UNIVERSIDADE FEDERAL DE OURO PRETO
NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS
Tratamento com HP
β
CD/Ang-(1-7) Reverte Distúrbios
da Síndrome Metabólica Através da Redução do
Processo Inflamatório do Tecido Adiposo, da Melhora
da via da Insulina e do Aumento da Termogênese
Adaptativa
Uberdan Guilherme Mendes de Castro
II
Uberdan Guilherme Mendes de Castro
Tratamento com HP
β
CD/Ang-(1-7) Reverte Distúrbios
da Síndrome Metabólica Através da Redução do
Processo Inflamatório do Tecido Adiposo, da Melhora
da via da Insulina e do Aumento da Termogênese
Adaptativa
Orientadora: Andréia Carvalho Alzamora
V
Uberdan Guilherme Mendes de Castro
Tese de Doutoarado desenvolvida no Laboratório de
Hipertensão da Universidade Federal de Ouro Preto com
VI Deus é a Lei e o legislador do Universo”
“Você não pode provar uma definição. O que você pode fazer é mostrar que ela faz sentido”
VIII AGRADECIMENTOS
Agradeço a todos que colaboraram para a realização deste trabalho, em especial:
À Profª. Drª. Andréia Carvalho Alzamora, minha orientadora, por ter me descoberto em sua turma de fisiologia e me dado a oportunidade de integrar sua equipe há 12 anos. Reconheço que estar sobre sua orientação foi uma oportunidade incrível que tentei aproveitar ao máximo, respeitando minhas limitações. As lições que aprendi sob a orientação da Prof.ª Andreia Alzamora vão além do meio acadêmico, visto que me trouxeram diversas competências, habilidades e conhecimentos que levarei para qualquer ambiente que eu estiver e em vários momentos de minha vida. Foram anos de muito aprendizado e conselhos, que levarei sempre comigo. Por todo ensinamento, convivência e paciência. O meu muito obrigado.
Ao Prof. Dr. Marcelo Eustáquio Silva, que sempre acreditou em minhas ideias e colaborou com a parte inicial de meu trabalho de Doutorado. O Prof. Marcelo ajudou-me em mais de cinco experimentos consecutivos na tentativa de encontrar um modelo dietético que mais se aproximasse da SM humana. E isso tudo ocorreu após a conclusão do mestrado, quando ainda não era aluno de Doutorado. Foi mais de um ano de repetições de experimentos, onde o Prof. Marcelo Eustáquio por vezes gastou de seu próprio orçamento para comprar materiais das dietas testadas e ainda contribuiu com o fornecimento dos animais deste estudo e dos estudos de padronização das dietas. Logo, a definição da dieta de síndrome metabólica e este estudo, não seriam possíveis sem esse grande conhecedor de modelos experimentais dietéticos e principalmente, desde grande amigo de anos.
À Prof.ª Renata Guerra que abriu as portas de seu laboratório (Laboratório de Bioquímica e Biologia Molecular) e por vezes me ensinou pessoalmente técnicas de biologia molecular. Além disso, a Prof.ª Renata Guerra ainda forneceu materiais e emprestou seus equipamentos para que grande parte desse trabalho fosse realizado. Sem a contribuição da Prof.ª Renata, também seria impossível a conclusão do presente trabalho.
IX À Karina Barbosa Queiroz que dedicou grande parte de seu tempo de doutoranda para me ensinar técnicas de biologia molecular e esclarecer dúvidas. Ajuda também imprescindível na conclusão do presente trabalho.
À Prof.ª. Drª. Maria José Campagnole-Santos e ao Prof. Dr. Robson Santos do Laboratório de Hipertensão do departamento de Fisiologia e Biofísica da Universidade Federal de Minas Gerais, agradeço por terem sempre acreditado no trabalho que desenvolvi e por fornecer recursos financeiros e materiais indispensáveis para o desenvolvimento deste trabalho.
À Maria Andrea, à Aline, à Lívia, à Denise, à Claudiane e ao Luiz pelo companheirismo de laboratório, pela boa convivência e amizade e por terem participado do mutirão de experimentos realizados neste trabalho que tardaram até quase meia noite em vários dias.
A todos demais colegas que integraram o Laboratório de Hipertensão da UFOP durante todos esses anos.
A Todos Professores do Núcleo de Pesquisas em Ciências Biológicas (NUPEB/UFOP), que com o trabalho de cada um em sua área, desenvolveram e mantém o respeito e prestígio desse programa de pós-graduação.
Aos financiadores deste trabalho: REDE TOXIFAR-INCT-FAPEMIG-CNPQ, CAPES, PROPP-NUPEB-UFOP.
À Universidade Federal de Ouro Preto e ao ensino público brasileiro que me deram oportunidade de concluir este trabalho.
X À minha irmã Narjara Suzana pelo companheirismo, amizade, amor de irmã e convivência. A família unida fortalece cada um de seus membros individualmente; Minha irmã é um dos pilares de minha família e a ela sou muito agradecido.
“São etapas difíceis que passamos em vários momentos da vida. São momentos de grande escuridão, porém também importantes, porque são as adversidades que despertam em nós, a grande força interior capaz de nos fazer superar os desafios da vida e também são nesses momentos percebemos quem realmente está ao nosso lado. O amor de meus pais Clodoveu Penha de Castro e Marly Mendes de Castro em todos momentos da vida sempre foi parte de minha força interior para superar esses desafios. É um amor que sempre me foi dado e ainda é dado e que a ciência jamais poderá explicar. Meus pais me deram a vida e partir daí tudo que esteve ao alcance deles, principalmente esse grande amor. Incentivaram-me a estudar, a ser um homem honesto e um ser humano de bem. Se depois de mais de 30 anos alcanço este nível acadêmico e de ser humano é porque meus pais em todo esse tempo dedicaram sua vida a mim. Não seria possível chegar onde estou e ser a pessoa que sou se não fosse os verdadeiros Doutores de minha vida Marly Mendes de Castro e Clodoveu Penha de Castro. A eles agradeço pelo carinho, compreensão, apoio, amizade e amor incondicional.
XI RESUMO
O objetivo do presente estudo foi avaliar o potencial terapêutico da formulação oral de Ang-(1-7), a HPβCD/Ang-(1-7), sobre o processo inflamatório do tecido adiposo branco visceral (TAV) e tecido adiposo marrom (TAM), sobre as vias de sinalização intracelular da insulina e sobre a termogênese adaptativa, na reversão de distúrbios metabólicos da síndrome metabólica (SM), induzidos pela dieta hiperlipídica. Ratos recém-desmamados com 4 semanas de idade, foram submetidas à dieta controle AIN-93 (dieta CT) ou à dieta hiperlipídica (dieta SM) por 7 semanas para indução da SM. Após este período, os grupos foram subdivididos em ratos tratados com HPβCD/Ang-(1-7) ou com ciclodextrina vazia (V) por mais 6 semanas. Ao final das 13 semanas de experimento foram realizadas avaliações biométricas, bioquímicas, Elisa e expressões gênicas por qRT-PCR. Os ratos SM-V apresentaram aumento da expressão de citocinas inflamatórias no TAV retroperitoneal e no TAM, redução da expressão do PPARγ2 no fígado, piora da via intracelular da insulina no fígado, no TAV retroperitoneal e no músculo gastrocnêmico, redução na expressão de PGC1-α, PGC1-β, UCP-2 e UCP-3 no gastrocnêmico e aumento da expressão de UCP-1 no TAM. Esses eventos ocorreram simultaneamente com a elevação da glicemia de jejum, da insulina, do HOMA-IR e do HOMA-ß e com o aumento do peso corporal, do índice de adiposidade, do colesterol total, do LDL e da alanina transferase (ALT). O tratamento com HPβCD/Ang-(1-7) reduziu a expressão das adipocinas pró-inflamatórias do TAV e TAM, normalizou a expressão de PPARγ2 no fígado, melhorou a via
intracelular da insulina no fígado, no TAB retroperitoneal e no músculo gastrocnêmico e aumentou a expressão de UCP-3 no músculo gastrocnêmico e a expressão de UCP-1 no TAM, quando comparado ao grupo controle CT-V. Esses eventos impediram o aumento do peso corporal, reduziram o índice de adiposidade e os níveis de colesterol total, além de normalizar a glicemia de jejum, os níveis de insulina, o HOMA-IR, o HOMA-ß, e os níveis de ALT. Nossos dados demonstram a eficácia do tratamento com HPβCD-Ang-(1-7) ao mostrar que essa formulação reverte distúrbios metabólicos da SM em ratos, a partir de alterações benéficas teciduais.
