hands and feet with extension to the pre-ti-bial region. A left foot dorsiflexion and right hand fle-xion deficit was found, associated with symptoms of hypoesthesia. A complete blood count revealed leuko-cytosis of 18.8x10^9/L with hypereosinophilia (59.8%), the ESR was 35mm/1st hour and the CRP of 147.2mg/L. Despite the absence of cardiac symptoms, serum troponin I level was markedly increased to 11429ng/L (normal <16) and also muscle enzymes were elevated - AST 133U/L, ALT 51U/L, LDH 782U/L, CK 1579U/L, myoglobin 575ng/mL, CK-MB 123ng/mL. BNP was increased by 543pg/mL and ANCA were undetectable. ECG documented sinus ta-chycardia of 114bpm and left bundle branch block. Transthoracic echocardiography revealed moderate to severe left ventri cular systolic dysfunction (LVSD) with ejection fraction ~32%, mild to moderate mitral re-gurgitation and a small volume pericardial effusion. A cardiac ca theterization was performed, which ruled out coronary disease. Cardiac magnetic resonance confir-med the severe LVSD and showed a late gadolinium enhancement pattern of subendocardial predominan-ce, compatible with fibrosis and suggestive of a vasculitis. Electromyography recorded findings of mul-LES ou de Doença de Behçet. Evoluiu com
recupera-ção completa dos défices neurológicos sob dupla anti-agregação plaquetária, tendo alta para a consulta de Neurologia. Em Agosto de 2016, repetiu angio-RM ce-rebral e, por agravamento da redução do calibre e da in-flamação na ACM esquerda, foi reinternada com o diagnós tico de VPSNC. Efetuou ciclo de metilpredni-solona 1g/dia (5 dias) por decisão conjunta de Neurologia-Reumatologia, tendo alta sob prednisolona 60mg id. Em Outubro de 2016, foi novamente inter-nada por AVC isquémico da ACM esquerda e subme-tida a novo ciclo de metilprednisolona 1g/dia (5 dias) e à 1ª administração e.v. de ciclofosfamida. Dada a per-sistência de défices neurológicos (hemiplegia direita), foi orientada para reabilitação motora, medicada com prednisolona 70mg id e pulsos mensais de ciclofosfa-mida. Após o 3º pulso de ciclofosfamida, a angio-RM cerebral mostrou acentuação do processo inflamatório vascular. Em Março de 2017 e novamente por decisão multidisplinar, iniciou tratamento mensal com tocili-zumab e.v. (8mg/Kg). Após 6 meses de terapêutica, a angio-RM cerebral mostrou pela primeira vez uma es-tabilização das lesões, permitindo doses de prednisolo-na de 7.5mg id.
Conclusão: A VPSNC constitui um desafio pois, dada
a ausência de estudos randomizados, a terapêutica re-sulta da experiência noutras vasculites: corticosteróides como âncora, sendo possível a associação de outros imunossupressores. Descrevemos um caso de VPSNC refratária à corticoterapia e à ciclofosfamida. Face à nos-sa má experiência prévia com o rituximab num caso semelhante de VPSNC, e tendo em conta os resultados positivos que o tocilizumab tem vindo a apresentar noutras vasculites, optou-se por este fármaco. Após a introdução do tocilizumab, verificou-se estabilização das lesões de vasculite cerebral, apesar do seguimento ainda curto. Do nosso conhecimento, este é o primei-ro caso descrito na literatura de utilização do tocilizu-mab no contexto de VPSNC. O aparente sucesso deste caso abre perspetivas para o tocilizumab como nova arma terapêutica no tratamento desta entidade. CC66 – sIlEnt ACUtE myoCARdItIs In EosInophIlIC GRAnUlomAtosIs wIth polyAnGIItIs
Raquel Miriam Ferreira1, Sara Ganhão1,
Miguel Guerra2, Pedro Madureira1, Sofia Pimenta1, Lúcia Costa1
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
tiple mononeuropathy and diagnosis was confirmed by biopsy. Due to typical clinical picture of EGPA, pul-ses of me thylprednisolone were started on the 2nd day of hospitalization, followed by oral prednisolone and cyclophosphamide (for 6 months). Montelukast was suspended. A rapid clinical and analytical improve-ment was noticed. After 5 months of follow-up, the echocardiographic reevaluation demonstrated the left ventricular function preserved (ejection fraction ~55%). Maintenance therapy with azathioprine was instituted, with continuous normalization of eosino -philia and inflammatory parameters.
Conclusion: As cardiomyopathy and congestive heart
failure can occur in EGPA but a significant proportion of patients are asymptomatic, complementary cardiac investigation are mandatory in any patient with suspi-cion of this disorder. Early detection and the appro-priate treatment are crucial due to the possible life-threatening manifestations.
CC71 – sÍndRomE hEmoFAGoCÍtICA no lÚpUs ERItEmAtoso sIstÉmICo: dEsAFIo Ao lImItE
Raquel Miriam Ferreira1, Teresa Martins-Rocha1, Sara Ganhão1, Miguel Guerra2,
Ana Filipa Rocha Águeda3, Eva Mariz1, José Pinto1, José Brito1, Sofia Pimenta1, Lúcia Costa1
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
2. Rheumatology, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal
3. Serviço de Reumatologia, Centro Hospitalar do Baixo Vouga, E.P.E., Aveiro, Portugal
Introdução: A síndrome de ativação macrofágica é uma
complicação rara e potencialmente fatal de algumas doenças reumatológicas nomeadamente do Lúpus eri-tematoso sistémico (LES). O seu diagnóstico e trata-mento revelam-se como um verdadeiro desafio.
