Primeira submissão em 23/01/12 Última submissão em 16/05/12 Aceito para publicação em 27/05/12 Publicado em 20/08/12
Sirenomelia associada a defeitos congênitos raros:
relato de três casos
Sirenomelia associated with rare congenital defects: report of three cases
Maria Angélica F. D. Lima1; Heloisa Novaes Machado2; Dione Correa de Araújo Dock3; Maria Auxiliadora Monteiro Villar4; Juan Clinton Llerena Junior5
Sirenomelia é um defeito congênito muito raro do campo primário do desenvolvimento, deinido pela substituição dos membros inferiores, normalmente pareados por um único membro mediano. Geralmente, associa-se a graus variados de anomalias gênito-urinárias. Relatamos três casos necropsiados dessa entidade, incluindo estudo radiológico do membro inferior único, associados a agenesia renal bilateral, de ureteres e da bexiga, atresia retal, ânus imperfurado, testículos abdominais e ausência de genitália externa, além de outros defeitos congênitos infrequentemente observados, que somente puderam ter seus diagnósticos irmados por meio da necropsia.
resumo
unitermosSirenomelia
Malformação
Patologia
abstract
Sirenomelia, an extremely rare congenital defect, is defined as a limb abnormality in which the normally paired lower limbs are replaced by a single midline limb. It is commonly associated with varied genitourinary anomalies. We report three cases of sirenomelia including x-ray documentation of the lower limb. Other associated aspects, whose diagnoses were established exclusively through autopsy, included bilateral renal, ureteral and bladder agenesis, rectal atresia, imperforate anus, intra-abdominal testis, absence of external genitalia and other rare congenital abnormalities.
key words
Sirenomelia
Malformation
Pathology
1. Mestra em Ciências; médica do Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/Fundação Oswaldo Cruz (FIOCRUZ). 2. Doutora em Ciências; médica do Departamento de Anatomia Patológica do Instituto Nacional da Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/FIOCRUZ.
3. Especialista em Anatomia Patológica; médica do Departamento de Anatomia Patológica do Instituto Nacional da Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/FIOCRUZ. 4. Doutora em Ciências; médica do Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/FIOCRUZ; pediatra do Departamento de Genética Médica do Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/FIOCRUZ.
Introdução
Sirenomelia é um defeito congênito raro com prevalên-cia estimada de 0,98:100.000 nascimentos(7, 13). É deinida como defeito caracterizado pela substituição dos membros inferiores, normalmente pareados por um único membro mediano(8, 13). Ocorre em torno da terceira semana de vida intrauterina(12) e associa-se a anomalias gênito-urinárias, incluindo agenesia renal, de ureteres e da bexiga, ânus im-perfurado e ausência da genitália externa(15), consideradas malformações graves, levando à morte no período perinatal. Aproximadamente, 50% dos casos estão associados a outros defeitos congênitos não relacionados com o defeito básico da sirenomelia, como o do tubo neural e os cardíacos e anomalias do trato gastrointestinal superior(1, 12-14).
Relatamos três casos de sirenomelia associados a outras malformações raramente observadas, diagnosticados no Insti-tuto Nacional de Saúde da Mulher, da Criança e do Adolescen-te Fernandes Figueira/Fundação Oswaldo Cruz (FIOCRUZ).