XII ABSTRACT
The aim of this study was to evaluate the therapeutic potential of the oral formulation of Ang-(1-7), the HPβCD/Ang-(1-7), on the inflammatory process of visceral white adipose tissue (WAT) and brown adipose tissue (BAT) and on intracellular signaling pathways of insulin and on the adaptive thermogenesis, in reversion of metabolic syndrome (MS) and related disorders induced by high-fat diet. Rats weaned at 4 weeks of age, were subjected to AIN-93 diet control (CT diet) or high fat diet (SM diet ) for 7 weeks for the induction of MS. After this period, the rats were divided into groups treated with HPβCD/Ang- (1-7) or empty cyclodextrin (V) for further six weeks. At the end of the experiment biometric, biochemical, Elisa and qRT-PCR assays were performed. SM-V rats showed increase in inflammatory cytokines expression in the retroperitoneal WAT and BAT, reduction in PPARγ2 expression in the liver, worsening in insulin signaling pathways in the liver, retroperitoneal WAT and gastrocnemius, reduction in PGC1- α, PGC1-β, UCP-2 and UCP-3 expression in gastrocnemius and an increase in UCP-1 expression in the TAM. These events occur simultaneously with the elevation of fasting glucose, insulin, HOMA-IR and HOMA-ß and with increasing the body weight, adiposity index, total cholesterol, LDL and alanine aminotransferase (ALT). Treatment with HPβCD/Ang-(1-7) reduced the inflammatory adipokines expression in retroperitoneal WAT and BAT, normalized PPARγ2 expression in the liver, improved the insulin signaling pathway in the liver, retroperitoneal WAT and gastrocnemius, increased UCP-3 expression in gastrocnemius and increased UCP-1 expression in TAM compared to CT-V group. These events prevented the increase in body weight, reduced adiposity index and total cholesterol, and normalized fasting plasma glucose, insulin, HOMA-IR, HOMA-ß, and ALT. Our data demonstrate the efficacy of HPβCD/Ang-(1-7) treatment and show that this formulation reverses MS and related disorders through of tissue beneficial changes.
XIII
LISTA DE FIGURAS
Figura 01: Demonstração do processo inflamatório crônico do tecido adiposo visceral. ... 24
Figura 02: visão esquemática do Sistema Renina Angiotensina... 35
Figura 03: Delineamento experimental ... 44
Figura 04: Glicemia de jejum e teste de tolerância oral a glicose ... 51
Figura 05: Expressão gênica das adipocinas do tecido adiposo retroperitoneal ... 55
Figura 06: Expressão gênica da Leptina e TNF-α no tecido adiposo marrom (TAM) ... 56
Figura 07: Expressão gênica de PPARγ2 no fígado e no TAM ... 57
Figura 08: Expressão gênica da via da insulina no tecido adiposo retroperitoneal ... 59
Figura 09: Expressão gênica da via da insulina no fígado ... 61
Figura 10: Expressão gênica da via da insulina no músculo gastrocnêmico ... 63
Figura 11: Expressão gênica da via da insulina no TAM ... 65
Figura 12: Expressão gênica do PGC-1α, PGC-1β, UCP-2 e 3 no músculo gastrocnêmico ... 66
Figura 13: Expressão gênica da UCP-1 no tecido adiposo marrom ... 67
XIV
LISTA DE TABELAS
Tabela I: Composição das dietas do experimento ... 42
Tabela II: Primers utilizados (Ford e Reverse) ... 47
Tabela III: Peso corporal e peso relativo de órgãos e tecidos dos ratos ... 52
Tabela IV: Parâmetros bioquímicos dos ratos ... 53
XV
LISTA DE ANEXOS
ANEXO I: Artigo Publicado em 2013 (revista LIPIDS IN HEALT AND DISEASE)... 116 ANEXO II: Documento de Aprovação do Projeto pelo Comitê de Ética Animal da Universidade Federal de Ouro Preto ... 117
XVI
LISTA DE ABREVIATURAS E SIGLAS
AACE –“American Association of Clinica Endocrinologists” AG – ácidos graxos
AGT – angiotensinogênio
AIN-93M – dieta controle de manutenção para roedores AKT – proteína cinase B
AKT-2 – proteína quinase B – beta (serina (Ser) / treonina (Thr) quinase) ALT – alanina aminotransferase
Ang I – angiotensina I Ang II – angiotensina II
Ang-(1-7) – angiotensina de 1 a 7 aminoácidos AS160 – substrato de 160KDa da Akt
AST – aspartato aminotransferase AT1 – receptor 1 da angiotensina II AT2 – receptor 2 da angiotensina II cDNA – DNA complementar CDs – ciclodextrinas
COX-1 – enzima ciclooxigenase 1 COX-2 – enzima ciclooxigenase 2 CT – dieta controle AIN-93 Dieta SM – dieta hiperlipídica DM2 – Diabetes Mellitus do tipo 2 DNA – ácido desoxirribonucleico
ECA – enzima conversora de angiotensina ECA2 – enzima conversora de angiotensina dois
EGIR –“European Group for Study of Insulin Resistance” GCN5 – histona acetiltransferase
GLUT-4, transportador de glicose do tipo 4 HAS – hipertensão arterial sistêmica
HOMA-IR – modelo de avaliação da homeostase da resistência à insulina
XVII HPβCD/Ang-(1-7) – Ang-(1-7) inserida na β-ciclodextrina
I.P – intraperitoneal
IDF –“International Diabetes Federation” IL-1 – interleucina 1
IL-10 – interleucina 10 IL-13 – interleucina 13 IL-1b – interleucina 1b IL-4 – interleucina 4 IL-6 – interleucina 6
IMC – índice de massa corporal Insr – receptor da insulina (primer) IR – receptor da insulina
IRS-1 – substrato do receptor de insulina 1 IRS-2 – substrato do receptor de insulina 2 JAK2 – tirosina quinase da via da insulina JNK – quinase c-Jun-NH2-terminal LPL – lipase lipoprotéica
LDL – Lipoproteína de baixa densidade
MAS – receptor da Angiotensina de 1 a 7 aminoácidos MCP-1 – proteína quimiotática de monócitos-1
MHO – obeso metabolicamente saudável MUO – obeso metabolicamente não saudável NCEP – "National Cholesterol Education Program” NFKβ – fator de transcrição nuclear kappa B
NKT – células T natural killer
OMS – Organização Mundial de Saúde PG – prostaglandina
PGC1 – coativador 1 do receptor ativado por proliferador do peroxissoma
PGC-1α– coativador 1 alfa dos receptores ativados por proliferadores dos peroxissomais PGC-1β– coativador 1 beta dos receptores ativados por proliferadores dos peroxissomais PGG2 – prostaglandina G2
PGH2 – prostaglandina H2
XVIII pO2 – pressão de oxigênio
PPARγ– coativador gamma dos receptores ativados por proliferadores dos peroxissomais Pprc1 – coativador 1 do receptor ativado por proliferadores de peroxissoma gama
qRT- PCR – Reação em Cadeia da Polimerase em Tempo Real RI – resistência à Insulina
SM – síndrome metabólica
SNS – sistema nervoso simpático
SRA – sistema renina angiotensina-aldosterona TA – tecido adiposo
TAB – tecido adiposo branco TAM – tecido adiposo marrom TAV – tecido adiposo branco visceral TG – triacilgliceróis ou triglicérides
TGF-β–fator de transformação do crescimento β TLR4, receptor Toll-like 4
TNF-α– fator de necrose tumoral α