Caso 1: Mulher de 38 anos, seguida em consulta por
LES desde os 16 anos de idade, foi enviada ao SU por quadro de febre, cefaleias intensas, edema palpebral e dor retroesternal. Realizou TC cerebral, punção lombar e hemoculturas que não revelaram alterações. O ecocardiograma era normal. Por cefaleias crónicas, te-ria já realizado previamente uma Angio RMN cerebral, que tinha excluído alterações do parênquima ou ano-malias vasculares. Por febre persistente, tumefação de novo das glândulas salivares e úlceras orais foi
inter-nada. À admissão, apresentava bicitopenia, VS e fibri-nogénio normal, PCR 12mg/L, elevação das transami-nases, LDH, triglicerídeos e ferritina. As serologias ví-ricas eram negativas. A ecografia revelou sialoadenite crónica das parótidas e submandibulares, cuja biópsia aspirativa foi inconclusiva. Foi submetida a biópsia ós-sea que relevou lesões de hemofagocitose. Foram instituídos pulsos de metilprednisolona (MP), seguido de prednisolona 30mg id e ciclosporina 2.5mg/kg/id. Ao 13º dia de internamento, a doente apresentou afun-damento do estado de consciência, traduzido imagio-logicamente por hemorragia parenquimatosa cerebral. Foi realizada uma derivação ventricular externa sem sucesso, tendo a doente falecido. Foi isolado, a poste-riori, na cultura do líquor uma Kingella Kingae.
Caso 2: Mulher de 54 anos, com LES com 16 anos de
evolução, com envolvimento articular, cutâneo e renal, sob azatioprina 150mg id, com má adesão terapêutica nos últimos 9 meses, altura em que desenvolve quadro de febre, hepatoesplenomegalia e adenopatias múlti-plas. Analiticamente, apresentava Hb 8g/dL e linfope-nia, citocolestase, ferritina: 1865ng/mL, elevação mar-cada da PCR e VS e consumo de C4. Apresentava DNA sérico positivo para EBV, com serologia IgG+/IgM-. A pesquisa de DNA vírico em biópsia hepática foi nega-tiva, sendo a histologia compatível com hepatite auto-imune. A biópsia medular excluiu sinais de hemofago-citose ou doença linfoproliferativa. O restante rastreio séptico e estudo neoplásico foi negativo. Foram insti-tuídos pulsos de MP, seguido de prednisolona, com me-lhoria clínica e analítica. Iniciou ciclosporina 200mg id, tendo tido alta para posterior reavaliação. Foi rein-ternada 23 dias depois por febre e anemia grave com Hb 4g/dL, com evidência inicial de hemólise. O envol-vimento hematológico revelou-se refratário à optimi-zação de prednisolona e IgIV. Após suporte transfusio-nal e pulsos de MP, obteve-se subida de Hb. Posterior-mente iniciou rituximab 1g, tendo tido alta. Após 1 mês, recorreu ao SU por febre persistente e dispneia. Apresentava pancitopenia e ferritina >22000ng/mL. A nova biópsia óssea documentou sinais de hemogafoci-tose, pelo que a dose de ciclosporina foi otimizada para 500mg id. Por infeção respiratória e genital nosocomial foi instituída antibioterapia. Ao 22º dia de interna-mento, a doente apresentou agravamento neurológico progressivo, vindo a falecer.
Conclusão: Um alto nível de suspeição é necessário
para o diagnóstico precoce desta síndrome. Estes casos clínicos, apesar do desfecho fatal, retratam a dificulda-de diagnóstica e a necessidadificulda-de urgente da instituição do
tratamento. Verificamos também, que o despiste de ou-tras causas associadas, nomeadamente infeciosas, é fun-damental para o tratamento adequado e atempado, po-dendo influenciar o prognóstico vital.
CC72 – ARtRItE sÉptICA ComplICAdA Com AbCEssos dA pAREdE toRÁCICA E ostEomIElItE do mAnUbRIo – RElAto dE Um CAso
Cristiana Lopes Martins1, Miguel Varela2, Natércia Joaquim2, Mihail Mogildea2, Ignacio Moreno2, Ana Lopes2
1. Serviço de Medicina Física e de Reabilitação, Centro Hospitalar do Algarve, Faro, Portugal
2. Medicina Interna, Centro Hospitalar e Universitário do Algarve, Faro, Portugal
Introdução: A artrite séptica (AS) é uma emergência
reumatológica, que pode culminar em dano irreversí-vel da articulação. O diagnóstico e tratamento preco-ces são imperativos. A AS na articulação esternoclavi-cular (AEC) pode ter um início insidioso, com irradia-ção da dor para outros locais, dificultando o diagnós-tico. As complicações graves são comuns, com desfecho potencialmente fatal
Objetivo: Relato de um caso clínico de artrite
séptica da articulação esternoclavicular (AS-AEC), complicada com abcessos da parede torácica e osteo-mielite.