Relato de casos
Caso 1
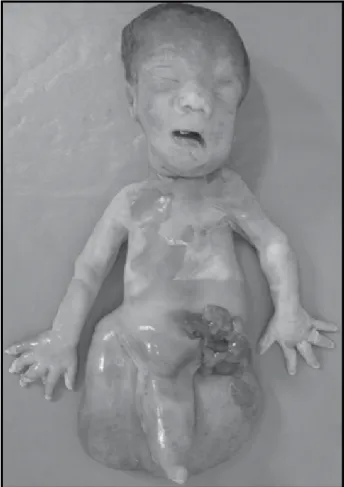
Primigesta, 30 anos de idade, 23 semanas de gestação, com ultrassonograia mostrando adramnia, feto com fêmures curtos e higroma cístico. Casal não consanguíneo com história familiar de defeito de tubo neural. Gestante com hipertensão arterial crônica, em uso de inibidor de enzima conversora de angiotensina (ECA) no primeiro trimestre. Res-sonância nuclear magnética (RM) revelou mielomeningocele e agenesia renal bilateral, não sendo identiicados os mem-bros inferiores. Interrupção da gestação na 29ª semana, após autorização judicial. O natimorto pesou 1.120 g. À necropsia
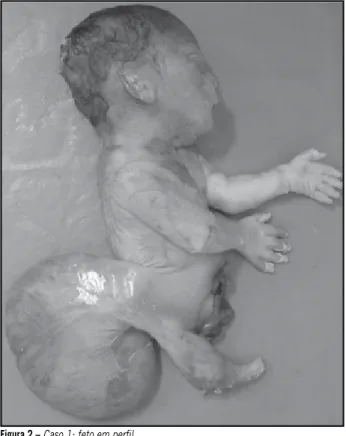
(Figuras 1 e 2), feto macerado com membro inferior único e
rudimentar, ausência de genitália externa, mielomeningocele lombossacra, gastrosquise expondo alças intestinais, ânus imperfurado, agenesia renal bilateral, comunicação inter-ventricular e artéria umbilical única. O exame radiológico do feto mostrou fêmur único, tíbia e fíbula ausentes (sirenomelia tipo VII, classiicação de Stocker e Heifetz[15]).
Caso 2
Paciente de 28 anos, com diagnóstico ultrassonográi-co de fêmur e artéria umbilical úniultrassonográi-cos na 12ª semana de
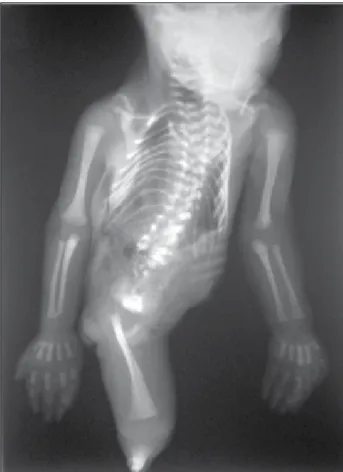
gestação. História familiar negativa para malformações. Sangramento vaginal de pequena monta na 15ª semana. O estudo fetal detalhado por RM mostrou membro inferior único. Parto cesáreo na 30ª semana. O recém-nascido pe-sou 1. 010 g, recebeu Apgar 1/4/7 e faleceu após 6 horas. A necropsia (Figura 3) mostrou, externamente, membro inferior único e rudimentar, ausência de genitália externa e ânus imperfurado. Internamente (Figuras 4 e 5), atresia do esôfago com fístula distal para traqueia, agenesia dos rins, dos ureteres e da bexiga, atresia retal e testículos abdomi-nais. Cordão umbilical curto com única artéria. Ao exame radiológico (Figura 6), fêmur único e tíbia rudimentar única, classiicada como tipo VI de Stocker e Heifetz(15).
Caso 3
Gestante de 23 anos de idade, referida na 25ª semana com diagnóstico ultrassonográico de adramnia, feto com membros inferiores fusionados, agenesia dos rins, dos ure-teres e da bexiga. Terceira gestação de casal não consanguí-neo, sem história familiar de malformações. Sangramento
Figura 2 – Caso 1: feto em perfil
Figura 3 – Caso 2: recém-nascido com membro inferior único. Agenesia de genitália externa
Figura 4 – Caso 2: atresia do esôfago com fístula distal para traqueia
vaginal na décima semana após exposição a misoprostol. Parto cesáreo a termo, recém-nascido com 2. 010 g e óbito com 20 horas. A necropsia (Figura 7) evidenciou fenótipo de sirenomelia. Internamente, os achados ultrassonográi-cos de agenesia dos rins, dos ureteres e da bexiga foram conirmados, acrescidos de lobulação pulmonar invertida
(Figura 8), comunicação interatrial, atresia do reto com
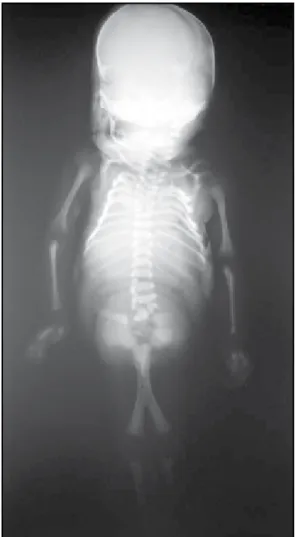
imperfuração anal e artéria umbilical única. Genitália in-terna representada por testículos abdominais. O exame radiológico post mortem (Figura 9) mostrou fusão parcial dos fêmures, duas tíbias e uma fíbula, caracterizando o tipo IV de Stocker e Heifetz(15).
