Authors
Rodrigo Bueno de Oliveira1 Hirokazu Okazaki2 Andrea E. Marques Stinghen3 Tilman B. Drüeke4
Ziad A. Massy5 Vanda Jorgetti6
1 INSERM U-1088, UFR de Médecine et de Pharmacie, Université de Picardie Jules Verne, Amiens, France and Division of Nephrology, Univer-sity of São Paulo, São Paulo, Brazil. 2 INSERM U-1088, UFR de Médecine et de Pharmacie, Université de Picardie Jules Verne, Amiens, France and Department of Metabolism, Endocrinology and Molecular Medicine, Osaka City University, Graduate School of Medicine, Osaka, Japan.
3 INSERM U-1088, UFR de Médecine et de Pharmacie, Université de Picardie Jules Verne, Amiens, France and Department of Basic Pathology, Experimental Nephrology Laboratory, Universidade Federal do Paraná, Curitiba, Brazil. 4 INSERM U-1088, UFR de Méde-cine et de Pharmacie, Universi-té de Picardie Jules Verne, Amiens, France.
5 Division of Nephrology, Ambroise Paré Hospital, Paris Ile de France Ouest (UVSQ) University, Boulogne Billancourt/Paris, and INSERM U-1088, Université de Picardie Jules Verne, Amiens, France.
6 Division of Nephrology, University of São Paulo, São Paulo, Brazil.
Submitted on: 01/30/2013. Approved on: 03/21/2013.
Correspondence to:
Vanda Jorgetti.
Division of Nephrology, University of São Paulo, São Paulo, Brazil. Av. Dr. Arnaldo, nº 455, 3º andar, sala 334, São Paulo, SP, Brazil. CEP: 01246-903.
Tel: +55 (11) 3061-8351.
We acknowledge funding by a grant from University of Picardie-Jules Verne. R.B. de Oliveira and H. Okazaki received postdoctoral grants from Picardy Regional Council/ University of Picardy Jules Verne, Amiens, France. R.B. de Oliveira and A.E.M. Stinghen received postdoctoral scholarships from the National Council of Technological and Scientific Development (CNPq), Brasília, Brazil.
Vascular calcification in chronic kidney disease: a review
Keywords: chronic, renal insufficiency; review; vascular calcification.
Vascular calcification (VC), an indepen-dent and strong predictor of cardiovascu-lar risk, is often found in CKD patients. The degree of VC is providing incremen-tal prognostic value over traditional risk markers. There is interest in improving our understanding of mechanisms, esta-blishing diagnostic methods and effective prevention and treatment modalities. The abnormal mineral metabolism of CKD is known to facilitate the progression of VC, in concert with altered activities of VC inhibitors. Possible measures to prevent VC include the control of serum calcium and phosphate as well as other factors involved in its progression, including vi-tamin D sterols, parathyroid hormone, fibroblast growth factor-23, klotho, and VC inhibitors. In addition, we discuss new possible therapeutic approaches to halt VC or reverse its progression. The principal aim of this review is to provi-de an updated overview of VC in patients with CKD, with particular focus on pa-thophysiology, diagnosis, prevention and treatment.
A
BSTRACTI
NTRODUCTIONBased on the Health Science Descriptors
definition,1 vascular calcification (VC)
is a pathologic process characterized by thickening and loss of elasticity of the muscular arteries walls due to calcification of the tunica media and/or intima.
The majority of authors refer to VC as a general term for both intima and media calcification. However, there are differences between them. Intima calci-fication, which typically occurs within
atherosclerotic plaque in the aorta, coro-nary arteries, and other large arteries, is an indicator of advanced stages of athe-rosclerosis. Media calcification, which is often found in patients with metabolic syndrome, diabetes and/or chronic kidney disease (CKD), is characterized by diffuse mineral deposition along elastic fibers in both elastic-type conduit arteries and muscle-type resistance arteries.
VC is a common problem among CKD patients. Its prevalence increases with the progressive decrease of kidney function.2
The association between soft-tissue calci-fication and uremia has been recognized more than 100 years ago. At that time, Rudolf Virchow (1821-1902) described the presence of extraosseous calcifications in a group of patients with non-metabolic bone disease, uremia, primary hyperpa-rathyroidism and vitamin D intoxication. Virchow proposed an interesting hypothe-sis to explain this phenomenon: he suppo-sed that calcium salts were dissolved from bone and carried by the blood stream to be deposited at some distant site to form “cal-cium metastases”, a process analogous to the dissemination of malignant cells from a primary neoplasm.3 Contemporaneously,
Karl Rokitansky (1804-1878) made ano-ther important contribution by describing the presence of atherosclerotic lesions.4
Some years later, in 1903, Johann Georg Mönckeberg described the medial type of VC which he named calcific sclerosis.5
past decades, studies in CKD patients showed that the degree of VC was an independent and strong predictor of death in association with vessel stiffness, arterial hypertension, left ventricular hypertrophy and cardiomyopathy.6,7
Therefore, great efforts have been made to improve our understanding of the pathogenesis, diagnosis, prevention and treatment of VC. The purpose of this review is to provide an update of our present insight into the VC related to CKD.
P
ATHOGENESISGENERALMECHANISMSOFVASCULARCALCIFICATION
VC is a complex process, involving not only simple precipitation of supersaturated phosphate and cal-cium concentrations in the extracellular milieu (mi-neral step) but also a tightly regulated, cell mediated process including apoptosis, osteochondrogenic diffe-rentiation, and elastin degradation (cellular step). The time course of these two steps in vivo, in particular to know which one appears first, remains poorly de-fined. The main pathogenetic events are summarized below.
APOPTOSIS
Vascular smooth muscle cells (VSMC) apoptosis is regarded as an important contributor in the initiation of VC. Of note, VSMC within atheromatous plaques exhibit increased sensitivity to apoptosis compared with those in normal vessel wall.8,9 Apoptotic
bodies derived from VSMC are thought to play a role as nucleating structures for calcium crystal formation such as matrix vesicles in the initiation of calcification.10,11
OSTEOCHONDROGENICDIFFERENTIATION
VSCM phenotype change to osteochondrogenic cells is characterized by the appearance of matrix vesicles containing apatite and calcifying collagen fibrils on the surface of VSMC. As in bone, these vesicles act as early nucleation sites for calcification. Moreover, VSMC synthesize bony-associated proteins and promote crystal formation and deposition.12In vitro
studies have demonstrated this phenotypic change, featured by the expression of bone-associated proteins including alkaline phosphatase (ALP), osteocalcin, and osteopontin. Runt-related transcription factor 2 (Runx2) and Msh homeobox 2, which are obligate transcription factors in normal bone development,
also have been shown to be associated with VSMC osteochondrogenic differentiation.13
The phenotypic change of VSMC has also been de-monstrated in in vivo studies. Apolipoprotein-E and matrix Gla protein (MGP) knockout mice exhibit oste-ochondrocyte-like cells flanking the calcium deposits in the vessel wall.14,15 In human calcified arteries, the
expression of collagen II and sex determining region Y related high-mobility group box 9 (sox9), which are keys transcription factors for chondrogenesis, has been documented as well.16
The VSMC nature of osteochondroblast-like cells in VC has recently been questioned by Tang Z et al.17
These authors claimed that in response to vascular injuries, multipotent vascular stem cells, not mature VSMC, differentiate into osteochondrogenic cells and thereby initiate VC. This hypothesis has been immediately refuted by Nguyen et al.18 who stated
that there is compelling evidence that VSMC are not terminally differentiated and are capable of transition into a phenotype characterized by cell proliferation and loss of differentiation markers.
ELASTINDEGRADATION
Elastic lamellae consist mainly of amorphous elastin, the major component of the aortic medial layer, in addition to the concentric layers of helically arranged smooth muscle cells. Elastocalcinosis is characterized by deposition of hydroxyapatite on the elastic lamel-lae of the arteries. Elastin degradation is known to play an important role in the initiation and progres-sion of VC. It is induced by elastase, metalloprotein-ases and other protemetalloprotein-ases including cysteine and serine. Degraded elastin has a high affinity for calcium, fa-cilitating growth of hydroxyapatite along the elastic lamellae. Moreover, elastin derived peptides binding to elastin laminin receptors on the surface of VSMC and through transforming growth factor-β signaling can increase proliferation and upregulate Runx-2, resulting in osteochondrogenic differentiation.19,20
MOLECULARMECHANISMSOFVASCULARCALCIFICATION
In addition to the mechanisms described above, it has become increasingly clear that there are several inhibitory proteins and promoters involved in the process of VC (Table 1).21-25 A complex interplay
pathway, and thereby favors VSMC apoptosis.29
Phosphate transport into the cell is primarily me-diated by sodium-dependent phosphate (Na/Pi) co-transporters. Its uptake into VSMC occurs mainly via phosphate transporter 1 (Pit-1), a member of the type III Na/Pi co-transporters, leading to osteochon-drogenic differentiation.30 Of note, Pit-1 knockdown
has been shown to suppress phosphate-induced calcification and to block induction of Runx2 and osteopontin.31
PYROPHOSPHATE (PPI)
PPi is a major physiological inhibitor of hydroxyapatite formation. It also potently inhibits VC. PPi is generated by hydrolysis of ATP induced by the enzyme ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1), an extracellular membrane-bound glycopro-tein. When the Enpp1 gene is mutated and the enzyme is inactive, like in the rare clinical syndrome called “idiopathic infantile arterial calcification”, severe calcification of the internal elastic lamina of muscle arteries ensues. This represents one among the nume-rous demonstrations of the important role of PPi in preventing VC.32 In contrast, ALP, whose expression
is induced by VSMC osteochondrogenic transforma-tion, promotes vessel wall matrix mineralization. One of the main roles of ALP is to hydrolyze PPi, which in turn generates phosphate ions and thereby favors the development of VC.