Caso Clínico: Doente do sexo masculino, 77 anos,
com antecedentes de diabetes mellitus e doença renal crónica. Foi transferido para o nosso hospital com diagnós tico de pneumonia, no 3º dia de antibiótico de primeira linha. O doente referia astenia e omalgia di-reita, com duração de 3 semanas, com ausência de trau-ma. À nossa observação, encontrava-se hipoxémico, fe-bril e apresentava uma tumefação sobre a AEC direita, indolor à palpação. Analiticamente, havia aumento dos parâmetros inflamatórios. A radiografia de tórax mos-trava opacificação do terço superior do pulmão direi-to, sugerindo pneumonia. No entandirei-to, a tomografia computorizada (TC) do tórax revelou uma AS-AEC di-reita, com extensão para a região posterior ao manú-brio, à 2ª costela direita e ao 1º espaço intercostal es-querdo. As imagens eram sugestivas de abcessos extra-pleurais. Nas hemoculturas foi isolado S. aureus. O doente iniciou tratamento conservador, inicialmen-te com vancomicina e piperacilina-tazobactam. Não houve indicação para tratamento cirúrgico pela Cirur-gia Toracica, dada a melhoria imagiológica inicial. Uma
das TC de reavaliação revelou osteomielite do manú-brio, antes inexistente. Apesar do tratamento e reava-liações periódicas, o doente acabou por falecer de cho-que séptico
Discussão: A AS-AEC é rara, constituindo até 1% de
to-das as AS. É mais prevalente no sexo masculino. Os fa-tores predisponentes incluem uso de drogas endove-nosas, artrite inflamatória, infeções à distância, diabetes mellitus, doença renal crónica, entre outros. É infre-quente em imunocompetentes. As queixas álgicas loca-lizam-se geralmente no tórax (78%) e ombro (24%). A tumefação na AE é sugestiva da patologia, mas rara (4%). A instalação do quadro é subaguda, diferente das restantes AS. Os agentes mais comummente isolados são o S. aureus, N. gonorrhea e P. aeruginosa.
As complicações graves são frequentes, nomeada-mente osteomielite (55% dos casos), abcesso (25%) da parece torácica e mediastinite (13%), com desfecho po-tencialmente fatal. Por isso, a severidade da infecção tem de ser avaliada ab initio com recurso a TC ou ressonân-cia magnética. As radiografias simples não têm sensibi-lidade suficiente para detetar alterações ósseas na AEC
No caso relatado, o doente apresentava alguns fato-res de risco (diabetes mellitus, doença renal crónica). Não foi identificada uma porta de entrada para infeção nem imunodeficiência. O agente isolado foi o mais usual. O diagnóstico e deteção das complicações ocor-reu quando o quadro tinha quase um mês. Apesar do tratamento conservador dirigido, o paciente acabou por falecer por complicações do quadro
Conclusão: A AS-AEC é uma patologia rara, com uma
clínica diferente dentro das AS. O seu diagnóstico re-quer um elevado índice de suspeição e o recurso a exa-FIGURA 1.A imagem mostra artrite septica da articulação esternoclavicular (AEC) (seta amarela) e o abcesso intra-torácico contíguo (seta vermelha)
mes imagiológicos específicos. Um diagnóstico preco-ce é essencial para evitar as complicações potencial-mente fatais de uma infeção prolongada
CC76 – FlARE dE ARtRItE REUmAtÓIdE ApÓs ARtRoplAstIA totAl do joElho: RElAto dE Um CAso
Joana Ramos Rodrigues1, Daniela Santos-Faria1, Joana Sousa-Neves1, Joana Leite Silva1,
Daniela Peixoto1, Sérgio Alcino 1, Carmo Afonso1, José Tavares-Costa1, Filipa Teixeira1
1. Rheumatology Department, Hospital Conde de Bertiandos (ULSAM), Ponte de Lima, Portugal
Introdução: A Artrite Reumatóide (AR) é uma doença
imunomediada, crónica e progressiva, que pode, em estadios avançados, resultar na perda de massa óssea e deformidade articular1.
Com a utilização crescente dos fármacos biotecnoló-gicos, são cada vez menos os doentes que evoluem para fases avançadas da doença2. Contudo, cerca de 20-25% dos doentes com AR desenvolvem dano arti-cular importante, sendo o joelho envolvido em mais de 90% dos casos3. A artroplastia total do joelho (ATJ) pro-vou ser um tratamento de sucesso na AR avançada4. Po-rém, existem relatos de casos de recidiva da artrite do joelho após ATJ5ou artroplastia unicompartimental6. As principais causas de artrite pós-ATJ são infeção, sinovi-te induzida por partículas, sensibilidade ao metal, si-novite não específica e flare da doença inflamatória de base, sendo esta última a causa mais comum4. Achados histológicos típicos de reativação da AR foram encon-trados em 26% dos doentes com AR e falência da pró-tese7. A sinovectomia radical deve ser realizada para re-duzir o risco de ocorrência desta complicação7.
Objetivo: Apresentação de um caso de reativação da
AR num joelho com prótese.
Caso Clínico: Doente de 71 anos de idade, com AR de
longa evolução, já submetida a artroplastia total bilate-ral dos joelhos e a artroplastia bilatebilate-ral da anca.
Recorreu à consulta por dor, tumefacção e aumento da temperatura local do joelho esquerdo. Na avaliação, constatada sinovite deste joelho, tendo-se suspeitado, inicialmente, de infecção da prótese ou de artrite por microcristais. No entanto, a pesquisa de cristais e o exa-me bacteriológico do líquido sinovial foram negativos, tornando estes diagnósticos menos prováveis. A doen-te melhorou com optimização da doen-terapêutica para a AR, apoiando um quadro de monoartrite aguda no contex-to de flare da doença.