UCP-1 – isoforma 1 da proteína de desacoplamento da cadeia respiratória UCP-2 – isoforma 2 da proteína de desacoplamento da cadeia respiratória UCP-3 – isoforma 3 da proteína de desacoplamento da cadeia respiratória UCPs – proteínas de desacoplamento da cadeia respiratória
XIX
SUMÁRIO
INTRODUÇÃO ... 19
Etiologia e Classificações da Síndrome Metabólica ... 19
Fisiopatologia do Tecido Adiposo Branco Visceral na Síndrome Metabólica ... 21
Adipocinas ... 24
Leptina ... 25
Fator de Necrose Tumoral α (TNF-α) ... 25
Ciclooxigenase 2 (COX-2) ... 27
Resistina ... 28
Adipsina ... 28
Adiponectina ... 29
Fisiopatologia da Síndrome Metabólica ... 30
Resistência à Insulina ... 30
Receptores Ativados por Proliferadores de Peroxissoma (PPARs) ... 31
Sistema Renina Angiotensina (SRA) e Síndrome Metabólica ... 32
Tecido adiposo Marrom (TAM) e Termogênese ... 36
Coativador 1 do Receptor Ativado por Proliferador do Peroxissoma (PGC1) ... 37
Potencial Terapêutico da HPβCD/Ang-(1-7) na Síndrome Metabólica ... 38
OBJETIVO ... 40
Objetivo Geral ... 40
Objetivos Específicos ... 40
MATERIAIS E MÉTODOS ... 41
Animais ... 41
Definição das Dietas ... 41
Cálculo da Concentração de Ang-(1-7) na HPβCD/Ang-(1-7) ... 42
Protocolo Experimental e Tratamento com HPβCD/Ang-(1-7) ... 43
Descrição dos Procedimentos Experimentais ... 44
Glicemia de Jejum e Teste de Tolerância Oral à Glicose na 7ª Semana das Dietas ... 44
Peso dos Animais, Índice de Lee, Índice de Adiposidade e Pesos dos Órgãos ... 44
Análises Bioquímicas ... 45
Dosagem da Insulina por ELISA... 45
Cálculo HOMA IR e HOMA β ... 46
XX
Oligonucleotídeos Iniciadores (primers) ... 46
Extração de RNA Total ... 47
Síntese de cDNA ... 48
Expressão Gênica por qRT- PCR ... 49
Curva de Eficiência e Amplificação dos Primers ... 49
ANÁLISE ESTATÍSTICA ... 50
RESULTADOS ... 50
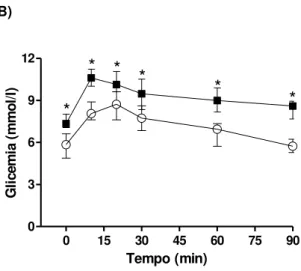
Glicemia de Jejum e Teste de Tolerância Oral a Glicose na 7ª Semana das Dietas ... 50
Avaliação do Peso Corporal e dos Órgãos ... 51
Análises Bioquímicas ... 52
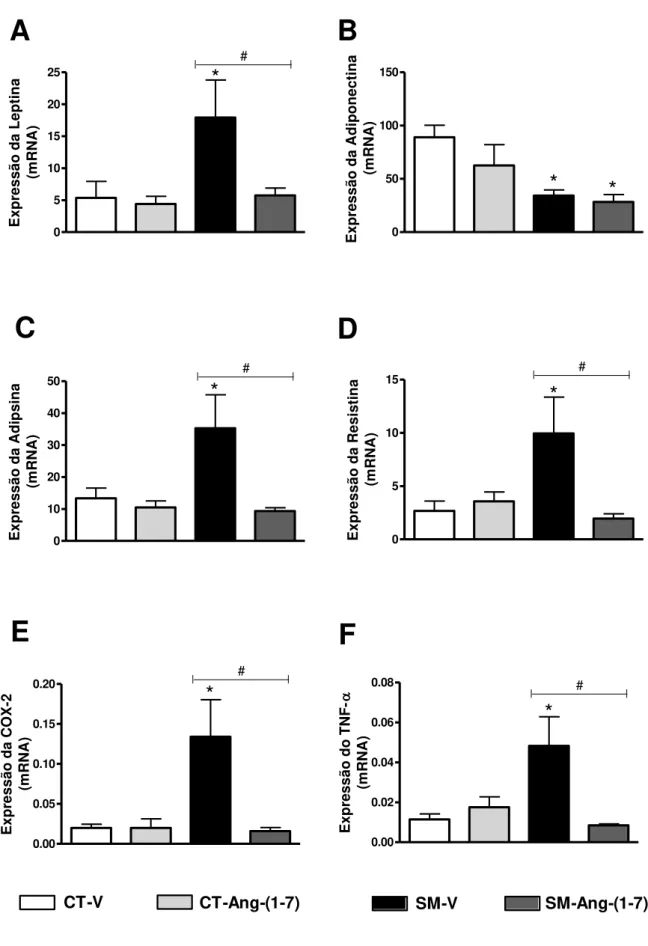
Expressão Gênica das Adipocinas no Tecido Adiposo Retroperitoneal e TAM por qPT-PCR .... 54
Expressão gênica das PPARγ2 nofígado e TAM por qPT-PCR... 56
Glicemia de jejum e Resistência à Insulina ... 57
Expressão da Via de Sinalização da Insulina no TAB Retroperitoneal qRT-PCR ... 58
Expressão da Via de Sinalização da Insulina no Fígado por qRT-PCR ... 60
Expressão da Via de Sinalização da Insulina no Gastrocnêmico por qRT-PCR ... 62
Expressão da Via de Sinalização da Insulina no TAM por qRT-PCR ... 64
Expressão Gênica de PGC-1α, PGC-1β, UCP-2 e UCP-3 no Músculo Gastrocnêmico ... 66
Expressão Gênica de UCP-1 no TAM ... 67
DISCUSSÃO ... 68
REFERÊNCIAS BIBLIOGRÁFICAS ... 84
19
1. INTRODUÇÃO
1.1) Etiologia e Classificações da Síndrome Metabólica
O estilo de vida moderno caracterizado pelo estresse, sedentarismo e hábitos alimentares inadequados, como o consumo de dietas ricas em lipídeos, carboidratos e alimentos industrializados tem aumentado a ocorrência da síndrome metabólica (SM) (Adeoye et al., 2015; Farhangi et al., 2015). Embora a SM tenha sido reconhecida em diferentes formas desde 1927, somente em 1988, que Gerald M. Reaven (Reaven, 1988) definiu a SM como uma manifestação simultânea de resistência à insulina, hiperinsulinemia, diabetes do tipo 2 (DM2), dislipidemias, obesidade central, hiperuricemia (elevação da concentração de ácido úrico no sangue) e pressão arterial elevada, denominando esse conjunto de distúrbios como “Sindrome X”. Em seu
trabalho, Reaven observou que estes disturbios metabólicos precedem outras complicações mais severas como doença isquémica cardíaca, disfunção ventricular esquerda e insuficiência cardíaca, aumentando o risco de morte por causas cardiovasculares. Esta síndrome também foi conhecida pelo nome de Síndrome de Reaven, em sua honra.
Já em 1998 a Organização Mundial da Saúde (OMS) (Alberti e Zimmet, 1998) utilizou o termo SM em substituição à “Síndrome X”, sendo que o principal fator de risco que se exigia para o diagnóstico era a resistência à insulina (RI). Nessa classificação, caso houvesse evidência de um dos diversos marcadores de resistência insulínica (glicemia de jejum, glicemia pós-prandial, DM2) associada a dois fatores de risco adicionais como obesidade, hipertensão arterial sistêmica (HAS), hipertrigliceridemia, HDL baixo ou microalbuminúria, estabelecia-se o diagnóstico da SM. Embora nem todos marcadores de resistência insulínica fossem de aplicabilidade prática para o diagnóstico de SM no cotidiano, foram considerados primordiais pela OMS. Os pacientes diabéticos também eram rotulados como portadores de SM.
20
fatores de risco adicionais como obesidade, HAS e dislipidemia. Aqui, o critério de obesidade era circunferência abdominal, enquanto o da OMS incluía a relação abdômen/quadril ou o índice de massa corporal (IMC). A microalbuminúria foi excluída desta classificação.
Em 2001, o National Cholesterol Education Program (NCEP) Adult Treatment Panel III (Hunt et al., 2004) formulou um conceito novo, introduzindo uma variável clínica para o diagnóstico da SM. A presença de três, dentre os fatores básicos estabelecidos (obesidade abdominal, hipertrigliceridemia, HDL baixo, HAS, glicemia de jejum elevada ou ainda presença de DM2) confirmava o diagnóstico de SM.
Em 2003, a American Association of Clinica Endocrinologists (AACE) retoma a presença de resistência insulínica como o fator principal da síndrome, propondo novamente a introdução do termo RI. Esse grupo reinstituía a importância das variáveis laboratoriais, HAS e obesidade, sem estabelecer quantidade de fatores presentes para o diagnóstico (Kahn et al., 2005). Os pacientes diabéticos foram novamente excluídos. A essa avaliação foram somadas a presença de síndrome de ovário policístico, hiperuricemia, história familiar de DM2 ou cardiopatia.
Em 2005, a International Diabetes Federation (IDF) publicou novos critérios para a SM, estabelecendo a obesidade visceral como fator crucial para o diagnóstico, considerando esta a principal evidência relacionada à SM. Nesse caso, a obesidade visceral associada a dois outros fatores listados no NCEP confirmava o diagnóstico da SM (National Cholesterol Education Program (Ncep) Expert Panel on Detection, 2002; Alberti et al., 2009).
Atualmente o diagnóstico clínico da SM é feito quando o portador apresenta três ou mais distúrbios destes distúrbios de acordo com a definição de Alberti e colaboradores de 2009 (Alberti et al., 2009). Logo, exames simples que comprovarem a associação da redução do HDL, aumento dos TG e da glicemia de jejum e elevação da pressão arterial já são suficientes para confirmar a presença da SM na prática clínica.