KLOTHO - FIBROBLASTGROWTHFACTOR-23 AXIS
In addition to its well-known anti-aging action, Klotho serves as a co-factor of FGF23 in conferring FGF23 specificity for FGF receptor activation. The two together play an essential role in the control of phosphate and vitamin D metabolism, by enhancing urinary phosphate excretion and suppressing renal 1α-25OH vitamin D hydroxylase activity. Klotho-deficient mice and FGF23-null mice exhibit a similar phenotype, characterized by an accelerated aging process with shortened life span, atheroscle-rosis, and soft-tissue calcification including VC.33,34
Interestingly, Klotho overexpression in CKD mice strikingly reduces aortic calcification, as compared to Klotho-deficient and wild-type mice with similar renal function impairment. Klotho also is capable of directly inhibiting phosphate-induced calcification of VSMC in vitro.35 Regarding FGF-23, its role in VC
is less clear in the context of CKD. As an apparent TABLE 1 SUMMARYOFTHEMOSTCOMMONINHIBITORY
ANDSTIMULATORYFACTORSINVOLVEDINTHE PATHOGENESISOFVASCULARCALCIFICATION
Inhibitors Promoters
Matrix Gla protein (MGP) Hyperphosphatemia
Osteopontin (OPN) Hypercalcemia
Bone morphogenic protein 7 (BMP-7)
Bone morphogenic protein 2 (BMP-2)
Magnesium Receptor activator of nuclear
factor-kappa B ligand (RANKL)
Fetuin-A
Osteoprotegerin (OPG)
Pyrophosphate (PPi)
Klotho
FGF-23 (?) FGF-23 (?)
including MGP, BMP-7, osteoprotegerin, fetuin-A, and osteopontin, regulates this process. Actually, it has also been speculated that promoter molecules can act via microRNA (miR) regulation. Recently, Balderman
et al.26 shown that human VSMC treated by BMP-2
downregulate miR-30b and miR-30c to increase Runx2 expression and promote mineralization.
The following paragraphs describe the main molecular mechanisms involved in VC.
PHOSPHATEMETABOLICPATHWAY
Phosphate homeostasis is maintained by the hormonal control of its transport in intestine, bone, and kidney. The most active form of vitamin D, 1,25-dihydroxyvitamin D [1,25 diOH D], which is synthesized in the kidney, increases the intestinal absorption of phosphate and stimulates osteoclastogenesis in bone, leading to an increase in extracellular phosphate concentration. PTH acts on the kidney to stimulate both 1,25 diOH D synthesis through activation of 1α-25OH D hydroxylase, and urinary phosphate excretion.27 In addition to PTH and 1,25
diOH D, FGF23 and Klotho have been discovered more recently as novel factors involved in phosphate metabolism. The importance of the Klotho - FGF23 axis will be discussed below.
Phosphate is a well-known inducer of VSMC apop-tosis and osteochondrogenic differentiation. Increased phosphate levels suppress both the expression of growth arrest specific gene 6 (Gas6) and its receptor in VSMC.28 This inhibition leads to the suppression
paradox, high serum FGF23 were also found to be associated with the severity of atherosclerosis36 and
VC in patients with CKD.37 Curiously, other study
involving non-CKD patients observed a negative association between FGF-23 and the presence of ath-erosclerotic lesion.38 Several recent studies, but not all,
showed an involvement of both Klotho and FGF23 in VC, both in interaction and separately from each other, in addition to calcium, phosphate, PTH and 1,25 diOH D.39,40 However, their exact and respective
roles in this process require further study.
CALCIUM-SENSINGRECEPTOR
The CaR is a G protein-coupled receptor. It is expressed in tissues which are involved in the regulation of cal-cium metabolism, including the parathyroid, thyroid, kidney, bone and intestine.41 In addition, the CaR has
also shown to be expressed in blood vessels, both in en-dothelial cells42 and VSMC.43 Alam et al.44 showed that
a reduction in CaR expression in VSMC is associated with increased mineralization, and that calcimimetics can attenuate mineral deposition. Ivanovski et al.45
showed a protective effect of calcimimetics against the progression of VC and atherosclerosis in uremic apoli-poprotein-E knockout mice. They also showed a direct inhibition by calcimimetics of phosphate-induced hu-man VSMC calcification in vitro. This inhibitory effect could be abolished by suppression of CaR expression. Koleganova et al.46 reported that calcimimetics
incre-ase MGP expression and decreincre-ase Pit1 expression in uremic rats, preventing vessel wall remodeling. These observations suggest that calcimimetics play an impor-tant role in VC not only by indirect effects, such as improved control of serum PTH, calcium and phos-phate levels, but also by a direct effect on vascular cells. These findings underline the importance of research into the role of CaR in the pathogenesis of of VC.
OXIDATIVESTRESS
Oxidative stress can be defined as a disturbance of nor-mal cellular and molecular function caused by an imbal-ance between the excessive production of reactive oxy-gen species (ROS) and the natural defense ability of cells against oxidation.47 As an example, oxidative
modifica-tions of proteins by ROS can induce nitrotyrosine ex-pression by endothelial cells and high-level exex-pression of receptors for uremic toxins such as advanced glycation end products (AGE). In the cardiovascular system, AGE accumulation contributes to arterial stiffening due to
its binding to collagen and elastin in a disorderly way. Moreover, uremic toxicity leads to an impairment of endothelial nitric oxide (NO) synthesis, which plays a crucial role in vascular protection since NO inhibits the proliferation and migration of VSMC, expression of adhesion molecules, and platelet aggregation.48
The precise relationship of oxidative stress with VC is not yet well established. Yamada et al.49
observed in rats with CKD induced by adenine-rich diet a progressive development of arterial me-dial calcification, which was accompanied by a time-dependent increase in both aortic and systemic oxidative stress. Time-course studies indicated that both oxidative stress and hyperphosphatemia were correlated with arterial medial calcification. Our group showed a significant increase in nitrotyrosine immunostaining in aortas of uremic as compared to non-uremic apoE knock-out mice,50 and Guilgen et al.51 reported same finding in arteries from CKD
pa-tients as compared to healthy donors, demonstrating the effective participation of oxidative stress in VC.
INFLAMMATION
Chronic systemic inflammation is a common feature in CKD, caused both by the accumulation of pro-inflammatory compounds related to the markedly decreased glomerular filtration rate, and enhanced production and release of inflammatory cytokines.52
Previous in vivo and in vitro studies showed that inflammation induced intracellular lipid accumulation and foam cell formation by disrupting low-density li-poprotein receptor feedback regulation, exacerbating the progression of atherosclerosis and VC.53,54 Another
interesting classical observation was reported by Ketteler et al. in a group of 312 stable patients on he-modialysis the authors observed that serum fetuin-A concentration was lower than in healthy controls and was inversely associated with serum C-reactive protein, and that both were associated with enhanced cardiovascular and all-cause mortality.55
These observations exemplify the essential role of inflammation in vascular calcification and its links with mortality in patients on CKD patients.
PATHOPHYSIOLOGICLINKSBETWEENBONEANDVASCULAR CALCIFICATION
demonstrated an association between the progres-sion of aortic calcification and decreased bone mi-neral density. The authors also found that the risk of vertebral and hip fractures was greater in wo-men with aortic calcification. Several studies ha-ve extended these findings to patients with CKD in whom coronary and aortic calcification is appa-rently even more closely associated with reduced bone volume and disturbances in bone remodeling, especially in those with low bone turnover.60,63-65
It has long been known that aging, diabetes mellitus,66 dyslipidemia,67 smoking, and alcohol abuse
contribute to both decreased bone mineral density and VC increase.68 However, the association persists
after adjusting for some of these factors, suggesting the existence of other mechanisms that have not yet been fully elucidated.
Among the mechanisms proposed some point to various vascular pathologies as the underlying cause. Others have suggested that changes in bone cells could affect the vascular tissue. A third mechanism would include metabolic disorders common to a variety of diseases, either alone or in conjunction with inflammation, such as diabetes mellitus and dyslipidemia which would stimulate both bone resorption and vascular disease.