Discussão/Conclusão: A ocorrência de sinovite após
ATJ é uma complicação infrequente. A exclusão de infe-ção ou artrite por deposiinfe-ção de cristais é importante no diagnóstico diferencial4. Nos doentes em que a sinovite ocorre por atividade inflamatória da AR, esta pode ser controlada com repouso e otimização da terapêutica imunossupressora4, como verificado no caso descrito.
bIblIoGRAFIA
1. Mirza R, Ishaq S, Khan M, Memon A. Rituximab therapy for fla-re-up rheumatoid arthritis after total knee replacement surge-ry. J Pak Med Assoc, 2012;62:1120-3
2. Danoff J, Moss G, Liabaud B, Geller J. Total Knee Arthroplasty Considerations in Rheumatoid Arthritis. Knee Surg Relat Res, 2012;24:1–6
3. Akpancar S, Turgut H, Akyildiz F, Ekinci S. Orthopedic Mana-gement of Total Knee Arthroplasty in the Patients with Rheu-matoid Arthritis. J Arthritis, 2016;5:1
4. Niki Y, Matsumoto H, Otani T et. al. Five types of inflammato-ry arthritis following total knee arthroplasty. Biomed Mater Res A, 2007;81:1005-10
5. Shinomiya F, Okada M, Hamada Y, et al. Indications of total ankle arthroplasty for rheumatoid arthritis: evaluation at 5 years or more after the operation. Mod Rheumatol, 2003;13:153-159 6. Hayakawa K, Date H, Nojiri S, Yamada H. Revision Total Knee Arthroplasty due to Rheumatoid Arthritis after Unicomparti-mental Knee Arthroplasty: A Case Report. J Arthritis, 2016;5:1 7. Fink B, Berger I, Segmüller C, Fassbender H et al. Recurring sy-novitis as a possible reason for aseptic loosening of knee endo-prostheses in patients with rheumatoid arthritis. J Bone Joint Surg, 2001;83:604-8.
CC79 – CUtAnEoUs And pERIphERAl nERvoUs systEm vAsCUlItIs – A pREsEntInG mAnIFEstAtIon oF slE/sjÖGREn ovERlAp
Miguel Guerra1, Sara Ganhão2,
Raquel Miriam Ferreira2, Teresa Martins-Rocha2, Ana Filipa Rocha Águeda3, Francisca Aguiar2, Eva Mariz2, José Pinto2, José Brito2,
Miguel Bernardes2, Lúcia Costa2
1. Rheumatology, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal
2. Rheumatology Department, Centro Hospitalar de São João, Porto, Portugal
3. Rheumatology Department, Centro Hospitalar do Baixo Vouga, Aveiro, Portugal
Introduction: Systemic Lupus Erythematosus (SLE)
and Sjögren Syndrome (SS) are closely related, not only due to their frequent coexistence, but also to overlap in their clinical and immunological expression. Prevalen-ce of secondary SS in SLE patients ranges between 9-9%. On the other hand, patients with primary SS
of-ten present clinical and immunological manifestations included in the SLE classification criteria. The authors describe the case of a patient presenting with cuta-neous/peripheral nervous system (PNS) vasculitis with overlapping immunology for SLE and SS.
Case report: A 42 years old female was referred to the
Rheumatology inpatient department with complaints of lower extremities hypo/dysesthesias for over a month and petechial skin lesions in both legs for a week. When questioned about other complaints, she refer-red xerostomia and polyarthalgia for more than a year. She also had a family history of small vessel vasculitis (2 uncles), SLE and Rheumatoid Arthritis (2 monozy-gotic twin cousins). On observation, she presented bi-lateral lower limb hypoesthesia (sock pattern), with di-minished bilateral foot dorsiflexion strength and uni-lateral foot plantar flexion. Petechial lesions were pre-sent bilaterally below the knee, coalescing into pur puric plaques, more evident on the posterior sur-face of legs. Laboratory evaluation showed raised ESR (62mm/h) and CRP (104,1mg/L), with normal blood counts. ANAs titre was >1/1000 (homogenous pat-tern), with positive anti-DsDNA (>800UI/ml by ELISA and >1/80 by indirect immunofluorescence), antiSSA (>200UI/ml), antiSSB (49UI/ML) and antinucleosso-me antibodies; direct coombs test was also positive; cryoglobulins were negative. Lower limbs electromyo-graphy was compatible with multiplex mononeuritis, highly suggestive of vasculitis. Moreover, the labial mi-nor salivary gland biopsy showed focus score of 2/4mm2. At this point, a diagnosis of vasculitis secon-dary to SLE/SS overlap was assumed and the patient started treatment with monthly iv cyclophosphamide (1gram) after three daily methylprednisolone pulses (1gram each). Later, skin histology confirmed vasculi-tis and also revealed epidermis necrolysis. Unfortuna-tely, the nerve biopsy was not representative. Despite a favourable response to treatment, the purpuric plaques evolved to ulcers, with the need of skin grafting to al-low skin healing.
Conclusion: The case presented fulfils classification
criteria both for SLE (SLICC) and SS (ACR/EULAR 2016), allowing the diagnosis of SLE/SS Overlap syn-drome. Despite controversial, literature suggests that SS occurring in association with SLE represents a dis-tinct entity. For example, age of onset is later than in SLE and sicca complaints precede lupoid manifesta-tions, as in the case described, in a 42 years’ female that presented poliarthalgia and xerostomia before mani-festing skin/PNS vasculitis.