Discussão
Sirenomelia é considerada um defeito do campo pri-mário do desenvolvimento que afeta múltiplas estruturas primordiais da linha média(11), podendo ter etiologia variada. Do ponto de vista etiopatogênico, foram aventadas duas hipóteses para explicar o desenvolvimento anormal dos membros inferiores(5, 11). A primeira relaciona-se com erro primário da blastogênese e a segunda, com evento vascular
Figura 6 – Caso 2: radiografia mostrando fêmur único e tíbia rudimentar tipo VI de Sotcker e Heifetz(15)
Figura 7 – Caso 3: recém-nascido a termo com membros inferiores fusionados e agenesia de genitália externa
Figura 9 – Caso 3: radiografia mostrando fusão parcial dos fêmures, duas tíbias e uma fíbula tipo IV de Stocker e Heifetz(15)
com base no desenvolvimento anormal dos vasos umbi-licais, resultando em aporte sanguíneo insuiciente para suprir a porção caudal do embrião(6). Garrido-Allepuz et al.(5) defendem que as duas teorias não se excluem mutuamente, embora a hipótese do defeito na blastogênese explique ambas as hipóteses. Esses mesmos autores(5) referem uma possível base genética para explicar o fenótipo de sireno-melia em modelos experimentais, nos quais a deiciência da enzima Cyp26a1, que degrada o ácido retinoico, e a redução da proteína morfogenética do osso (pmo) afetariam o desenvolvimento normal da região caudal do embrião de camundongos. No entanto, até o momento, não há relatos em fetos humanos da associação da exposição ao ácido retinoico e à sirenomelia(5).
Em 1987, Stocker e Heifetz(15) classiicaram os casos de sirenomelia, de acordo com a fusão e/ou agenesia dos ossos dos membros inferiores, em sete tipos. No tipo I, não há comprometimento numérico dos ossos, apenas fusão de partes moles, enquanto no tipo VII, está presente um fêmur
único, não sendo observadas tíbia e fíbula. A classiicação completa caracteriza-se da seguinte forma:
• tipo I – fêmures, tíbias e fíbulas em número habitual; • tipo II – fíbula única fusionada;
• tipo III – ausência de fíbula;
• tipo IV – fêmures parcialmente fusionados e fíbula única;
• tipo V – fêmures parcialmente fusionados e fíbula ausente;
• tipo VI: fêmur e tíbia únicos;
• tipo VII: fêmur único, tíbia e fíbula ausentes. Na casuística ora apresentada, utilizamos essa clas-siicação. No primeiro caso, embora defeitos cardíacos tenham sido relatados em associação à sirenomelia(3, 12), a exposição do feto aos inibidores da ECA poderia justiicar a ocorrência da cardiopatia(2). Os defeitos de fechamento da porção inferior do tubo neural podem estar patogeni-camente associados, uma vez que a coluna lombossacra está comprometida na sirenomelia(13). Já a associação com gastrosquise é rara, tendo sido relatada por Mastroiacovo
et al.(10). Feldkamp et al.(4) sugeriram que a gastrosquise seria um defeito primário e não um evento disruptivo, ocorrendo entre a terceira e a quinta semanas do desenvolvimento embrionário, mesmo período em que a sirenomelia supos-tamente ocorreria.