All organs are endowed with a vascular tree assuring the entry of nutrients and oxygen. Bone tissue is no exception to this rule. In addition, the blood transports bone cell precursors which are involved in bone remodeling and thereby con-tribute to the integrity of the skeleton. Ischemia caused by intraosseous atherosclerosis can compro-mise vascularization and favor osteoporosis.69 An
association has also been shown between decreased bone mineral density and the presence of peripheral arterial disease.70 A study in women showed that the
rate of bone perfusion was markedly reduced in those with osteoporosis as compared to osteopenic women or those with normal bone mass.71
Recently, novel functions of bone tissue have been discovered in addition to already well-known functions such as locomotion, protection of internal organs and participation in the regulation of mineral metabolism. Osteoblasts, through the production of osteocalcin, have been found to participate in the regulation of fat metabolism, energy homeostasis, and insulin se-cretion and sensitivity, all of which are essential for proper functioning and integrity of the cardiovascular
system.72 Osteocalcin regulates insulin gene expression,
β-cell proliferation, and adiponectin expression and secretion in adipocytes.73 Both in general population
and in patients with CKD serum osteocalcin has been shown to be positively correlated with serum adiponec-tin.74,75 Moreover, both correlate inversely with arterial
stiffness and progression of coronary calcification.74
Leptin, a hormone that regulates adipose tissue mass, is a potent inhibitor of bone formation and also pro-motes VC.76 In advanced stages of CKD serum leptin
levels are generally high. Coen et al.77 demonstrated
that uremic patients with high serum leptin levels and low serum PTH levels were prone to develop VC.
The identification of circulating bone cells with potential for VC raised questions as to whether this could be another link.78-80 Both mesenchymal cells
from bone marrow and those of hematological lineage can give rise to circulating bone cells with osteogenic potential that could, for example, also home in atherosclerotic lesions and contribute to intima calcification. The ability of these cells to promote VC has as yet only been demonstrated in vitro but not
in vivo.81
Recently, two comprehensive reviews about the bone-vascular axis were published by Fadini et al.81
and London.82
D
IAGNOSISThe clinical diagnosis of media calcification is practically impossible by physical examination alone. Its presence is suggested when palpable arteries are detectable when the sphygmomanometer is inflated to a higher level than the true systolic blood pressure. This propaedeutic ma-neuver is known as the Osler’s sign.83
At present, there is no reliable, sufficiently sensitive and specific biomarker for the diagnosis of VC. As described above, in the “Molecular mechanisms of vascular calcification” section, several biomarkers have been shown to be associated with the initiation and/or development of VC. However, it remains unproven that any of these markers reflects calcium phosphate deposition in the arterial wall. It is therefo-re unclear at ptherefo-resent whether they atherefo-re of any use for clinical practice.
are the most frequently used methods for a precise assessment of the severity of VC and its progression. Results of coronary artery calcification (CAC) are typically reported using the Agatston CAC score, which is based on the product of calcified plaque area and density coefficient. This score has been shown to be predictive of cardiac events. A limitation is the inability to distinguish between intima and media calcification. Moreover, Kristanto et al.84 pointed out
that the early onset of calcium deposition remains invisible even with these quantitative techniques and a zero Agatston score does not exclude the presence of incipient coronary calcification.
The Kidney Disease Improving Global Outcomes (KDIGO) 2009 guideline suggests to use cheap lateral abdominal radiography for the semiquantitative assessment of VC and an echocardiogram for the detec-tion of valvular calcificadetec-tion in patients with CKD stages 3 to 5. In clinical routine this technique is a reasonable alternative to costly CT-based imaging methods.85
Kauppila et al.86 developed an abdominal
calcifi-cation severity scoring system, using lateral lumbar films. The severity of calcified deposits is graded from 0 to 3, separately for the posterior and anterior wall at each vertebral segment, from the first to the fourth lumbar vertebra. Adragao et al.87 developed another
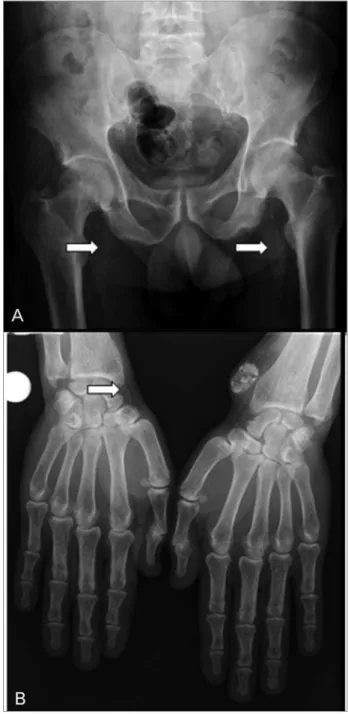
scoring system, using pelvis films for iliac and femoral arteries, and hand films for radial and digital arteries. They divided pelvis and hand films into eight sections and graded from 0 to 8 (maximum), depending on the presence or absence of VC (Figure 1). Although these semi-quantitative methods have limited ability to distinguish between the extension and degree of severity of calcification, they are more widely available and less expensive and can be used for cardiovascular risk stratification.
P
REVENTION ANDT
REATMENTTo date, none among a variety of available treatments has been definitively proven to prevent or reverse VC and it is widely admitted that this complication, once established, is irreversible. However, some drugs, including phosphate binders and cinacalcet, have been shown to halt or at least slow the progression of the disease in patients with CKD. Whether this translates into an improvement of clinical outcomes has yet to be demonstrated.85
Therefore, major efforts should be directed at prevention as the main option.
Figure 1. A: Arterial calcifications of pelvic arteries of a chronic hemodialysis patient; B: Arterial calcifications of hand arteries of a chronic hemodialysis patient. White arrows show extended vascular calcification areas (images from Dr. V. Jorgetti's personal archive).
In observational studies hyperphosphatemia, hypercalcemia and extremely high as well as low serum PTH values have been associated with poor outcomes in CKD. As to serum phosphate, even when its serum levels are in the high normal range, it may be associated with VC and mortality, as shown in patients with no known kidney disease and in CKD stage 3 patients.88,89
have been found to be associated with lower rates of VC (Table 2)85 and risks of mortality (Figure 2).90
However, in early stages of CKD these parameters are in the normal range the majority of patients, in contrast to what is observed in later stages.91
Nevertheless, some groups have examined the effects of reducing phosphate overload and/or bringing PTH levels towards the range of normal values in CKD stage 3-4 patients.92
The following paragraphs describe the main options which are presently available for the prevention of VC, with particular focus on phosphate control, as well as the supposedly protective role of some drugs in current use for the treatment of CKD-associated mineral and bone disorder (CKD-MBD). In addition, we discuss therapeutic modalities for new potential targets.
CONTROLOFSERUMPHOSPHATE
Currently, the options available to lower serum phosphate in CKD patients with hyperphosphatemia are (i) limiting dietary phosphate intake (while ensuring adequate protein intake), (ii) increasing the frequency or duration of dialysis in CKD stage 5D, and (iii) using phosphate binders and calcimimetics.
DIET
The KDIGO guideline suggests, in patients with CKD stages 3-5D, to limit dietary phosphate intake in the treatment of hyperphosphatemia alone or in combination with other treatments but this sugges-tion is mainly based on expert opinion, not on hard evidence.85 In patients with CKD stage 5, phosphate
intake should not exceed 1.000 mg per day. Recently, increasing attention has been paid not only to the
amount of phosphate in the diet, but also to its quali-ty. Inorganic phosphate in food additives is frequently found in processed food and “fast food”. In contrast to organic phosphate, inorganic phosphate is more effectively absorbed and therefore leads to phosphate overload more easily.93 Therefore, the
multiprofessio-nal clinical staff who has in charge the treatment of CKD patients should have two goals in mind with res-pect to recommendations on phosphate intake: avoid inorganic phosphate contained in food additives and limit daily phosphate intake to less than 1.000 mg/ day. Note that these goals should be attained without inducing protein malnutrition.
PHOSPHATEREMOVALBYDIALYSIS
Intradialytic plasma phosphate kinetics obeys to a 2-compartment model, thus differing from urea kinetics. The elimination mode of phosphate appears to resemble more that of typical middle molecules than that of small molecules such as urea.94 Phosphate
clearance during hemodialysis is affected by several factors, including blood and dialysate flow rate, dialyzer membrane surface area, ultrafiltration rate, and dialysis session frequency and length.