CC80 – slE pREsEntInG wIth myoCARdItIs – A CAsE REpoRt
Miguel Guerra1, Raquel Miriam Ferreira2, Sara Ganhão2, Teresa Martins-Rocha2, Ana Filipa Rocha Águeda3, Francisca Aguiar2, Eva Mariz2, José Pinto2, José Brito2, Pedro Madureira2, Lúcia Costa2
1. Rheumatology Department, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal 2. Rheumatology Department, Centro Hospitalar São João, Porto, Portugal
3. Rheumatology Department, Centro Hospitalar do Baixo Vouga, Aveiro, Portugal
Introduction: Systemic Lupus Erythematosus (SLE)
has myriad of presentations with highly variable cour-se, ranging from indolent to a fulminant course. Car-diac manifestations are protean with myocarditis being usually subclinical and rarely presenting with left ven-tricular dysfunction.
Case description: A 21 years old man was referred to
the inpatient department of Rheumatology from ano -ther institution, with skin rash, fever and polyarthralgias, for over a month. At observation, he had peri -pheral symmetric polyarthritis (metacarpal-phalangeal and proximal interphalangeal joints), tachycardia, sys-tolic pulmonary murmur and fever (38,1ºC). He also presented diffuse alopecia and erythematous skin rash of trunk and arms. Study from previous institution ex-cluded infectious aetiology and showed bicitopenia (Haemoglobin 8.4g/dl; 2160 leucocytes/mm3) and small pericardial effusion on cardiac ultrasound; it also documented ANA titre >1/1000 (homogenous), posi-tive dsDNA antibodies (522 UI/ml), posiposi-tive anti--histones/anti-nucleossome antibodies and low C3c (50,6mlg/dl) and C4 (9,2mg/dl). On admission at our department, besides anaemia (haemoglobin 9,5g/dl), leukopenia (2940/mm3 with 450 lymphocytes/mm3) and raised ESR (94mm/h) and CRP (23,3mg/L), he also presented leukocyturia and proteinuria (2g/L, spot uri-ne), with normal serum creatinine and chest x-ray. As-suming the diagnosis of SLE, transthoracic cardiac ul-trasound was repeated and reported systolic depres-sion (ejection fraction EF 45%) and 24 h urine sample revealed a proteinuria of 1,27g. Despite normal range troponin I, BNP was slightly raised (109,4pg/ml). Car-diac MRI showed late sub-epicardial enhancement pat-tern compatible with myocarditis. After performing a renal biopsy, treatment was started, with 3 methyl-prednisolone pulses of 500mg, intravenous monthly
cyclophosphamide (CYC) and hydroxychloroquine. Renal histology confirmed lupus nephritis class IV. At discharge, the patient was feverless, without arthritis or skin rash and resolved anemia/leukopenia; new car-diac ultrasound revealed slight EF improvement (49%) and spot urine with 0,7g/L proteins. The patient was administered a total dose of 6g of CYC the following 6 months, with posterior conversion to mycophenolate mofetil (2g/day). One year after disease onset, there is no evidence of disease flare, with renal remission (spot urine proteins <0,5g/L, inactive sediment, normal se-rum creatinine) and no cardiopulmonary complaints. Conclusion - myocardial involvement is not uncom-mon in SLE, described in up to 50-80% patients on ne-cropsy studies; however, most cases are subclinical. Symptomatic myocarditis is rare, seen in only up to 9% of cases. The case described, despite almost asympto-matic (mild persistent tachycardia), had systolic dys-function in an inaugural multi-systemic picture (in-cluding renal, cutaneous, articular and haematologi-cal). A prompt and adequate diagnostic approach allo-wed early treatment with rapid disease control, avoiding irreversible sequelae in a young adult patient. CC81 – bElImUmAb no tRAtAmEnto
do lÚpUs ERItEmAtoso sIstÉmICo: A ExpERIÊnCIA dE Um CEntRo Bruno Miguel Fernandes1, Salomé Garcia1, Miguel Bernardes1, Francisca Aguiar1, Raquel Miriam Ferreira1, Pedro Madureira1, Iva Brito1, Lúcia Costa1
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
Introdução: O Lúpus Eritematoso Sistémico (LES) é
uma doença autoimune crónica, que afeta mais fre-quentemente as mulheres jovens. Tem caráter intermi-tente, caracterizando-se pela alternância entre perío-dos de remissão e de atividade da doença. A terapêuti-ca instituída é em função da sua gravidade e dos ór-gãos atingidos, reservando-se os imunossupressores para os casos mais graves. Em 2011, foi aprovado o pri-meiro fármaco biotecnológico para o tratamento do LES, o belimumab – um anticorpo monoclonal huma-nizado, que inibe especificamente o BlyS (estimulador de linfócitos B). Os seus ensaios clínicos revelaram: di-minuição da atividade da doença, redução do número e gravidade dos flares, efeito poupador de corticoste-róides e melhoria na qualidade de vida.
Objetivo: Descrever os doentes com LES que
inicia-ram belimumab e permanecem em seguimento num Serviço de Reumatologia de um Centro Hospitalar Uni-versitário.
Metodologia: Para os doentes incluídos na revisão,
descrevem-se vários aspetos da doença lúpica (tempo de evolução, grau de atingimento de orgão e imunolo-gia) e do seu tratamento (terapêuticas prévias, mo-mento e motivo(s) de início do belimumab assim como a sua efetividade pela evolução do SLEDAI).