21
e por estarem presentes na grande maioria dos pacientes portadores da SM. Sendo assim, é de fundamental importância o entendimento dos distúrbios apresentados por este tecido durante patológias como obesidade e SM.
1.2) Fisiopatologia do Tecido Adiposo Branco Visceral na Síndrome Metabólica
O TAB é o um órgão complexo, multifuncional com implicações autócrina, parácrina e endócrina (Sethi, 2007; Gollisch et al., 2009). É composto de adipócitos, matriz extracelular, vasos sanguíneos, nervos e vários outros tipos de células, incluindo pré-adipócitos, células estaminais e células imunitárias (Kalupahana et al., 2011). Os adipócitos são as únicas células especializadas no armazenamento de lipídios na forma de triacilgliceróis ou triglicérides (TG) em seu citoplasma. Essas células possuem todas as enzimas e proteínas reguladoras necessárias para síntese dos ácidos graxos (AG) e TG a partir substratos oriundos dos quilomícrons e das lipoproteínas circulantes como lipoproteínas de muito baixa densidade (VLDL) (Tacken et al., 2000). Os adipócitos maduros armazenam os TG em uma única e grande gota lipídica que pode ocupar até 85% da massa total do tecido (Pond, 2001) e apresentam menos inervação e vascularização, quando comparado a outros tecidos endógenos (Ryu e Bartness, 2014).
Durante décadas, o TAB foi considerado apenas como um tecido de armazenamento de energia para o corpo, defesa mecânica contra lesões, e um termorregulador (Greenberg e Obin, 2006). A descoberta da adipocina leptina, em 1994, e da adiponectina um ano após ampliou significativamente o conhecimento sobre a contribuição do TAB para homeostase corporal e atraiu a atenção de cientistas de diversas especialidades. Embora, inicialmente, as adipocinas tenham sido associadas somente com disturbios alimentares, estudos posteriores revelaram que as adipocinas desempenham um papel importante na regulação da resposta imune, nos sistemas reprodutivo e endócrino, nos processos inflamatórios sistemicos e na sensibilidade à insulina (Fantuzzi, 2005). Atualmente, o TAB é reconhecido como um órgão endócrino que produz, armazena e libera uma variedade de hormônios e adipocinas incluindo leptina, adiponectina, angiotensina II e fator de necrose tumoral α (TNF-α) (Trayhurn, 2005; Galic, Oakhill e Steinberg, 2010).
22
desenvolvimento de alterações metabólicas, como a RI (Chan et al., 2004; Berg e Scherer, 2005), DM2 e aterosclerose, que o TAB subcutâneo (Gollisch et al., 2009; Bjørndal et al., 2011). A obesidade visceral patológica é caracterizada pela hipertrofia e hiperplasia dos adipócitos do TAV e um subsequente desenvolvimento de um processo inflamatório crônico de baixo grau no qual ocorre uma infiltração de macrófagos que secretam citocinas pró-inflamatórias (Kalupahana, Moustaid-Moussa e Claycombe, 2012). Entretanto, nem todo obeso pode ser considerado como portador da SM. Nesse sentido, foram identificados dois perfis de obesos denominados obeso metabolicamente saudável ou obeso metabolicamente normal (MHO) e o obeso metabolicamente não saudável (MUO) (O'connell et al., 2010; Primeau et al., 2011). Geralmente a obesidade metabolicamente normal é definida como aquela que apresenta ausência de qualquer desordem metabólica incluindo DM2, dislipidemia e HAS em indivíduo com IMC ≥
30kg/m2 (Blüher, 2010). No MHO, os depósitos de gordura viscerais ainda não apresentam processos inflamatórios significativos. O TAV apresenta-se com um perfil anti-inflamatório eutrófico, como irrigação sanguínea normal, predominância de adipocinas anti-inflamatórias como adiponectina e macrófagos residentes M2 (macrófagos alternativamente ativados), que expressam outros fatores anti-inflamatórios como interleucinas 4, 10 e 13 (IL-4, IL-10 e IL-13) e fator de transformação do crescimento beta (TGF-β), que contribuem para a função metabólica normal do TAB visceral (Esser et al., 2013).
23
categorizar indivíduos em metabolicamente normais ou não (Pataky, Bobbioni-Harsch e Golay, 2010).
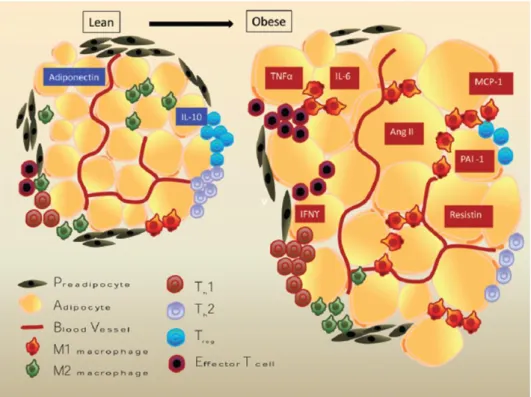
Com a progressão da obesidade e do processo inflamatório do TAB, o adipócito hipertrofiado secreta quimiocinas que atraem mais células imunes. Os macrófagos estão entre as primeiras células a infiltrarem no TAV (Patel, Buras e Balasubramanyam, 2013). Fatores secretados por leucócitos influenciam reciprocamente a atividade de adipócitos, bem como de células endócrinas e musculares, o que por sua vez interfere nos níveis de nutrientes (particularmente a glicose), insulina e outras adipocinas (Kaminski e Randall, 2010). Assim, outras células do sistema imune como células T reguladoras, células T efetoras CD8+, Células T CD4+ (Th1 e Th2), células T natural killer (NKT), células B, células dendríticas, eosinófilos, neutrófilos e mastócitos presentes no TAB também têm sido implicados na patogênese da RI relacionada à obesidade (Suganami e Ogawa, 2010; Bertola et al., 2012; Patel, Buras e Balasubramanyam, 2013). A ativação e recrutamento de outras células do sistema imune aumentam a necrose e apoptose de adipócitos, causam elevação do recrutamento de macrófagos para o TAV e induzem mudança do fenótipo de macrófagos residentes M2, para macrófagos pró-inflamatórios M1, que são responsáveis por mais produção de citocinas pró-inflamatórias como TNF-α, caracterizando o perfil pró-inflamatório crônico de baixa intensidade do TAV durante obesidade e SM (Engström et al., 2003), com produção elevada de leptina, resistina, adipsina, ciclooxigenase-se 2 (COX-2), proteína quimiotática de monócitos-1 (MCP-1), interleucina 6 (IL-6), (De Ferranti e Mozaffarian, 2008) e baixa produção de adipocinas anti-inflamatórias como adiponectina, como mostrado na figura 1 abaixo.
24
Figura 01: Demonstração do processo inflamatório crônico do tecido adiposo visceral durante a obesidade e SM (Kalupahana, Moustaid-Moussa e Claycombe, 2012).
Esse desequilíbrio na produção de adipocinas pró-inflamatórias e anti-inflamatórias contribui para o surgimento dos distúrbios subsequentes da SM (Meshkani e Adeli, 2009; Van Beek et al., 2014), visto que adipocinas pró-inflamatórias, como
TNF-α são secretadas na corrente sanguínea e provocam disturbios metabólicos
secundários, como a RI em tecidos insulino-dependentes durante a SM.
1.3) Adipocinas
25
1.3.1) Leptina
A leptina é uma hormonio polipeptídico não glicosilado de 16 kDa produzida principalmente pelo TAB, descoberto por uma única mutação no gene ob/ob de camundongos. Nos seres humanos, é codificada pelo gene Lep, um equivalente do gene ob em camundongos (Zhang et al., 1994) e é classicamente conhecida como hormônio da saciedade, que ao ser liberado pelo TAB, atua no centro da fome induzindo redução da ingesta alimentar (Yu et al., 1997). Após atravessar a barreira hemato-encefálica, a leptina se liga a receptores específicos no hipotálamo, onde atua como reguladora fundamental da ingesta alimentar, através do aumento da expressão de fatores anorexígenos e redução da produção de peptídeos orexígenos pelo hipotálamo. Logo, a leptina atua como importante regulador do consumo alimentar em longo prazo (Otero et al., 2005) e sua produção e concentração sérica está relacionada com o tamanho da massa do TAB e depende ainda dos níveis de insulina, estado energético, hormonios sexuais, e de uma grande variedade de mediadores inflamatórios, incluindo interleucina-1 (IL-interleucina-1) e TNF-α (Krysiak, Okopień e Herman, 2005; Lago et al., 2007). A leptina também foi identificada em locais fora do TAB, como estômago, placenta, tecido adiposo marrom (TAM) e no músculo esquelético (Badman e Flier, 2005; Galic, Oakhill e Steinberg, 2010).