Increasing dialysis frequency (e.g., short daily dialysis, 6 times a week, for 2.5-3 hr each session) or dialysis length (e.g., nocturnal hemodialysis, 6 times a week, 8 hr each session) are helpful strategies to treat hyperphosphatemia in CKD stage 5D.95,96
Hemodiafiltration can increase phosphate mass removal further.97 In peritoneal dialysis (PD), a
cross-sectional comparative study between automated PD and continuous ambulatory PD showed that weekly total phosphate mass removal was similar with both methods.98 Mass transfer could be increased
TABLE 2 OPTIMALRANGERECOMMENDATIONSFORSERUMCONCENTRATIONSOFTOTALCALCIUM, PHOSPHATEAND
PARATHYROIDHORMONE (PTH), ACCORDINGTO CKD STAGES§
CKD stages 3 to 5 CKD stage 5D Normal range*
Phosphate (mg/dl) Normal range Lowering elevated P levels
toward the normal range 2.7-4.5
Total calcium (mg/dl) Normal range Normal range 8.6-10.2
[Cad] (mEq/l) NA 2.5 to 3.0 NA
PTH (pg/ml) < Upper normal range
(for range, see +)
2-9 x Upper normal range**
(for range, see +) 10-65
Cad: Dialysate Ca concentration; NA: Not applicable; § Slightly modified from text of reference 85 by the authors; * Normal range can show
by increasing total daily peritoneal fluid infusion.99
An interesting review on phosphate removal using various hemodialysis and PD treatment modalities was recently published by Kuhlmann.100
ORALPHOSPHATEBINDERS
Phosphate binder choice should be individualized depending on patients’ preference and tolerance. The greatest problem with all phosphate binders is not lack of efficacy, but lack of patient compliance. All currently available phosphate binders, such as calcium or magnesium salts, sevelamer hydrochloride or carbonate, and lanthanum carbonate are effective in lowering serum phosphate.85 Of note, there is
insufficient evidence that any specific phosphate binder significantly impacts patient-level outcomes.
Although there is some published evidence sug-gesting that sevelamer compared with calcium-based phosphate binders attenuates the progression of VC in patients with CKD stages 3-5101 and 5D,102,103
other studies failed to confirm these results.104,105
Sevelamer’s effects on VC could be both direct and indirect. This phosphate binder exerts pleiotropic effects, including an attenuation of oxidative stress and inflammation and a decrease in circulating levels of uremic toxins.106,107
Regarding calcium-based phosphate binders, they can lead to calcium overload when taken in excessive amounts.108,109 They should be restricted to no more
than a total of 1.500 mg elemental Ca intake per day, especially in presence of VC, hypercalcemia and/or adynamic bone disease,85 the two latter conditions
being associated with VC. Magnesium/calcium-based phosphate binders have recently been shown to be an acceptable alternative.110
CALCIMIMETICS
Both experimental and clinical evidence indicates that the calcium-sensing receptor (CaR) is not only expressed in kidney and parathyroid tissue, but also in vascular cells, and that plays a role in VC. In a mouse model of CKD, calcimimetics delayed the progression of aortic calcification and atherosclerosis,111 and in
a uremic rat model calcimimetics attenuated media calcification and proliferation of VSMC.112
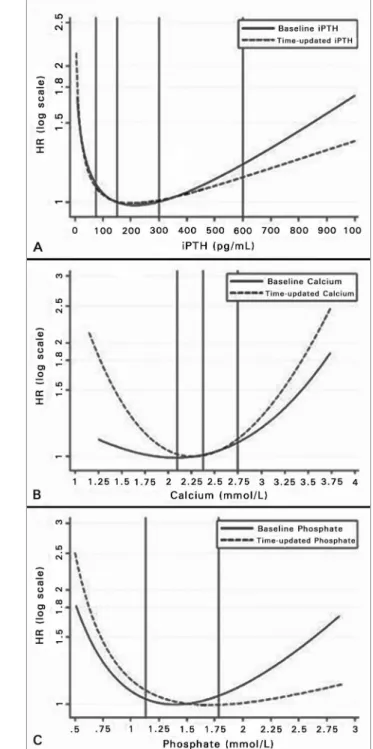
There is more limited evidence for a positive effect of calcimimetics on VC in the clinical setting. The admi-nistration of calcimimetics to dialysis patients with se-condary hyperparathyroidism, in addition to lowering Figure 2. Relative risk (RR) of all-cause mortality in chronic hemodialysis
serum PTH, calcium and phosphate,113 can also
re-duce the progression of VC, as shown in the recent ADVANCE study. The authors examined the effect of cinacalcet plus low-dose vitamin D on coronary ar-tery and cardiac valve calcification in 360 prevalent HD patients with secondary hyperparathyroidism, as compared to placebo with optimal standard therapy and flexible doses of vitamin D. In the cinacalcet group there was a 24% increase in Agatston CAC score, as compared to a 31% increase in placebo group (p = 0.073), with corresponding changes in the more recently developed volume CAC score of 22% and 30%, respectively (p = 0.009).114
One of the mechanisms by which a calcimimetic may slow VC progression is its serum PTH, calcium and phosphate lowering action. Other potential mechanisms are a direct stimulation of the CaR expressed in VSMC and an increase in MGP expression in the arterial wall, as shown both in
in vivo and in vitro experiments.45,115
VITAMIN D
Both native vitamin D and active vitamin D sterols, also called vitamin D receptor activators (VDRA), may be use-ful to treat secondary hyperparathyroidism, a condition associated with VC. However, pharmacological doses can result in undesirable effects such as the development of adynamic bone disease,116 hypercalcemia, and/or
hyperphosphatemia,117 which all favor the development
of VC. Newer VDRAs, such as paricalcitol, have been said to be more selective in suppressing PTH secretion and to be less hypercalcemic and hyperphosphatemic.118
Some experimental studies provided evidence in favor of this claim.119-121 An interesting observation
came from the experimental study of Lim et al.40 The
authors observed that CKD induces vascular klotho deficiency. They further demonstrated that both calcitriol and paricalcitol significantly upregulated klotho and restored FGF receptor-1 mRNA expression in VSMC of arteries from patients with CKD, whi-ch were cultured in procalcific media. They propo-sed that klotho restoration by vitamin D receptor activation confers VSMC FGF23 responsiveness and unmasks FGF23 calcification inhibitory effects.
Despite these exciting news from experimental models, there is so far no convincing study demonstrating that any vitamin D derivative is less prone than the native parent compounds to induce VC in patients with CKD.
NEWPOTENTIAL TREATMENTMODALITIES PYROPHOSPHATE (PPI)
PPi is a potent calcification inhibitor in vitro and an inhibitor of arterial media calcification in vivo, exerting its effects through direct physicochemical inhibition of hydroxyapatite crystal formation.122,123
Despite its known protective effects against the progression of VC, intravenous administration has been considered to be problematic due to short-half life and complications such as skin necrosis.124
Renewed interest in the calcification inhibitory effects of PPi has been raised by recent experimental studies.125,126 Since hemodialysis patients have low
circulating PPi levels,127 an interesting therapeutic
option could be to deliver PPi into the peritoneal ca-vity from where PPi would be slowly transported in-to the circulation. Riser et al.125 and O’Neill et al.126
have recently shown that daily administration of so-dium PPi by peritoneal route was able to prevent the development of aorta calcification in two different animal models of CKD. The next step could be exploratory studies in PD patients.
VITAMIN K
MGP, which is synthesized by VSMC in the arterial media, is a vitamin K-dependent inhibitor of calcium and phosphate precipitation and crystal formation in the vessel wall. In addition, it suppresses the activity of bone morphogenetic proteins 2 and 4.128-130
Vitamin K deficiency affects the activation of MGP by gamma-carboxylation of glutamic acid residues. Undercarboxylated and/or nonphosphorylated MGP loses its inhibitory action on the development and progression of VC and associates with mortality risk in CKD patients.131,132 A recent study showed that
the majority of HD patients have a poor vitamin K status and low vitamin K intake compared with he-althy subjects.133 Another recent study showed that
vitamin K deficiency was associated with fractures and VC in general population.134 In patients with
CKD, low vitamin K intake may be related, at least in part, to the dietary regimen generally prescribed, which is restricted in green vegetables and other foods containing large amounts of potassium, but also vitamin K.
that inactive MGP concentration can be markedly de-creased by daily vitamin K supplementation during 6 weeks.136
To date, there are some trials registered on the
U.S. National Institutes of Health website (www.cli-nicaltrials.gov) that are related with vitamin K, CKD and VC. A search with the input keywords “Vitamin K” AND “Chronic kidney disease” resulted in three ongoing trials: “Warfarin and Coronary Calcification Project (WACC)”, “Vitamin K to Attenuate Coronary Artery Calcification in Hemodialysis Patients (iPACK HD)”, and “Vitamin K2 and Vessel Calcification in Chronic Kidney Disease Patients (CACSK2)”. Also, we should mention the European study VITAVASC. We expect to have, in the near future, more information on potentially beneficial effects of vitamin K supplementation on VC risk and outcomes in patients with CKD.
G
ENERAL CONCLUSIONVC is a complex disease process. It involves not only the local precipitation of calcium and phosphate in the vessel wall, but it also is a regulated cell mediated pro-cess, which is under the control of both inhibitory and stimulatory proteins and nonpeptidic factors. This nor-mal, physiological equilibrium is disturbed by CKD, favouring the initiation and progression of VC in pa-rallel with the progressive decline in kidney function.