No segundo caso, a atresia do esôfago, outra associa-ção rara, foi observada previamente por Duncan et al.(3) e é também exemplo de defeito do campo primário de desenvolvimento(9).
No terceiro caso, a alteração da lobulação pulmonar pode ser considerada parte do fenótipo de situs inversus,
igualmente ao outro defeito da blastogênese(9).
Esses três casos representam exemplos de associação de defeitos do campo primário de desenvolvimento, raramente observados com sirenomelia, e somente puderam ter seus diagnósticos irmados por meio da necropsia perinatal.
Referências
Endereço para correspondência
Heloisa Novaes
Av. Atlântica, 3772/1001 – Copacabana CEP: 22070-001 – Rio de Janeiro-RJ
sejam rotineiramente diagnosticadas pela ultrassonograia, outras malformações associadas podem não ser detectadas. Assim, torna-se oportuno para melhor delinear o espectro
de anomalias associadas à sirenomelia e, consequentemen-te, a investigação etiopatogênica desse defeito tão raro, a realização sistemática de necropsia dos afetados.
1. CHEN, C. P. et al. Sirenomelia with uncommon osseous fusion associated with a neural tube defect. Pediatr
Radiol, v. 28, n. 5, p. 293-6, 1998.
2. COOPER, W. O. et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med, v. 354, n. 23, p. 2443-51, 2006.
3. DUNCAN, P. A.; SHAPIRO, L. R. Interrelationships of the hemifacial-microsomia-VATER, VATER and sirenomelia phenotypes. Am J Med Genet, v. 47, n. 1, p. 75-84, 1993. 4. FELDKAMP, M. L.; CAREY, J. C.; SADLER, T. W.
Development of gastroschisis: review of hypotheses, a novel hypothesis, and implications for research. Am J
Med Genet, v. 143A, n. 7, p. 639-52, 2007.
5. GARRIDO-ALLEPUZ, C. et al. A clinical and experimental overview of sirenomelia: insight into the mechanisms of congenital limb malformations. Dis Models Mech, v. 4, n. 3, p. 289-99, 2011.
6. HENTSCHEL, J. et al. Caudal regression sequence: vascular origin? J Perinatol, v. 26, n. 7, p. 445-7, 2006.
7. KÄLLEN, B. et al. The cyclops and the mermaid: an
epidemiological study of two types of rare malformation.
J Med Genet, v. 29, n. 1, p. 30-5, 1992.
8. LADURE, H. et al. Diagnostic antenatal d’une sirénomélie.
J Gynecol Obstet Biol Reprod, v. 35, n. 2, p. 181-5,
2006.
9. MARTINEZ-FRIAS, M. L.; FRIAS, J. L. Primary
developmental field III: clinical and epidemiological study of blastogenetic anomalies and their relationship to different MCD patterns. Am J Med Genet, v. 70, n. 1, p. 11-5, 1997.
10. MASTROIACOVO, P. et al. Gastroschisis and associated defects: an international study. Am J Med Genet, v. 143A, n. 7, p. 660-71, 2007.
11. OPITZ, J. M. et al. Defects of blastogenesis. Am J Med
Genet, v. 115, n. 4, p. 269-86, 2002.
12. OPITZ, J. M.; WILSON, G. N.; GILBERT-BARNESS, E. Analysis of developmental pathology. In: Potter’s
Pathology of the fetus, infant and child. 2. ed.
Philadelphia, PA: Mosby-Elsevier, 2007. Cap. 3, p. 97-133.
13. ORIOLI, I. M. et al. Sirenomelia: an epidemiologic study in a large dataset from the International Clearinghouse of Birth Defects Surveillance and Research, and literature
review. Am J Med Genet Part C Semin Med Genet,
v. 157, n. 4, p. 358-78, 2011.
14. RODRIGUEZ, J. I.; PALACIOS, J. Craniorachischisis totalis and sirenomelia. Am J Med Genet, v. 43, n. 4, p. 732-6, 1992.