Em condições normais os níveis circulantes de leptina correlacionam-se com o conteúdo de gordura corporal, como ja mencionado. Entretanto durante a obesidade e SM, observa-se que o aumento da leptina não é mais capaz de regular corretamente a ingestão alimentar, caracterizando a redução da sensibilidade à leptina ou resistencia a leptina e hiperleptinemia. Em adição, diversos estudos mostraram que o aumento dos níveis séricos de leptina durante processos infecciosos e inflamatórios (Howard et al., 1999; Faggioni, Feingold e Grunfeld, 2001), podem ser atribuídos também ao aumento de adipocinas pró-inflamatórias como interleucina- 1b (IL-1b), TNF-α e IL-6, o que mostra que a atuação da leptina durante processos inflamatórios como a SM pode ser regulada pela liberação de outras adipocinas (Fantuzzi, 2005).
1.3.2) Fator de Necrose Tumoral α (TNF-α)
O TNF-α é uma citocina imunomodulatória e pró-inflamatória com efeitos autócrinos, parácrinos e endócrinos e influencia substancialmente a síntese de adipocinas pró-inflamatórias, por meio da ativação do fator de transcrição nuclear kappa
26
a expressão de adiponectina (Gualillo, González-Juanatey e Lago, 2007; Bray et al., 2009). Como já mencionado, durante a SM ocorre um aumento do TNF-α no TAV (Shimamoto e Miura, 2009) que é secretado na corrente sanguínea, o que interfere em diversos processos fisiológicos, como a via intracelular da insulina no fígado, no musculos esqueléticos e no próprio TAB, o que altera a homeostase glicêmica e o metabolismo dos lipídios (Hotamisligil, Shargill e Spiegelman, 1993; Das e Balakrishnan, 2011). O TNF-α foi descrito como o primeiro fator derivado do TAB que representa associação entre obesidade, inflamação e diabetes. Hotamisligil, Shargill e Spiegelman em 1993, demonstraram expressão aumentada de RNAm da TNF-α na obesidade e desde então está adipocina tem sido fortemente associada à patogênese da RI (Hotamisligil, Shargill e Spiegelman, 1993). Anteriormente, acreditava-se que o adipócito fosse a única fonte de TNF-α na obesidade. Entretanto tem sido reconhecido que macrófagos do estroma vascular, principalmente do tipo M1 são responsáveis também por grande produção de TNF-α no TAV de obesos (Galic, Oakhill e Steinberg, 2010; Coelho, Oliveira e Fernandes, 2013). A expressão aumentada de RNAm do
TNF-α no tecido adiposo (TA) é altamente induzida pela obesidade, e é maior no TAV em relação ao TAB subcutâneo (Zou e Shao, 2008; Maury e Brichard, 2010).
27
músculos (Guilherme et al., 2008; Hermsdorff et al., 2008). Esses dados corroboram com o estudo de Lee e colaboradores (2013) que demonstraram que a RI induzida por dieta ou obesidade é mediada por citocinas pró-inflamatórias, incluindo o TNF-α, de uma forma dependente de NFkB (Lee, 2013). Todas estas evidências mostraram que o TNF-α é uma das principais adipocinas do TAV responsável pelos distúrbios metabólicos que ocorrem na SM.
1.3.3) Ciclooxigenase 2 (COX-2)
Sobre a ação de estímulos químicos, traumáticos, mitogênicos e principalmente inflamatórios a prostaglandina (PG) G/H sintase citosólica, também denominada Ciclooxigenase (COX), converte o ácido araquidônico oriundo da clivagem dos fosfolipídios de membrana pela enzima fosfolipase A2, nos compostos intermediários PGG2 e PGH2 (Gryglewski et al., 2001). A enzima ciclooxigenase apresenta duas isoformas intituladas COX-1 e COX-2. A COX-1 foi rotulada como fisiologicamente constitutiva, agindo como citoprotetora gástrica e sendo importante para a homeostase renal e plaquetária. Já a COX-2 ou indutiva, foi inicialmente identificada como induzida apenas em situações de trauma tissular e processos inflamatórios (Perazella e Tray, 2001; Yaksh et al., 2001). Estudos mostraram que a COX-2 apresenta atividade e expressão alterada também durante dirturbios metabólicos e fisiológicos como, alterações do metabolismo lipídico e da glicose, hipertrofia e hiperplasia dos adipócitos, redução da sensibilidade à insulina, hipertensão e inflamação (Hsieh et al., 2009; Hsieh et al., 2010), que são ocorrências que fazem parte da SM. Durante a inflamação crônica no TAV que ocorre na SM, os adipócitos liberam MCP-1, que promove o recrutamento de macrófagos para o TAV. Esses macrófagos, uma vez ativados, elevam a produção da COX-2 juntamente com uma série de mediadores inflamatórios como a enzima óxido nítrico sintase induzível (iNOS) e junto com outras adipocinas pró-inflamatórias que contribuem para a RI e outros distúrbios da SM (Jungbauer e Medjakovic, 2012; Santilli et al., 2012; Sarı et al., 2015). A expressão de COX-2 nos macrófagos e nas células endoteliais pode ser induzida por TNF-α (Wang et al., 2006; Wu e Wu, 2006).
1.3.4) Resistina
28
al., 2005). Em camundongos, a resistina é sintetizada pelo TAB (Bokarewa et al., 2005). Em humanos, o TAB produz somente pequenas quantias dessa proteína (Savage et al., 2001), enquanto níveis relativamente elevados de mRNA de resistina são detectáveis em celulas mononucleares circulantes periféricas (CMSPs) (Patel et al., 2003). A função fisiológica da resistina pode diferir entre humanos e roedores (Ghosh et al., 2003), devido ao fato da resistina humana e de camundongos apresentarem apenas cerca de 64% de sequência homologica em nível de mRNA e somente 59% de identidade para a estrutura de aminoácidos (Steppan e Lazar, 2004). Entretanto, nos seres humanos, o papel mais documentado da resistina é na regulação de processos metabólicos, da adipogênese e participação em processos pró-inflamatórias como a SM (Degawa-Yamauchi et al., 2003; Norata et al., 2007). Alguns estudos encontraram que os níveis plasmáticos de resistina correlacionam-se positivamente com a obesidade e outros fatores caracteristicos da SM (Takeishi et al., 2007) Em adição, níveis elevados de resistina foram correlacionados com aumento do risco de eventos cardíacos (Takeishi et al., 2007). Nos seres humanos, a resistina induz a produção de IL-6, IL-1b, TNF-α
por células CMSPs durante processos inflamatórios (Patel et al., 2003; Nagaev et al., 2006) e estudos mostraram que células do fluido sinovial expressam a IL-6 e TNF-α
quando estimulados por resistina e, de forma semelhante, citocinas pró-inflamatórias aumentam a expressão de resistina nas CMSPs, o que confirma seus efeitos pró-inflamatórios (Bokarewa et al., 2005).
1.3.5) Adipsina
A Adipsina foi descrita em 1987 (Cook et al., 1987) e posteriormente foi identificada como (fator de complemento D) (Rosen et al., 1989; White et al., 1992), que catalisa o passo limitante da velocidade da via alternativa de ativação do complemento (Xu et al., 2001). A adipsina desempenha um papel central em modelos de isquemia-reperfusão (Stahl et al., 2003) e sépsis (Dahlke et al., 2011). As funções desta molécula incluem a formação da C5-C9 complexo de ataque à membrana e a geração de um número de moléculas de sinalização incluindo os anafilatoxinas C3a e C5a (Ricklin et al., 2010). Os receptores para complemento péptidicos são amplamente expressos em vários tipos de células imunitárias (Ricklin et al., 2010).
29
adipsina em roedores obesos (Flier et al., 1987). Posteriormente, foi observado que a adipsina é um dos componentes oriundos da ativação do sistema imunológico no TAV, que participa do metabolismo lipídico e da glicose (Cianflone, Xia e Chen, 2003). Estudos mais recentes (Kwon et al., 2012; Lee et al., 2013) mostram que a adipsina correlaciona-se positivamente com a adiposidade, resistência à insulina, dislipidemia e doenças cardiovasculares. Por outro lado, outros estudos mostram que a adipisina possui um efeito anti-inflamatório, protetor contra a obesidade, atuando na melhora da sensibilidade a insulina (Mamane et al., 2009; Lim et al., 2013; Lo et al., 2014). Logo, a participação da adipsina em eventos metabólicos permanece ainda obscura e necessita ainda de mais estudos.