Resultados: Encontrámos 4 doentes com LES tratados
com belimumab, do sexo feminino, com uma idade média atual de 36 anos (23-55 anos) e com uma dura-ção média da doença de 5 anos (2-11 anos). Todas se apresentavam com envolvimento articular, 3 com en-volvimento cutâneo (3/4) e 3 com atingimento hema-tológico (3/4). No estudo imunológico prévio à intro-dução do belimumab, 3 doentes tinham positividade para anticorpos anti-dsDNA (142.1-716.4 UI/mL) e to-das exibiam hipocomplementemia (C3c: 6-10mg/dL; C4: 55-152mg/dL). A doente que apresentava negati-vidade para os anticorpos anti-dsDNA, tinha realizado previamente 2 ciclos de rituximab. Das terapêuticas prévias para o LES, todos as doentes já tinham sido tra-tadas com hidroxicloroquina, com descontinuação em apenas uma delas e por toxicodermia. Duas doentes já teriam realizado tratamento com azatioprina, 1 doen-te com micofenolato de mofetilo (MFM) e rituximab e outra ainda com MFM isoladamente. Metade da amos-tra iniciou belimumab por atingimento mucocutâneo grave/refratário; 1 doente por envolvimento hemato-lógico e articular graves e 1 doente por envolvimento hematológico grave. Foram comparados os valores de SLEDAI à data do início do belimumab, em média de 11 (7- 15), com os valores aos 6 e 12 meses. Uma doen-te indoen-terrompeu o belimumab aos 6 meses por nefridoen-te lú-pica; as restantes mantêm-se sob terapêutica. As varia-ções de SLEDAI nas 3 doentes ainda sob belimumab fo-ram em média de 8 (4-15) aos 6 meses e de 8 (2-15) aos 12 meses. Duas doentes apresentam-se atualmen-te em remissão (SLEDAI de 0). (ver Tabela 1).
Conclusão: Apesar do tamanho da amostra não
per-mitir realizar grandes inferências, os nossos resultados são positivos e vão de encontro à literatura. Esta últi-ma suporta a utilização de belimuúlti-mab em doentes com LES, com doença musculoesquelética e/ou cutânea ati-vas e refratárias à terapêutica convencional. Em con-formidade, das duas doentes que iniciaram belimumab por envolvimento cutâneo grave, uma doente apresen-ta-se atualmente em remissão SLEDAI e a outra teve uma boa resposta SLEDAI aos 6 meses. Por outro lado,
a nossa pequena série fornece um dado novo e mais in-teressante, que se prende com uma inusitada resposta ao belimumab numa indicação pouco formal, ou seja, numa trombocitopenia grave e refratária ao rituximab no contexto do LES.
CC82 – ARtRItE dA ARtICUlAÇÃo
tEmpoRo-mAndIbUlAR: A mAnIFEstAÇÃo InICIAl dE EspondIlItE AnqUIlosAntE? Nathalie Madeira1, Manuela Micaelo1, 2,
Sandra Bitoque3, António Vilar2, José Vaz Patto1
1. Rheumatology Department, Instituto Português de
Reumatologia, Lisboa, Portugal
2. Reumatologia, Hospital dos Lusíadas, Lisboa, Portugal 3. Cirurgia Maxilo-Facial, Hospital dos Lusíadas, Lisboa, Portugal Introdução: A artrite é a condição dolorosa mais
co-mum que afeta a articulação temporo-mandibular (ATM). Embora o seu envolvimento esteja descrito mais frequentemente nos casos de osteoartrose e de artrite reumatóide, o comprometimento da ATM como parte das espondilartropatias também tem sido relatado. Na espondilite anquilosante (EA), a ATM tem sido repor-tada como atingida em 10 a 15% dos casos e em doen-tAbElA I. CARACtERIzAÇÃo dos doEntEs Com lEs mEdICAdos Com bElImUmAb E Em sEGUImEnto nUm sERvIÇo dE REUmAtoloGIA dE Um CEntRo hospItAlAR UnIvERsItÁRIo
L.C.S.C (Doente 1) M.P.P.F (Doente 2) M.A.O.M. (Doente 3) L.C.C. (Doente 4)
Sexo Feminino Feminino Feminino Feminino
Idade atual 36 23 55 29
Idade ao diagnóstico 25 20 49 27
Tempo de doença (anos) 11 3 6 2
Tipo de envolvimento articular, cutâneo, articular, cutâneo, articular, articular, cutâneo, imunológico hematológico, hematológico, hematológico,
imunológico imunológico imunológico, serosas Imunologia pré-belimumab:
• anticorpos anti-dsDNA 278,2 UI/mL 142,1 UI/mL 17,1 UI/mL 716,4 UI/mL (N <100UI/mL)
• complemento
C3c (N 83-177mg/dL) 74.0 mg/dL 152 mg/dL 104 mg/dL 55 mg/dL C4 (N 12-36 mg/dL) 10 mg/dL 10 mg/dL 9 mg/dL 6 mg/dL Terapêuticas prévias prednisolona, hidroxicloroquina prednisolona, prednisolona, para o LES hidroxicloroquina, (suspensa por hidroxicloroquina, naproxeno,
nifedipina, toxicodermia), azatioprima, hidroxicloroquina, pentoxifilina, resoquina micofenolato de mofetil, azatioprina micofenolato de mofetil rituximab, IgIV
Início do belimumab
• Data: Motivo(s): Maio/2016 Junho/2015 Março/2015 Agosto/2017 envolvimento envolvimento trombocitopenia Envolvimento mucocutâneo grave cutâneo refratário refratária articular e
hematológico refratários
Tempo sob belimumab 21 32 35 6
(meses) SLEDAI
pré-belimumab 10 15 7 14
aos 6 meses 6 0 3 18 (nefrite lúpica)
aos 12 meses 8 0 1 –
atualmente 4 0 0 –
tes com longa evolução. Apresenta-se um caso raro de artrite da ATM em doente sem diagnóstico prévio de doença reumática inflamatória.