Based on available experimental and clinical evidence, several drugs currently used for the treatment of CKD-MBD, including sevelamer and calcimimetics, appear to exert at least partially protective effects against the VC process associated with CKD. Drugs such as PPi and vitamin K may represent new avenues for a hopefully more efficient prevention and treatment of VC and its dramatic cardiovascular complications. However, the protective effects of these latter drugs have yet to be demonstrated by future randomized, controlled trials in patients with CKD.
A
CKNOWLEDGEMENTSFinancial support. We aknowledge funding by a grant from University of Picardie-Jules Verne. R.B. de Oliveira and H. Okazaki received postdoctoral grants from Picardy Regional Council/University of Picardy Jules Verne, Amiens, France. R.B. de Oliveira and A.E.M. Stinghen received postdoctoral scholar-ships from the National Council of Technological and Scientific Development (CNPq), Brasília, Brazil.
C
ONFLICT OF INTEREST STATEMENTT.B. Drüeke declares having received speaker ho-noraria, consulting fees and/or research funding from Abbott, Amgen, Fresenius, Genzyme, Kirin, Leo, Mitsubishi, Shire and Theraclion. Z.A. Massy declares having received speaker honoraria, con-sulting fees and/or research funding from Amgen, Fresenius, Genzyme and Shire. V. Jorgetti declares having received speaker honoraria, consulting fees and/or research funding from Amgen, Abbott and Genzyme.
R
EFERENCES1. Health Science Descriptors [cited 2012 december 07]; 1(1):[1 screen]. Available from: URL: http://decs.bvs.br/ 2. Temmar M, Liabeuf S, Renard C, Czernichow S, Esper NE,
Shahapuni I, et al. Pulse wave velocity and vascular cal-cification at different stages of chronic kidney disease. J Hypertens 2010;28:163-9. http://dx.doi.org/10.1097/ HJH.0b013e328331b81e PMid:19927012
3. Parfitt AM. Soft-tissue calcification in uremia. Arch In-tern Med 1969;124:544-56. http://dx.doi.org/10.1001/ archinte.124.5.544 http://dx.doi.org/10.1001/archin-te.1969.00300210026004 PMid:4899444
4. Steiner I, Laco J. Rokitansky on atherosclerosis. Cesk Patol 2008;44:23-4. PMid:18333331
5. Mönckeberg JG. Ueber die reine Mediaverkalkung der Extremi-taetenarterien und ihr Verhalten zur Arteriosklerose. Virchows Arch Pathol Anat 1903;171:141-67. http://dx.doi.org/10.1007/ BF01926946
6. London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003;18:1731-40. http://dx.doi.org/10.1093/ndt/ gfg414 PMid:12937218
7. Chiu YW, Adler SG, Budoff MJ, Takasu J, Ashai J, Mehrotra R. Coronary artery calcification and mortality in diabetic patients with proteinuria. Kidney Int 2010;77:1107-14. http://dx.doi. org/10.1038/ki.2010.70 PMid:20237457
8. Scott S, O'Sullivan M, Hafizi S, Shapiro LM, Bennett MR. Hu-man vascular smooth muscle cells from restenosis or in-stent ste-nosis sites demonstrate enhanced responses to p53: implications for brachytherapy and drug treatment for restenosis Circ Res 2002;90:398-404. http://dx.doi.org/10.1161/hh0402.105900 PMid:11884368
9. Littlewood TD, Bennett MR. Apoptotic cell death in atheros-clerosis. Curr Opin Lipidol 2003;14:469-75. http://dx.doi. org/10.1097/00041433-200310000-00007 PMid:14501585 10. Kockx MM. Apoptosis in the atherosclerotic plaque:
quanti-tative and qualiquanti-tative aspects. Arterioscler Thromb Vasc Biol 1998;18:1519-22. http://dx.doi.org/10.1161/01.ATV.18.10.1519 PMid:9763521
11. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calci-fication in vitro: evidence for initiation of vascular calcicalci-fication by apoptotic bodies. Circ Res 2000;87:1055-62. http://dx.doi. org/10.1161/01.RES.87.11.1055 PMid:11090552
13. Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arte-rioscler Thromb Vasc Biol 2006;26:1423-30. http://dx.doi. org/10.1161/01.ATV.0000220441.42041.20 PMid:16601233 14. Bea F, Blessing E, Bennett B, Levitz M, Wallace EP,
Rosenfeld ME. Simvastatin promotes atherosclerotic plaque stability in apoE-deficient mice independently of lipid lowering. Arterioscler Thromb Vasc Biol 2002;22:1832-7. http://dx.doi. org/10.1161/01.ATV.0000036081.01231.16 PMid:12426212 15. Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et
al. Smooth muscle cells give rise to osteochondrogenic pre-cursors and chondrocytes in calcifying arteries. Circ Res 2009;104:733-41. http://dx.doi.org/10.1161/CIRCRESA-HA.108.183053 PMid:19197075 PMCid:2716055
16. Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arte-rial calcification. Arterioscler Thromb Vasc Biol 2003;23:489-94. http://dx.doi.org/10.1161/01.ATV.0000059406.92165.31 PMid:12615658
17. Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, et al. Di-fferentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun 2012;3:875. http://dx.doi. org/10.1038/ncomms1867 PMid:22673902 PMCid:3538044 18. Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW,
Gabbiani G, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res 2013;112:17-22. http://dx.doi.org/10.1161/CIRCRE-SAHA.112.281048 PMid:23093573
19. Simionescu A, Philips K, Vyavahare N. Elastin-derived peptides and TGF-beta1 induce osteogenic responses in smooth muscle cells. Biochem Biophys Res Commun 2005;334:524-32. http:// dx.doi.org/10.1016/j.bbrc.2005.06.119 PMid:16005428 20. Hosaka N, Mizobuchi M, Ogata H, Kumata C, Kondo F,
Koiwa F, et al. Elastin degradation accelerates phosphate-in-duced mineralization of vascular smooth muscle cells. Calcif Tissue Int 2009;85:523-9. http://dx.doi.org/10.1007/s00223-009-9297-8 PMid:19806384
21. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997;386:78-81. http:// dx.doi.org/10.1038/386078a0 PMid:9052783
22. Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY, Tung E, et al. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evi-dence for osteopontin as an inducible inhibitor of vascular cal-cification in vivo. J Exp Med 2002;196:1047-55. http://dx.doi. org/10.1084/jem.20020911 PMid:12391016 PMCid:2194039 23. Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M,
Floege J, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ec-topic calcification. J Clin Invest 2003;112:357-66. http:// dx.doi.org/10.1172/JCI17202 http://dx.doi.org/10.1172/ JCI200317202 PMid:12897203 PMCid:166290
24. Collin-Osdoby P. Regulation of vascular calcification by os-teoclast regulatory factors RANKL and osteoprotegerin. Circ Res 2004;95:1046-57. http://dx.doi.org/10.1161/01. RES.0000149165.99974.12 PMid:15564564
25. Hruska KA, Mathew S, Saab G. Bone morphogenetic proteins in vascular calcification. Circ Res 2005;97:105-14. http://dx.doi. org/10.1161/01.RES.00000175571.53833.6c PMid:16037577 26. Balderman JA, Lee HY, Mahoney CE, Handy DE, White K,
Annis S, et al. Bone morphogenetic protein-2 decreases mi-croRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc 2012;1:e003905. PMid:23316327 PMCid:3540659
27. Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341-59. http://dx.doi.org/10.1146/annurev.physiol.69.040705.141729 PMid:17002592
28. Son BK, Akishita M, Iijima K, Eto M, Ouchi Y. Mecha-nism of pi-induced vascular calcification. J Atheroscler Thromb 2008;15:63-8. http://dx.doi.org/10.5551/jat.E545 PMid:18385534
29. Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, et al. Phosphate regulation of vascular smooth muscle cell calci-fication. Circ Res 2000;87:E10-7. http://dx.doi.org/10.1161/01. RES.87.7.e10 PMid:11009570
30. Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res 2006;98:905-12. http://dx.doi. org/10.1161/01.RES.0000216409.20863.e7 PMid:16527991 31. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W,
et al. Mutations in ENPP1 are associated with 'idiopathic' in-fantile arterial calcification. Nat Genet 2003;34:379-81. http:// dx.doi.org/10.1038/ng1221 PMid:12881724
32. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vi-tamin D metabolism. J Clin Invest 2004;113:561-8. http:// dx.doi.org/10.1172/JCI200419081 http://dx.doi.org/10.1172/ JCI19081 PMid:14966565 PMCid:338262
33. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45-51. http:// dx.doi.org/10.1038/36285 PMid:9363890
34. Hu MC, Shi M, Zhang J, Qui-ones H, Griffith C, Kuro-o M, et al. Klotho deficiency causes vascular calcification in chro-nic kidney disease. J Am Soc Nephrol 2011;22:124-36. http:// dx.doi.org/10.1681/ASN.2009121311 PMid:21115613 PM-Cid:3014041
35. Mirza MA, Hansen T, Johansson L, Ahlström H, Larsson A, Lind L, et al. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol Dial Trans-plant 2009;24:3125-31. http://dx.doi.org/10.1093/ndt/gfp205 PMid:19429932
36. Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis pa-tients. Nephrol Dial Transplant 2009;24:2792-6. http://dx.doi. org/10.1093/ndt/gfp191 PMid:19395730
37. Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, et al.; European Uremic Toxin (EUTox) Work Group. FGF23 is independently associated with vascular cal-cification but not bone mineral density in patients at various CKD stages. Osteoporos Int 2012;23:2017-25. http://dx.doi. org/10.1007/s00198-011-1838-0 PMid:22109743
38. Cancela AL, Santos RD, Titan SM, Goldenstein PT, Rochitte CE, Lemos PA, et al. Phosphorus is associated with coronary artery disease in patients with preserved renal function. PLoS One 2012;7:e36883. http://dx.doi.org/10.1371/journal.pone.0036883 PMid:22590632 PMCid:3349637
39. Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not as-sociated with and does not induce arterial calcification. Kidney Int 2013; doi: 10.1038/ki.2013.3. [Epub ahead of print] http:// dx.doi.org/10.1038/ki.2013.3
40. Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fi-broblast growth factor 23. Circulation 2012;125:2243-55. http://dx.doi.org/10.1161/CIRCULATIONAHA.111.053405 PMid:22492635
41. Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 2001;81:239-97. PMid:11152759
43. Ziegelstein RC, Xiong Y, He C, Hu Q. Expression of a functio-nal extracellular calcium-sensing receptor in human aortic endo-thelial cells. Biochem Biophys Res Commun 2006;342:153-63. http://dx.doi.org/10.1016/j.bbrc.2006.01.135 PMid:16472767 44. Alam MU, Kirton JP, Wilkinson FL, Towers E, Sinha S,
Rouhi M, et al. Calcification is associated with loss of functio-nal calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res 2009;81:260-8. http://dx.doi.org/10.1093/cvr/ cvn279 PMid:18852253
45. Ivanovski O, Nikolov IG, Joki N, Caudrillier A, Phan O, Mentaverri R, et al. The calcimimetic R-568 retards uremia--enhanced vascular calcification and atherosclerosis in apolipo-protein E deficient (apoE-/-) mice. Atherosclerosis 2009;205:55-62. http://dx.doi.org/10.1016/j.atherosclerosis.2008.10.043 PMid:19118829
46. Koleganova N, Piecha G, Ritz E, Schmitt CP, Gross ML. A calcimimetic (R-568), but not calcitriol, prevents vascular re-modeling in uremia. Kidney Int 2009;75:60-71. http://dx.doi. org/10.1038/ki.2008.490 PMid:19092814
47. Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 2012;17:311-21. http://dx.doi. org/10.1111/j.1440-1797.2012.01572.x PMid:22288610 48. Stinghen AE, Pecoits-Filho R. Vascular damage in kidney
disea-se: beyond hypertension. Int J Hypertens 2011;2011:232683. 49. Yamada S, Taniguchi M, Tokumoto M, Toyonaga J, Fujisaki K,
Suehiro T, et al. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidati-ve stress in the pathogenesis of vascular calcification in chro-nic kidney disease. J Bone Miner Res 2012;27:474-85. http:// dx.doi.org/10.1002/jbmr.539 PMid:21987400
50. Phan O, Ivanovski O, Nguyen-Khoa T, Mothu N, Angulo J, Westenfeld R, et al. Sevelamer prevents uremia-enhanced athe-rosclerosis progression in apolipoprotein E-deficient mice. Cir-culation 2005;112:2875-82. http://dx.doi.org/10.1161/CIR-CULATIONAHA105.541854 PMid:16267260
51. Guilgen G, Werneck ML, de Noronha L, Martins AP, Varela AM, Nakao LS, et al. Increased calcification and protein nitration in ar-teries of chronic kidney disease patients. Blood Purif 2011;32:296-302. http://dx.doi.org/10.1159/000330327 PMid:21876352 52. Kaysen GA. The microinflammatory state in uremia: causes
and potential consequences. J Am Soc Nephrol 2001;12:1549-57. PMid:11423586
53. Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH, et al. Mechanisms of dysregulation of low-den-sity lipoprotein receptor expression in vascular smooth mus-cle cells by inflammatory cytokines. Arteriosmus-cler Thromb Vasc Biol 2006;26:1150-5. http://dx.doi.org/10.1161/01. ATV.0000217957.93135.c2 PMid:16543490
54. Liu J, Ma KL, Gao M, Wang CX, Ni J, Zhang Y, et al. In-flammation disrupts the LDL receptor pathway and accelera-tes the progression of vascular calcification in ESRD patients. PLoS One 2012;7:e47217. http://dx.doi.org/10.1371/journal. pone.0047217 PMid:23115640 PMCid:3480367
55. Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, Mahnken AH, Böhm R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in pa-tients on dialysis: a cross-sectional study. Lancet 2003;361:827-33. http://dx.doi.org/10.1016/S0140-6736(03)12710-9
56. Price PA, Roublick AM, Williamson MK. Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int 2006;70:1577-83. http://dx.doi.org/10.1038/sj.ki.5001841 PMid:16955099 57. Price PA, Faus SA, Williamson MK. Bisphosphonates
alen-dronate and ibanalen-dronate inhibit artery calcification at do-ses comparable to those that inhibit bone resorption. Arte-rioscler Thromb Vasc Biol 2001;21:817-24. http://dx.doi. org/10.1161/01.ATV.21.5.817 PMid:11348880
58. Hamerman D. Osteoporosis and atherosclerosis: biological linkages and the emergence of dual-purpose therapies. QJM 2005;98:467-84. http://dx.doi.org/10.1093/qjmed/ hci077 PMid:15955801
59. Tankó LB, Christiansen C, Cox DA, Geiger MJ, McNabb MA, Cummings SR. Relationship between osteoporosis and cardio-vascular disease in postmenopausal women. J Bone Miner Res 2005;20:1912-20. http://dx.doi.org/10.1359/JBMR.050711 PMid:16234963
60. Barreto DV, Barreto FC, Carvalho AB, Cuppari L, Cendoroglo M, Draibe SA, et al. Coronary calcification in hemodialysis patients: the contribution of traditional and uremia-related risk factors. Kidney Int 2005;67:1576-82. http://dx.doi.org/10.1111/j.1523-1755.2005.00239.x PMid:15780114
61. London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, de Vernejoul MC. Arterial calcifications and bone histomorphome-try in end-stage renal disease. J Am Soc Nephrol 2004;15:1943-51. http://dx.doi.org/10.1097/01.ASN.0000129337.50739.48 PMid:15213285
62. Schulz E, Arfai K, Liu X, Sayre J, Gilsanz V. Aortic calcifica-tion and the risk of osteoporosis and fractures. J Clin Endocrin Metab 2004;89:4246-53. http://dx.doi.org/10.1210/jc.2003-030964 PMid:15356016
63. Adragao T, Herberth J, Monier-Faugere MC, Branscum AJ, Ferreira A, Frazao JM, et al. Low bone volume-a risk factor for coronary calcifications in hemodialysis patients. Clin J Am Soc Nephrol 2009;4:450-5. http://dx.doi.org/10.2215/ CJN.01870408 PMid:19158372 PMCid:2637600
64. London GM, Marchais SJ, Guérin AP, Boutouyrie P, Métivier F, de Vernejoul MC. Association of bone activity, calcium load, aortic stiffness, and calcifications in ESRD. J Am Soc Nephrol 2008;19:1827-35. http://dx.doi.org/10.1681/ASN.2007050622 PMid:18480316 PMCid:2518431
65. Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG. Associations between vascular calcification, arterial stiffness and bone mineral density in chronic kidney disease. Nephrol Dial Transplant 2008;23:586-93. http://dx.doi.org/10.1093/ ndt/gfm660 PMid:17933842