1.3.6) Adiponectina
30
cardiovasculares patológicos futuros em pacientes com doença arterial coronariana, e por esse motivo foi considerada um marcador da gravidade da SM e de doenças cardiovasculares (Inoue et al., 2007).
Dois receptores de adiponectina foram descritos, adipoR1 e adipoR2. Esses receptores são expressos no tecido adiposo, macrófagos e células beta pancreáticas (Whitehead et al., 2006). O receptor de adiponectina 1 (adipoR1) é altamente expresso no músculo esquelético, enquanto que o receptor de adiponectina 2 (adipoR2) é predominantemente expresso no fígado (Beylot, Pinteur e Peroni, 2006).
1.4) Fisiopatologia da Síndrome Metabólica
Como já mencionado, durante a SM o aumento da obesidade visceral e o desequilíbrio na produção entre adipocinas geram um estado pró-inflamatório crônico do TAV. Com a progressão da inflamação crônica do TAV, as adipocinas pró-inflamatórias são secretadas na corrente sanguínea e podem promover diversos distúrbios metabólicos sistêmicos, como a RI, apontada por diversos estudos como fator patológico central da SM (Akagiri et al., 2008; Saki e Karamizadeh, 2014; Hwang et al., 2015). Entretanto, outros fatores endógenos também alterados durante a SM, como receptores ativados por proliferadores de peroxissoma (PPARs) (Mohamed Youssef et al., 2013), os componentes do sistema renina angiotensina-aldosterona (SRA) sistêmico e locais (Kamide, 2014) e os genes envolvidos na biogênese mitocondrial e na função respiratória (Sacks et al., 2009; Liu, W. et al., 2012) participam do desenvolvimento e/ou na manutenção da SM, contribuindo de maneira importante para todo processo metabólico e fisiopatológico da doença.
1.4.1) Resistência à Insulina
A RI é um estado em que os tecidos sensíveis à insulina como TA, fígado e músculo não respondem adequadamente aos níveis circulantes normais de insulina. Como forma de compensação, as células-β pancreáticas aumentam a produção de insulina levando à hiperinsulinemia (Dominici et al., 2014). Logo, ocorrem diversas alterações na via intracelular da insulina nos tecidos alvo, que comprometem desde a ligação da insulina ao seu receptor nas células alvo (IR), até os estágios finais da via intracelular da insulina nesses tecidos (Boucher, Kleinridders e Kahn, 2014). O IR é
31
seguido pela autofosforilação dos resíduos de tirosina da subunidade β e a um aumento
na sua atividade cinase. A cinase do IR fosforila os resíduos de tirosina dos substratos do receptor de insulina (IRS 1 e 2). Quando os resíduos de tirosina do IRS-1 e 2 são fosforilados, os IRs ligam-se a fosfatidilinositol 3-quinase (PI3K), ativando-a. A P13K converte seu substrato, fosfatidilinositol 4,5-bifosfato (PIP2), no mensageiro lipídico fosfatidilinositol 3,4,5 - trifosfato (PIP3). O PIP3 então se liga a AKT. A AKT continua a propagação do sinal hormonal ao ativar a proteína AS160, que irá sensibilizar as pequenas proteínas ao redor do GLUT4, favorecendo a sua translocação até a membrana lipídica para captação da glicose (Jager et al., 2007; Gurriarán-Rodríguez et al., 2011). Os mecanismos moleculares que levam ao aparecimento da RI são diversos e ainda alvo de estudos. No entanto, sabe-se que o cross-talk entre adipocinas, como a leptina (Coppari e Bjørbæk, 2012), adiponectina (Awazawa et al., 2011) e angiotensina II (Kamide, 2014), e via de sinalização da insulina, pode aumentar ou diminuir a ação da insulina em seus tecidos alvos (Westhoff, Rau e Zink, 2007).
1.4.2) Receptores Ativados por Proliferadores de Peroxissoma (PPARs)
Os PPARs são um grupo de proteínas receptoras nucleares que funcionam como fatores de transcrição que regulam a expressão dos genes. Os PPARs desempenham um papel essencial na regulação de eventos como diferenciação celular, no desenvolvimento, no metabolismo de carboidratos, lipídios e proteínas. Os PPARs heterodimerizam com o receptor de retinoíde X (RRX) e se ligam a regiões específicas do DNA de genes alvo. Estas sequências de DNA são denominadas PPREs (elementos de resposta hormonal proliferador de peroxissoma). A sequência de consenso do DNA é AGGTCANAGGTCA, com N sendo qualquer nucleótido. De um modo geral, esta sequência ocorre na região do promotor de um gene, e, quando o PPAR se liga ao seu ligante, a transcrição dos genes alvo é aumentada ou diminuída, dependendo do gene. Portanto a atividade transcricional dos PPARs requer a ligação de ligantes específicos, heterodimerização com o RRX e a interação com coativadores de transcrição, tais como o coativador 1α dos receptores ativados por proliferadores dos peroxissomais (PGC-1α) e PGC-1β (Berger e Moller, 2002; Yu e Reddy, 2007), que participam da biogênese mitocondrial e termogênese adaptativa em diferentes tecidos (Hsieh et al., 2005; Espinoza et al., 2010)
32
adiposo, dentre outros tecidos (Tyagi et al., 2011). O PPARβ é expresso em muitos
tecidos, mas predominantemente no cérebro, no tecido adiposo e na pele. Já o PPARγ possui 3 subformas, são elas: PPARγ1 expresso em vários tecidos, incluindo coração,
músculo, cólon, rim, pâncreas e baço; O PPARγ2 expresso principalmente no fígado e
no TAB, enquanto que o PPARγ3 expresso em macrófagos, intestino grosso e TAB
(Asnani, Theuma e Fonseca, 2003; Galuppo et al., 2010)
Estudos demonstraram que o (PPAR)γ esta associado com a RI, com o metabolismo lipídico e com a regulação da inflamação durante a esteatose hepática (Zhao et al., 2004; Nan et al., 2009), constituindo a isoforma mais importante em estudos sobre a SM. Esses dados são corroborados por estudos que utilizaram pioglitazona (agonista do PPARγ) no tratamento de doenças como diabetes, esteatose hepática e observaram melhora no metabolismo da glicose e dos lipídeos, aumento da sensibilidade à insulina e aumento dos níveis de adiponectina, embora o exato mecanismo de ação em tecidos como o fígado permanece controverso (Harrison, 2010; Tripathy et al., 2013).
Estudos sugerem ainda uma relação entre o Sistema Renina Angiotensina (SRA) e PPARs, na medida em que antagonistas do receptor AT1 apresentaram efeitos benéficos no metabolismo, que são independentes de sua ligação ao receptor AT1 (Younis et al., 2010). Portanto, acredita-se que antagonistas de receptor AT1 possam atuar como agonistas na ativação de PPARs (Sugimoto et al., 2008; Rong et al., 2010; Younis et al., 2010), o que é corroborado pelo estudo de Sugimoto (2008) que mostraram que a ativação do PPARγ com telmisartan (antagonista de AT1) protegem animais contra a obesidade induzida pela dieta, através do aumento da oxidação dos ácidos graxos no músculo ex vivo (Sugimoto et al., 2008).
1.4.3) Sistema Renina Angiotensina (SRA) e Síndrome Metabólica
33
Santos et al., 2005). Entretanto, durante a ocorrência de doenças cardiovasculares (Julius, 1990), diabetes, obesidade e SM ocorre uma disfunção deste eixo, de forma que a Ang II apresenta-se como potente agente inflamatório, oxidante e pró-trombótico, contribuindo para a fisiopatologia destas doenças (Favre, Esnault e Van Obberghen, 2015).