Caso clínico: Doente do sexo masculino, 54 anos,
correu ao Serviço de Urgência por dor e edema da re-gião temporo-mandibular direita e limitação da aber-tura bucal, com cerca de 2 semanas de evolução. Rea-lizou exames que revelaram PCR de 4,84 mg/dL e ra-diografia da ATM direita com alargamento do espaço inter-articular. Foi encaminhado para consulta de Ci-rurgia Maxilo-Facial, onde realizou vários exames, no-meadamente TAC e RMN das ATM que revelaram irre-gularidade e deformação do contorno da cortical do côndilo mandibular direito, edema dos tecidos envol-ventes e derrame articular, compatível com artropatia inflamatória e biópsia de tecido capsular e aspiração do líquido sinovial cujo exame histopatológico confirmou a presença de infiltrado inflamatório crónico e inespe-cífico, sendo negativo para células neoplásicas e para bactérias potencialmente patogénicas. Foi medicado com deflazacort 12 mg e AINE em SOS e referenciado para consulta de Reumatologia. Quando observado pela primeira vez por Reumatologia, cerca de 3 meses após o início da artrite da ATM, apurou-se história de cervicalgia e lombalgia inflamatórias desde os 20 anos, que motivavam a toma de AINE nas crises, com alívio, e já tinham levado o doente a recorrer à consulta de Neurocirurgia e realizado TAC e RMN, mas sem qual-quer diagnóstico de doença reumática inflamatória. Ne-gou qualquer outra sintomatologia nomeadamente ar-tralgias e tumefações noutras localizações. À observa-ção destacava-se palpaobserva-ção dolorosa da ATM direita e tumefação elástica da mesma, manobras sacro-ilíacas positivas, sem limitação da coluna cervical e lombar. Colocou-se a hipótese de espondilartrite, solicitando--se estudo para confirmar o diagnóstico. Dos exames realizados destaca-se VS 48 mm/H e PCR 1,4 mg/dL, HLA B27 positivo, radiografia da bacia com te bilateral grau 2, TAC das sacro-ilíacas com sacro-ileí-te erosiva e radiografia da coluna dorsal com presença de sindemófitos. Admitiu-se o diagnóstico de EA. À te-rapêutica prévia adicionou-se salazopirina até à dose máxima de 2g por dia, verificando-se, 4 meses após o início do quadro, resolução da artrite, ausência de pre-juízo funcional da ATM, melhoria da lombalgia infla-matória e ausência de padrão inflamatório nas análises.
Discussão: O doente foi encaminhado por
monoartri-te da ATM, após exclusão de causa infecciosa e neo-plásica. Na primeira consulta de Reumatologia, verifi-cou-se a presença de lombalgia e cervicalgia
inflama-tória desde os 20 anos, com presença de sacroileite radiológica bilateral. Foi pelo envolvimento monoarti-cular da ATM, articulação não tão frequentemente atin-gida na EA que se procedeu ao diagnóstico, 34 anos após o seu início.
CC85 – mIopAtIA poR CoRpos dE InClUsÃo E sEU tRAtAmEnto: RElAto dE Um CAso ClÍnICo
Joana Leite Silva1, Daniela Santos-Faria1, Joana Sousa-Neves1, Joana Ramos Rodrigues1, Soraia Azevedo1, Sérgio Alcino 1, José Tavares-Costa1, Daniela Peixoto1, Carmo Afonso1, Filipa Teixeira1
1. Serviço de Reumatologia, Unidade Local de Saúde do Alto Minho, Ponte de Lima, Portugal
Introdução: A miopatia por corpos de inclusão (MCI)
é uma miopatia inflamatória idiopática, rara, mais co-mum em indivíduos com idade superior a 50 anos de idade e mais frequente no sexo masculino1. A sua etio-logia é desconhecida e a sua apresentação clínica é va-riável. Caracteriza-se, habitualmente, por fraqueza muscular, proximal e distal, assimétrica e progressiva, podendo evoluir para atrofia muscular e incapacidade funcional.
As enzimas musculares séricas podem encontrar-se em níveis normais ou moderadamente elevadas. His-tologicamente, a presença de vacúolos nas fibras mus-culares é característica desta doença.
Em geral, a MCI é descrita com sendo refratária ao tratamento com imunossupressores, incluindo os cor-ticosteroides. No entanto, há relatos de casos em que se verificou alguma resposta, justificando a sua utilização.
Objetivo: Relato de um caso clínico de MCI e
respos-ta a diferentes atitudes terapêuticas.
Caso clínico: Doente do sexo feminino, de 80 anos de
idade, internada no Serviço de Reumatologia em 2009 por quadro clínico de fraqueza muscular dos membros superiores e inferiores. Analiticamente apresentava ele-vação de parâmetros inflamatórios, eleele-vação da creati-noquinase (1115 UI/L) e da DHL (462 UI/L) e anticor-pos antinucleares negativos. A eletromiografia dos membros superiores (MS) e inferiores (MI) apoiava diagnóstico de miopatia inflamatória, pelo que iniciou prednisolona (PDN), na dose de 1mg/kg/dia, com res-posta favorável, clinica e analiticamente. No entanto, a histologia revelou alterações sugestivas de miopatia por corpos de inclusão, pelo que se questionou o diagnós-tico. A revisão da histologia confirmou o diagnóstico de MCI.