66. Carr JJ, Register TC, Hsu FC, Lohman K, Lenchik L, Bowden DW, et al. Calcified atherosclerotic plaque and bone mineral density in type 2 diabetes: the diabetes heart study. Bone 2008;42:43-52. http://dx.doi.org/10.1016/j.bone.2007.08.023 PMid:17964237 PMCid:2239236
67. Demer L, Tintut Y. The roles of lipid oxidation products and receptor activator of nuclear factor-κB signaling in atheroscle-rotic calcification. Circ Res 2011;108:1482-93. http://dx.doi. org/10.1161/CIRCRESAHA.110.234245 PMid:21659652 PMCid:3128471
68. Kulak CA, Borba VC, Jorgetti V, Dos Reis LM, Liu XS, Kimmel DB, et al. Skeletal microstructural abnormalities in postmenopausal women with chronic obstructive pulmona-ry disease. J Bone Miner Res 2010;25:1931-40. http://dx.doi. org/10.1002/jbmr.88 PMid:20564248
69. Bridgeman G, Brookes M. Blood supply to the human femoral diaphysis in youth and senescence. J Anat 1996;188:611-21. PMid:8763478 PMCid:1167489
70. Laroche M. Intraosseous circulation from physiology to disea-se. Joint Bone Spine 2002;69:262-9. http://dx.doi.org/10.1016/ S1297-319X(02)00391-3
71. Griffith JF, Yeung DK, Tsang PH, Choi KC, Kwok TC, Ahuja AT, et al. Compromised bone marrow perfusion in os-teoporosis. J Bone Miner Res 2008;23:1068-75. http://dx.doi. org/10.1359/jbmr.080233 PMid:18302498
73. Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin diffe-rentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A 2008;105:5266-70. http:// dx.doi.org/10.1073/pnas.0711119105 PMid:18362359 PM-Cid:2278202
74 Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Kurioka S, Yano S, et al. Serum osteocalcin level is associa-ted with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J Clin Endocrinol Metab 2009;94:45-9. http://dx.doi.org/10.1210/jc.2008-1455 PMid:18984661 75. Bacchetta J, Boutroy S, Guebre-Egziabher F, Juillard L, Drai J,
Pelletier S, et al. The relationship between adipokines, osteocal-cin and bone quality in chronic kidney disease. Nephrol Dial Transplant 2009;24:3120-5. http://dx.doi.org/10.1093/ndt/ gfp262 PMid:19515806
76. Elefteriou F, Takeda S, Ebihara K, Magre J, Patano N, Kim CA, et al. Serum leptin level is a regulator of bone mass. Proc Natl Acad Sci U S A 2004;101:3258-63. http://dx.doi.org/10.1073/ pnas.0308744101 PMid:14978271 PMCid:365777
77. Coen G, Ballanti P, Fischer MS, Balducci A, Calabria S, Colamarco L, et al. Serum leptin in dialysis renal osteodys-trophy. Am J Kidney Dis 2003;42:1036-42. http://dx.doi. org/10.1016/j.ajkd.2003.07.005 PMid:14582047
78. Eghbali-Fatourechi GZ, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N Engl J Med 2005;352:1959-66. http://dx.doi.org/10.1056/NEJ-Moa044264 PMid:15888696
79. Pirro M, Leli C, Fabbriciani G, Manfredelli MR, Callarelli L, Bagaglia F, et al. Association between circulating osteoprogeni-tor cell numbers and bone mineral density in postmenopausal osteoporosis. Osteoporos Int 2010;21:297-306. http://dx.doi. org/10.1007/s00198-009-0968-0 PMid:19484167
80. Pal SN, Rush C, Parr A, Van Campenhout A, Golledge J. Os-teocalcin positive mononuclear cells are associated with the severity of aortic calcification. Atherosclerosis 2010;210:88-93. http://dx.doi.org/10.1016/j.atherosclerosis.2009.11.001 PMid:20004897 PMCid:2862100
81. Fadini GP, Rattazzi M, Matsumoto T, Asahara T, Khosla S. Emerging role of circulating calcifying cells in the bone-vascular axis. Circulation 2012;125:2772-81. http://dx.doi.org/10.1161/ CIRCULATIONAHA.112.090860 PMid:22665885
82. London GM. Bone-vascular cross-talk. J Nephrol 2012;25:619-25. http://dx.doi.org/10.5301/jn.5000187 PMid:22711433 83. Cunha UGV, Valle EA, Melo RA. Peculiaridades do exame
fí-sico do idoso. Rev Med Minas Gerais 2011;21:181-5.
84. Kristanto W, van Ooijen PM, Groen JM, Vliegenthart R, Oudkerk M. Small calcified coronary atherosclerotic pla-que simulation model: minimal size and attenuation detec-table by 64-MDCT and MicroCT. Int J Cardiovasc Imaging 2012;28:843-53. http://dx.doi.org/10.1007/s10554-011-9869-3 PMid:215094http://dx.doi.org/10.1007/s10554-011-9869-30 PMCid:http://dx.doi.org/10.1007/s10554-011-9869-3http://dx.doi.org/10.1007/s10554-011-9869-360866
85. Kidney Disease: Improving Global Outcomes (KDIGO) CKD--MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kid-ney Int Suppl 2009;(113)S1:130.
86. Kauppila LI, Polak JF, Cupples LA, Hannan MT, Kiel DP, Wilson PW. New indices to classify location, severity and pro-gression of calcific lesions in the abdominal aorta: a 25-year follow-up study. Atherosclerosis 1997;132:245-50. http:// dx.doi.org/10.1016/S0021-9150(97)00106-8
87. Adragao T, Pires A, Lucas C, Birne R, Magalhaes L, Gonçalves M, et al. A simple vascular calcification score pre-dicts cardiovascular risk in haemodialysis patients. Nephrol Dial Transplant 2004;19:1480-8. http://dx.doi.org/10.1093/ndt/gfh217 PMid:15034154
88. Tomiyama C, Higa A, Dalboni MA, Cendoroglo M, Draibe SA, Cuppari L, et al. The impact of traditional and non-traditional risk factors on coronary calcification in pre-dialysis patients. Nephrol Dial Transplant 2006;21:2464-71. http://dx.doi. org/10.1093/ndt/gfl291 PMid:16735378
89. Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 2005;16:520-8. http://dx.doi.org/10.1681/ASN.2004070602 PMid:15615819
90. Floege J, Kim J, Ireland E, Chazot C, Drueke T, de Francisco A, et al.; ARO Investigators. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis popula-tion. Nephrol Dial Transplant 2011;26:1948-55. http://dx.doi. org/10.1093/ndt/gfq219 PMid:20466670 PMCid:3107766 91. Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA,
et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007;71:31-8. http://dx.doi.org/10.1038/sj.ki.5002009 PMid:17091124
92. Oliveira RB, Cancela AL, Graciolli FG, Dos Reis LM, Draibe SA, Cuppari L, et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD--MBD therapy? Clin J Am Soc Nephrol 2010;5:286-91. http:// dx.doi.org/10.2215/CJN.05420709 PMid:19965540 PM-Cid:2827593
93. Ritz E, Hahn K, Ketteler M, Kuhlmann MK, Mann J. Phosphate additives in food--a health risk. Dtsch Arztebl Int 2012;109:49-55. PMid:22334826 PMCid:3278747
94. Bammens B, Evenepoel P, Verbeke K, Vanrenterghem Y. Re-moval of middle molecules and protein-bound solutes by peritoneal dialysis and relation with uremic symptoms. Kid-ney Int 2003;64:2238-43. http://dx.doi.org/10.1046/j.1523-1755.2003.00310.x PMid:14633148
95. Ayus JC, Mizani MR, Achinger SG, Thadhani R, Go AS, Lee S. Effects of short daily versus conventional hemodialysis on left ventricular hypertrophy and inflammatory markers: a prospec-tive, controlled study. J Am Soc Nephrol 2005;16:2778-88. http://dx.doi.org/10.1681/ASN.2005040392 PMid:16033855 96. Pierratos A, Ouwendyk M, Francoeur R, Vas S, Raj DS,
Ecclestone AM, et al. Nocturnal hemodialysis: three-year expe-rience. J Am Soc Nephrol 1998;9:859-68. PMid:9596084 97. Feliciani A, Riva MA, Zerbi S, Ruggiero P, Plati AR, Cozzi G,
et al. New strategies in haemodiafiltration (HDF): prospective comparative analysis between on-line mixed HDF and mid-di-lution HDF. Nephrol Dial Transplant 2007;22:1672-9. http:// dx.doi.org/10.1093/ndt/gfm023 PMid:17347283
98. Evenepoel P, Bammens B, Verbeke K, Vanrenterghem Y. Su-perior dialytic clearance of beta(2)-microglobulin and p-cresol by high-flux hemodialysis as compared to peritoneal dialy-sis. Kidney Int 2006;70:794-9. http://dx.doi.org/10.1038/ sj.ki.5001640 PMid:16820785
99. Messa P, Gropuzzo M, Cleva M, Boscutti G, Mioni G, Cruciatti A, et al. Behaviour of phosphate removal with diffe-rent dialysis schedules. Nephrol Dial Transplant 1998;13:43-8. http://dx.doi.org/10.1093/ndt/13.suppl_6.43 PMid:9719204 100. Kuhlmann MK. Phosphate elimination in modalities of
hemo-dialysis and peritoneal hemo-dialysis. Blood Purif 2010;29:137-44. http://dx.doi.org/10.1159/000245640 PMid:20093819 101. Chertow GM, Burke SK, Raggi P.; Treat to Goal Working