Desde a década de 70 (Romero, Mak e Hoobler, 1974), estudos já haviam observado um envolvimento do SRA com distúrbios metabólicos como o diabetes (Ho e Michelakis, 1985; Mann e Ritz, 1988). Naquela época os estudos da atividade e eficácia de inibidores da ECA, mostraram que esses agentes apresentavam redução na pressão arterial através da diminuição da produção da Ang II de forma diferente um dos outros (Romero, Mak e Hoobler, 1974; Macgregor et al., 1985). De fato, a melhora do metabolismo da glicose foi apontada pelo estudo de Ferrière e colaboradores em 1990 (Ferrière et al., 1990). Nesta mesma época iniciavam-se estudos que correlacionavam o SRA e o diabetes (Hauger-Klevene e Barontini De Moyano, 1974). Estudos posteriores mostraram que Ang II participava do processo de glicosilação de proteínas renais durante o diabetes e do processo patológico da hiperfiltração glomerular em humanos e modelos animais diabéticos (Jenkins et al., 1990; Mathis e Banks, 1996) e em 1992, Donnelly demonstrou que a utilização de inibidores da ECA como captopril e enalapril promovia, além de seus efeitos cardiovasculares clássicos, uma melhora na RI que se acreditava estar associada ao aumento da captação da glicose pelos músculos esqueléticos (Donnelly, 1992). Da década de 90 até os anos 2000 diversos estudos confirmavam que a Ang II estava envolvida na fisiopatologia da diabetes, sendo parcialmente responsável pelos danos renais da doença (Anderson, 1997; Kennefick e Anderson, 1997; Anderson, 1998; Burns, 2000). Adicionalmente a utilização de bloqueadores do receptor AT1 da Ang II, como o losartan (Hebert et al., 1999; Kedziora-Kornatowska, 1999) e sua combinação com inibidores da ECA como captopril em pacientes com distúrbios metabólicos como diabetes (Zhang et al., 2007) ajudaram a fortalecer as primeiras evidências científicas da participação do SRA em doenças de origem metabólica. Em adição, Boustany e colaboradores (2004) mostraram que todos componentes do SRA são expressos no TAB e parecem ser regulados de forma independente do SRA sistêmico (Boustany et al., 2004).
34
2004). O SRA local no TAV exerce importantes funções autócrina e paracrina na modulação da lipogênse, lipólise, adipogênese, assim como em processos inflamatórios sistemicos (Boustany et al., 2004; Saiki et al., 2009). Estudos mais recentes realizados em animais e humanos sugerem que a obesidade visceral promova um desequilíbrio nos SRA endógenos, através da ativação do eixo ECA/Ang II/AT1 (Boustany et al., 2004; Cassis et al., 2008), que está associada a diversos distúrbios metabólicos, como prejuízos na via de sinalização intracelular de insulina (Rao, 1994; Richey et al., 1999; Kamide, 2014). Nesse contexto, foi observado que a Ang II e seu receptor AT1 estão superexpressos em vários tecidos em resposta à resistência a insulina/ hiperinsulinemia que ocorre durante a SM (Iyer, Raizada e Katovich, 1996; Giacchetti et al., 2000; Nyby et al., 2007). Outros trabalhos mostraram ainda, que o eixo ECA/Ang II/AT1 contribui para a gênese de outros eventos patológicos da SM, como dislipidemias (Richey et al., 1999), alterações hepáticas como a fibrose e esteatose (Uesugi et al., 2004; Yamamoto et al., 2008), alterações cardíacas como hipertrofia, fibrose e disfunção ventricular esquerda e alterações na estrutura e função renal (Toblli et al., 2004; Watanabe et al., 2009). No músculo, a Ang II diminui a sensibilidade à insulina, e subsequentemente diminui a ativação da proteína quinase B e proteína quinase C, reduzindo a translocação do GLUT4 para a membrana, e consequentemente, prejudicando o transporte da glicose (Thomas et al., 2011). Esses dados são corroborados pelos estudos que utilizaram inibidores de ECA e antagonistas do receptor AT1 e observaram aumento na translocação de GLUT-4 para a membrana celular e melhora na absorção da glicose pela musculatura esquelética de modelos animais (Luther e Brown, 2011).
35
ECA/Ang II/AT1. A representação esquemática do SRA está descrito na figura 2 abaixo:
Figura 02: visão esquemática do Sistema Renina Angiotensina e os receptores de ação das angiotensinas Adaptado de Ferrario e Strawn (Ferrario e Strawn, 2006).
O papel da Ang-(1-7) na regulação metabólica ainda não está totalmente estabelecido. Estudos recentes indicam que o eixo ECA2/Ang-(1-7)/MAS é capaz de produzir uma melhora do metabolismo dos lípidos e da glicose e reduzir a gordura corporal (Santos et al., 2008; Giani, Mayer, Munoz, et al., 2009; Silva et al., 2013; Oliveira Andrade et al., 2014). Também foi demonstrado que a Ang-(1-7), atuando através de seu receptor MAS acoplado à proteína G previne a disfunção cardiovascular induzida pela diabetes (Benter et al., 2007), e reverte a resistência à insulina em modelos de SM induzida por dieta rica em frutose (Giani, Mayer, Munoz, et al., 2009) . Estudos prévios demonstraram que a ausência do receptor MAS leva a alterações na glicemia e no metabolismo dos lípidos, induzindo um estado semelhante a SM (Santos et al., 2008). Adicionalmente, a elevação crônica dos níveis plasmáticos de Ang-(1-7)
Angiotensinogênio
Angiotensina I
Angiotensina II
Angiotensina- (1-9)
Angiotensina- (1-7)
Renina
AT1 11
AT2
MAS
Vasoconstrição
Pró-fibróticos
Pró-hipertróficos
Estresse oxidativo
Vasodilatação
Anti-hipertensivas
Anti-fibróticos
Anti-hipertróficos
Anti-trombóticas ECA
ECA2
36
melhora a sensibilidade à insulina, tolerância à glicose e aumenta a captação de glicose pelos adipócitos (Santos et al., 2010). Corroborando esses achados, Gaini e cols., (2009) (Giani, Mayer, Muñoz, et al., 2009) mostraram que a infusão crônica de Ang-(1-7) reverte a hipertrigliceridemia, hiperinsulinemia e o prejuízo na via de sinalização intracelular da insulina em um modelo de SM induzido por uma dieta rica em frutose. Esses dados sugerem que o eixo ACE2/Ang-(1-7)/MAS possua efeitos benéficos na prevenção e/ou tratamento de doenças de origem metabólica (Oliveira Andrade et al., 2014).
1.5) Tecido adiposo Marrom (TAM) e Termogênese
Em oposição ao observado com o TAB durante a SM, o tecido adiposo marrom (TAM) tem sido considerado como importante sistema na regulação metabólica. Estudos mostraram que o TAM atua como um tecido protetor contra os distúrbios causados pela obesidade e SM, através do aumento de diversas proteínas, como as UCPs, responsáveis pelo aumento da termogênese (Seale et al., 2011; Matsumura et al., 2014). Nos últimos 20 anos ocorreu um aumento do numero de estudos sobre a participação do TAM nas atividades metabólicas (Cannon e Nedergaard, 2004). O TAM é um depósito de lipídeos que se correlaciona inversamente com os distúrbios da obesidade e SM (Cypess et al., 2009; Saito et al., 2009), devido ao fato de apresentar um padrão metabólico diferenciado e ser altamente rico em mitocondrias. Os adipócitos marrons contêm várias gotículas lipídicas, grande numero de mitocôndrias grandes em seu citoplasma e ainda são ricamente vascularizados.
37
Estudos propõem que a UCP-1 seja um carreador da forma reduzida dos AG, que são oxidados no espaço intermembranar (Garlid et al., 2000; Kraus et al., 2005). Outra isoforma como a UCP-2 é expressa no músculo esquelético, coração, placenta, linfócitos, intestino, pulmão, fígado, rins, pâncreas e TAB (Kageyama et al., 1998; Ricquier e Bouillaud, 2000; Li et al., 2003). Já a UCP-3 é expressa no principalmente e músculo esquelético (Cunningham et al., 2003). Entre as funções das UCPs estão o controle da termogênese adaptativa em resposta à exposição ao frio e à dieta; controle da produção de espécies reativas de oxigênio na mitocôndria (Vidal et al., 1999; Barger, Barnes e Boyer, 2006), e regulação da síntese da ATP e regulação da oxidação de AG (Ricquier e Bouillaud, 2000; Ricquier et al., 2000; Tsuboyama-Kasaoka e Ezaki, 2001).
O desenvolvimento e a manutenção do TAM dependem da atividade do sistema nervoso simpático (SNS) através da atividade dos neurônios pós-ganglionares noradrenérgicos. A termogênese é iniciada com a ativação do SNS e dos receptores adrenérgicos no TAM (Nedergaard e Cannon, 2010). Estudos mostraram que os coativadores 1 dos receptores ativados por proliferadores dos peroxissomais (PGC-1) α
e β interagem com as UCPs e participam do aumento da atividade respiratória mitocondrial (Shimasaki et al., 2013). A noradrenalina interage com o receptor β que
aumenta a expressão de PGC-1α (Bachman et al., 2002; Robinson et al., 2010). O resultado global do aumento da atividade do TAM é a elevação do consumo de AG e glicose, que são utilizados como um combustível durante o aumento da termogênse (Rothwell e Stock, 1983; Van Marken Lichtenbelt et al., 2009; Bartelt et al., 2011), estimulada pela elevação da expressão de UCPs (Cannon e Nedergaard, 2004), com produção de calor ao invés de produzir ATPs. Esses fatores fazem do TAM um importante mecanismo de defesa contra situações adiversas como frio, obesidade (Spiegelman e Flier, 2001; Cannon e Nedergaard, 2004) e SM (Le et al., 2012) através da termogenese adaptativa. Atualmente, sabe-se que seres humanos adultos têm depósitos consideráveis de TAM metabolicamente ativos (Cypess et al., 2009; Saito et al., 2009; Virtanen et al., 2009), sugerindo que este tecido possa desempenhar um papel fundamental na manutenção de um fenótipo saudável também nesta fase da vida.