Esta doente manteve-se clinicamente estável com PDN, em doses entre 7,5-10mg/dia até novembro de 2013. Nesta altura, apresentou-se de novo com fra-queza muscular, grau 4/5, quer nos MS quer nos MI e, analiticamente constatou-se novo aumento de CK. Dada a resposta prévia à corticoterapia, aumentou-se a dose de PDN para 0,5mg/kg/dia e associou-se meto-trexato (MTX) (dose máxima de 25 mg/semana), ten-do-se conseguido reduzir de novo a dose de PDN até 5mg/dia e atingir a remissão clínica. O surgimento de pancitopenia grave, em Dezembro de 2016, conduziu à suspensão deste fármaco.
Em Novembro de 2017, por novo agravamento clí-nico e não podendo reiniciar MTX, tentou-se terapêu-tica com imunoglobulinas endovenosas, na dose de 400mg/Kg durante 5 dias, trimestralmente, mantendo PDN na dose de 5 mg/dia. Verificou-se, também, res-posta clínica favorável. Encontra-se, de momento, sem sinais de atividade da doença, com enzimas muscula-res e parâmetros de fase aguda normais.
Discussão/Conclusão: A MCI é uma miopatia
infla-matória rara, que cursa com fraqueza muscular difusa, proximal e distal, de início lento e insidioso, habitual-mente refratária a vários tratamentos.
Há alguns estudos que demonstram que uma baixa dose de metotrexato associado a corticóide pode retar-dar a progressão da doença, não sendo, no entanto, consensual a sua utilização2.
Com o presente caso clínico pretendeu-se salientar que, apesar das dificuldades de tratamento descritas para esta condição clínica, haverá casos com possibili-dade de resposta a diferentes terapêuticas.
REFERÊnCIAs
1. Monte et al. Miosite de corpos de inclusão: série de 30 casos de um centro terciário brasileiro. Acta Reumatol Port. 2013;38: 179-185.
2. Acharya et al. A Rare Case of Sporadic Inclusion Body Myositis (s-IBM). Journal of Clinical and Diagnostic Research. 2016 Jan, Vol-10(1).
CC108 – sARCoIdosE E doEnÇA dE stIll do AdUlto, UmA AssoCIAÇÃo InvUlGAR?
Sara Ganhão1, Raquel Miriam Ferreira1, Ana Filipa Rocha Águeda2, Miguel Guerra3, Eva Mariz1, Miguel Bernardes1, Lúcia Costa1
1. Serviço de Reumatologia, Centro Hospitalar de São João, Porto, Portugal
2. Serviço de Reumatologia, Centro Hospitalar do Baixo Vouga, E.P.E., Aveiro, Portugal
3. Rheumatology Department, Centro Hospitalar de Vila Nova de Gaia/Espinho, Vila Nova de Gaia, Portugal Introdução: A sarcoidose é uma doença inflamatória
crónica de etiologia desconhecida caracterizada pela formação de granulomas não-caseosos. A doença de Still do Adulto é uma doença inflamatória crónica que se manifesta tipicamente por febre, artrite e rash cutâ-neo evanescente. A associação entre ambas foi rara-mente reportada na literatura. Os autores apresentam um caso clínico de doença de Still do adulto numa doente com o diagnóstico prévio de sarcoidose, com resposta clínica e laboratorial exclusiva ao Tocilizumab (TCZ).
Caso Clínico: Mulher de 41 anos, previamente
assin-tomática até ao início de 2014, altura em que iniciou artralgias de ritmo inflamatório com tumefação dos joe-lhos e tornozelos e lesões cutâneas de eritema nodoso nos membros inferiores. A TC toracoabdominal reve-lou inúmeras adenomegalias mediastínicas e hilares bi-laterais. Fez broncofibroscopia e o estudo do lavado bronco-alveolar demonstrou aumento da relação CD4+/CD8+, com diminuição desta relação no sangue periférico; negatividade para células malignas e para os exames micobacteriológicos diretos e culturais. Reali-zou ainda biópsia de um gânglio mediastínico, cuja his-tologia confirmou a presença de processo granuloma-toso não-caseoso compatível com o diagnóstico de sar-coidose, tendo iniciado terapêutica com corticosteroi-des com boa resposta clínica. Em finais de 2015, apresentou-se com um novo quadro clínico, caracteri-zado por picos subfebris de predomínio vespertino, embora sem rash cutâneo evanescente associado, po-liartrite simétrica, aumento persistente da VS:114 mm e PCR:344 mg/L; anemia normocítica normocrómica (Hgb:10 g/dl); leucocitose com neutrofilia marcadas (17.000/ml e 15.000/ml, respetivamente) e franca ele-vação da ferritina (2.372 ng/mL). A TC torácica reve-lou a presença de várias formações ganglionares mediastínicas e a ecografia abdominal mostrou uma hepa -tomegalia discreta. Do estudo complementar efetuado destacam-se: imunologia para conectivites negativa; metabolismo fosfocálcico normal; hemoculturas nega-tivas, negatividade para Micobacterium tuberculosis,
M. Avium e M. intracelular e serologias víricas nega tivas.
A doente cumpria os Critérios Yamaguchi para doença de Still do Adulto: 2 critérios major (artrite ≥2 semanas e leucocitose (≥10.000/ml com ≥80% neutrófilos) + 3 critérios minor (linfadenopatia, hepatomegalia e negatividade para FR e ANAs). Foi estabelecido o dia