Group. Sevelamer attenuates the progression of corona-ry and aortic calcification in hemodialysis patients. Kid-ney Int 2002;62:245-52. http://dx.doi.org/10.1046/j.1523-1755.2002.00434.x PMid:12081584
103. Russo D, Miranda I, Ruocco C, Battaglia Y, Buonanno E, Manzi S, et al. The progression of coronary artery calcifica-tion in predialysis patients on calcium carbonate or sevela-mer. Kidney Int 2007;72:1255-61. http://dx.doi.org/10.1038/ sj.ki.5002518 PMid:17805238
104. Qunibi W, Moustafa M, Muenz LR, He DY, Kessler PD, Diaz-Buxo JA, et al. A 1-year randomized trial of calcium ace-tate versus sevelamer on progression of coronary artery calcifi-cation in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008;51:952-65. http://dx.doi.org/10.1053/j. ajkd.2008.02.298 PMid:18423809
105. Barreto DV, Barreto Fde C, de Carvalho AB, Cuppari L, Draibe SA, Dalboni MA, et al. Phosphate binder impact on bone remodeling and coronary calcification--results from the BRiC study. Nephron Clin Pract 2008;110c:273-83. http:// dx.doi.org/10.1159/000170783 PMid:19001830
106. Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, et al. Effects of sevelamer on HbA1c, inflammation, and ad-vanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol 2012;7:934-42. http://dx.doi.org/10.2215/ CJN.12891211 PMid:22461535
107. Nikolov IG, Joki N, Maizel J, Lacour B, Drüeke TB, Massy ZA. Pleiotropic effects of the non-calcium phosphate binder sevelamer. Kidney Int Suppl 2006;(105):S16-23. http:// dx.doi.org/10.1038/sj.ki.5001994 PMid:17136111
108. Spiegel DM, Brady K. Calcium balance in normal indivi-duals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int 2012;81:1116-22. http://dx.doi. org/10.1038/ki.2011.490 PMid:22297674 PMCid:3352985 109. Hill KM, Martin BR, Wastney ME, McCabe GP, Moe SM,
Weaver CM, et al. Oral calcium carbonate affects calcium but not phosphorus balance in stage 3-4 chronic kidney disease. Kidney Int 2012; doi: 10.1038/ki.2012.403. [Epub ahead of print] http://dx.doi.org/10.1038/ki.2012.403
110. Drüeke TB, Massy ZA. Phosphate binders in CKD: bad news or good news? J Am Soc Nephrol. 2012;23:1277-80. http:// dx.doi.org/10.1681/ASN.2012060569 PMid:22797178 111. Joki N, Nikolov IG, Caudrillier A, Mentaverri R, Massy ZA,
Drüeke TB. Effects of calcimimetic on vascular calcification and atherosclerosis in uremic mice. Bone 2009;45:S30-4. http:// dx.doi.org/10.1016/j.bone.2009.03.653 PMid:19303957 112. Koleganova N, Piecha G, Ritz E, Schmitt CP, Gross ML. A
calcimimetic (R-568), but not calcitriol, prevents vascular re-modeling in uremia. Kidney Int 2009;75:60-71. http://dx.doi. org/10.1038/ki.2008.490 PMid:19092814
113. Torres PA, De Broe M. Calcium-sensing receptor, calcimi-metics, and cardiovascular calcifications in chronic kidney disease. Kidney Int 2012;82:19-25. http://dx.doi.org/10.1038/ ki.2012.69 PMid:22437409
114. Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, et al.; ADVANCE Study Group. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 2011;26:1327-39. http://dx.doi.org/10.1093/ndt/gfq725 PMid:21148030 115. Mendoza FJ, Martinez-Moreno J, Almaden Y, Rodriguez-Ortiz
ME, Lopez I, Estepa JC, et al. Effect of calcium and the calci-mimetic AMG 641 on matrix-Gla protein in vascular smooth muscle cells. Calcif Tissue Int 2011;88:169-78. http://dx.doi. org/10.1007/s00223-010-9442-4 PMid:21161195
116. Goodman WG, Ramirez JA, Belin TR, Chon Y, Gales B, Segre GV, et al. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol the-rapy. Kidney Int 1994;46:1160-6. http://dx.doi.org/10.1038/ ki.1994.380 PMid:7861712
117. Quarles LD, Yohay DA, Carroll BA, Spritzer CE, Minda SA, Bartholomay D, et al. Prospective trial of pulse oral versus intravenous calcitriol treatment of hyperparathyroidism in ESRD. Kidney Int 1994;45:1710-21. http://dx.doi.org/10.1038/ ki.1994.223 PMid:7933819
118. Drüeke TB. Which vitamin D derivative to prescribe for re-nal patients. Curr Opin Nephrol Hypertens 2005;14:343-9. http://dx.doi.org/10.1097/01.mnh.0000172720.34229.39 PMid:15931002
119. Cardús A, Panizo S, Parisi E, Fernandez E, Valdivielso JM. Di-fferential effects of vitamin D analogs on vascular calcification. J Bone Miner Res 2007;22:860-6. http://dx.doi.org/10.1359/ jbmr.070305 PMid:17352647
120. Mizobuchi M, Finch JL, Martin DR, Slatopolsky E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int 2007;72:709-15. http:// dx.doi.org/10.1038/sj.ki.5002406 PMid:17597697
121. Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, et al. Vitamin D receptor agonists increase klotho and osteopon-tin while decreasing aortic calcification in mice with chronic kid-ney disease fed a high phosphate diet. Kidkid-ney Int 2012;82:1261-70. http://dx.doi.org/10.1038/ki.2012.322 PMid:22932118 122. Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI,
O'Neill WC. Phosphate-induced vascular calcification: role of pyro-phosphate and osteopontin. J Am Soc Nephrol 2004;15:1392-401. http://dx.doi.org/10.1097/01.ASN.0000128955.83129.9C PMid:15153550
123. Schibler D, Russell RG, Fleisch H. Inhibition by pyrophospha-te and polyphosphapyrophospha-te of aortic calcification induced by vitamin D3 in rats. Clin Sci 1968;35:363-72. PMid:4305530
124. Jung A, Russel RG, Bisaz S, Morgan DB, Fleisch H. Fate of in-travenously injected pyrophosphate-32P in dogs. Am J Physiol 1970;218:1757-64. PMid:4317203
125. Riser BL, Barreto FC, Rezg R, Valaitis PW, Cook CS, White JA, et al. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol Dial Transplant 2011;26:3349-57. http://dx.doi.org/10.1093/ ndt/gfr039 PMid:21398365
126. O'Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL. Treatment with pyrophosphate inhibits uremic vas-cular calcification. Kidney Int 2011;79:512-7. http://dx.doi. org/10.1038/ki.2010.461 PMid:21124302 PMCid:3183997 127. Lomashvili KA, Khawandi W, O'Neill WC. Reduced
plas-ma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol 2005;16:2495-500. http://dx.doi.org/10.1681/ ASN.2004080694 PMid:15958726
128. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997;386:78-81. http:// dx.doi.org/10.1038/386078a0 PMid:9052783
129. Wajih N, Borras T, Xue W, Hutson SM, Wallin R. Processing and transport of matrix gamma-carboxyglutamic acid protein and bone morphogenetic protein-2 in cultured human vascular smooth muscle cells: evidence for an uptake mechanism for serum fetuin. J Biol Chem 2004;279:43052-60. http://dx.doi. org/10.1074/jbc.M407180200 PMid:15280384
130. Murshed M, Schinke T, McKee MD, Karsenty G. Extrace-llular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol 2004;165:625-30. http://dx.doi.org/10.1083/jcb.200402046 PMid:15184399 PMCid:2172384
132. Schlieper G, Westenfeld R, Krüger T, Cranenburg EC, Magdeleyns EJ, Brandenburg VM, et al. Circulating nonphos-phorylated carboxylated matrix gla protein predicts sur-vival in ESRD. J Am Soc Nephrol 2011;22:387-95. http:// dx.doi.org/10.1681/ASN.2010040339 PMid:21289218 PM-Cid:3029911
133. Cranenburg EC, Schurgers LJ, Uiterwijk HH, Beulens JW, Dalmeijer GW, Westerhuis R, et al. Vitamin K intake and status are low in hemodialysis patients. Kidney Int 2012;82:605-10. http://dx.doi.org/10.1038/ki.2012.191 PMid:22648294 134. Fusaro M, Noale M, Viola V, Galli F, Tripepi G, Vajente N,
Plebani M, et al.; VItamin K Italian (VIKI) Dialysis Study In-vestigators. Vitamin K, vertebral fractures, vascular calcifica-tions, and mortality: VItamin K Italian (VIKI) dialysis study. J Bone Miner Res 2012;27:2271-8. http://dx.doi.org/10.1002/ jbmr.1677 PMid:22692665
135. Krueger T, Westenfeld R, Ketteler M, Schurgers LJ, Floege J. Vitamin K deficiency in CKD patients: a modifiable risk factor for vascular calcification? Kidney Int 2009;76:18-22. http:// dx.doi.org/10.1038/ki.2009.126 PMid:19387474