38
respiratória, promovendo aumento da capacidade enzimática para β-oxidação de ácidos graxos, ciclo de Krebs e fosforilação oxidativa (Soyal et al., 2006; Sadana et al., 2007). O PGC1-α e β são expressos no TA, no coração, nos rins, no fígado, no pâncreas e nos músculos (Staiger et al., 2005). Nos músculos, PGC1-α ativa a oxidação de ácidos graxos, a fosforilação oxidativa, a biogênese mitocondrial e a termogênese (Wu, Chou e Huang, 2014) durante protocolos de treinamento físico, nos quais sabidamente ocorre um aumento do metabolismo (Wu et al., 2014). Ambos PGC-1α e PGC-1β estão envolvidos na termogênese adaptativa que é um mecanismo de adaptação do organismo, que aumenta a atividade e a função respiratória mitocondrial em resposta a um estímulo adverso como exposição ao frio (Chang et al., 2012) ou variações na composição das dietas (Arçari et al., 2009), protegendo o organismo da hipotermia e atenuando distúrbios metabólicos (Pereira et al., 2015)
1.7) Potencial Terapêutico da HPβCD/Ang-(1-7) na Síndrome Metabólica
Nos últimos anos a SM tem sido alvo de muitos estudos devido ao aumento de sua incidência e devido ao fato dessa doença apresentar grande variabilidade na manifestação dos distúrbios entre pacientes e também por ser uma doença que pode permanecer silenciosa durante vários anos antes de provocar complicações tardias mais sérias. Os estudos realizados até o momento foram importantes no sentido de elucidar diversos mecanismos da SM. Entretanto, como se trata de uma doença multifatorial que envolve muitos sistemas endógenos, devido ao fato da doença apresentar múltiplas consequências para a saúde da população e aumentar os gastos públicos para seu tratamento e devido ao fato dos medicamentos atuais apresentarem efeitos colaterais e não serem totalmente eficientes e específicos faz-se necessário o estudo de ferramentas terapêuticas mais eficazes e/ou que tragam prejuízos menores a saúde dos portadores da SM, já que o avanço atual sobre o conhecimento da doença, não foi suficiente para evitar o aumento dos casos observados no Brasil e no mundo.
39
redução do colesterol HDL. Dentre as duas dietas testadas no trabalho publicado de 2013, a hiperlipídica foi a escolhida por induzir SM em ratos, semelhante à que ocorre nos seres humanos. E no presente estudo avaliamos o potencial terapêutico da formulação oral da Ang-(1-7), a ciclodextrina-Ang-(1-7) ou HPβCD/Ang-(1-7) sobre os distúrbios da SM induzida por esta dieta hiperlipídica em ratos.
As ciclodextrinas (CDs) são carboidratos complexos compostos de unidades de
glicose (α-D-glicopiranose) unidas por ligações tipo α-1,4, com estrutura semelhante a
um tronco de cone (Habon, Fritsch e Szejtli, 1984; Backensfeld et al., 1990; Cattaneo e Luong, 1994; Valero, Pérez-Revuelta e Rodríguez, 2003). As CDs são produtos resultantes da degradação do amido pela ação da enzima amilase ciclodextrina glicosil transferase 2 (CGTase 2)produzida pelo microorganismo Bacillus macerans, capaz de romper um segmento da hélice do amido e unir as duas porções terminais deste fragmento numa única molécula cíclica (Bender, 1977; Kobayashi, Kainuma e Suzuki, 1978; Stavn e Granum, 1979). Estas estruturas químicas, chamadas de compostos de inclusão ou ciclodextrinas, permitem o transporte de substâncias passíveis de degradação pelas secreções gástricas e intestinais, preservando, com isso, a estrutura dos componentes nelas inseridos por proteger os sítios catalíticos dos compostos da ação das enzimas digestivas. Atualmente, são encontradas inúmeras aplicações para as ciclodextrinas na indústria de agroquímicos, fragrâncias, alimentícia, farmacêutica, entre outras.
Como todo peptídeo, uma terapia com Ang-(1-7) via oral seria improvável devido a sua degradação no trato gastrointestinal. Nos últimos anos tem sido sugerido que a inclusão de Ang-(1-7) na cavidade do oligossacarídeo hidroxipropil-β -ciclodextrina [hydroxypropyl-β-cyclodextrin; HPβCD/Ang-(1-7)] protege o peptídeo durante a passagem no trato gastrointestinal quando administrado oralmente (Marques et al., 2011). Nesse sentido, a HPβCD/Ang-(1-7) tornou-se um agente em potencial para o tratamento de doenças cardiovasculares e distúrbios metabólicos como a SM (Fraga-Silva et al., 2011; Marques et al., 2011; Marques et al., 2012), devido às diversas evidências científicas já apresentadas que a Ang-(1-7), atuando sem seu receptor MAS, possua efeitos benéficos sobre o metabolismo. Dessa forma, o objetivo do presente estudo foi avaliar o potencial terapêutico da formulação oral da Ang-(1-7), a
HPβCD/Ang-(1-7), sobre a via da insulina e termogênese adapatativa e sobre os
40
pelo fato da HPβCD/Ang-(1-7) ser um medicamento formulado a partir de um peptídeo endógeno, de efeitos colaterais ainda não identificados e associados a diversos efeitos benéficos em diversas doenças, como doenças cardiovasculares e de origem metabólica.
2) OBJETIVOS
2.1) Objetivo Geral
Avaliar o efeito terapêutico da formulação oral de Ang-(1-7), a HPβ CD/Ang-(1-7) na atenuação ou reversão de distúrbios metabólicos da síndrome metabólica, induzidos pela dieta hiperlipídica em ratos.
2.2) Objetivos Específicos
Avaliar glicemia de jejum e a resistência à insulina através do teste de tolerância oral a glicose antes do início dos tratamentos (7ª semana das dietas) e;
Avaliar o efeito terapêutico da HPβCD/Ang-(1-7) sobre:
I. Parâmetros biométricos como peso corporal, índice de Lee e índice de adiposidade e sobre os pesos de fígado, rins, ventrículo esquerdo, músculos (gastrocnêmico e sóleo) e depósitos de tecido adiposo (inguinal, retroperitoneal e epididimal) em ratos com SM induzida por dieta hiperlipídica;
II. Parâmetros bioquímicos como colesterol total, lipoproteína de baixa densidade (LDL), lipoproteína de muito baixa densidade (VLDL), triglicérides, lipoproteína de alta densidade (HDL), alanina aminotransferase (ALT), aspartato aminotransferase (AST), amilase, creatinina, ureia, proteínas totais, albumina e globulina em ratos com SM induzida por dieta hiperlipídica;
41
IV. A expressão gênica das PPARγ2 no fígado e no TAM de ratos com SM
induzida por dieta hiperlipídica;
V. A glicemia de jejum, sobre a resistência à insulina e sobre a expressão gênica de componentes da via intracelular da insulina no tecido adiposo retroperitoneal, fígado, músculo gastrocnêmico e TAM de ratos com SM induzida por dieta hiperlipídica;
VI. Sobre a biogênese mitocondrial e termogênese adaptativa através da expressão gênica do PGC-1α, PGC-1β, UCP-2 e UCP-3 no músculo gastrocnêmico e através da expressão da UCP-1 no TAM β de ratos com SM induzida por dieta hiperlipídica.
3) MATERIAIS E MÉTODOS
3.1) Animais
Foram utilizados ratos Fisher, recém-desmamados, com 4 semanas de idade (40
– 60g) provenientes do Laboratório de Nutrição Experimental (LABNEX/UFOP) da Universidade Federal de Ouro Preto (UFOP, Brasil). Os animais permaneceram em gaiolas individuais e em ciclo claro-escuro 12h – 12h no Centro de Ciência Animal da UFOP (CCA/UFOP). Durante todo o experimento os animais tiveram livre acesso à água e às dietas. Todos os procedimentos foram realizados de acordo com as Diretrizes de Ética em Cuidados de Animais Experimentais. O projeto foi aprovado pelo comitê de ética animal da Universidade Federal de Ouro Preto nº do protocolo 2011/31 (Anexo II)
3.2) Definição das Dietas
42
Composição e conteúdo energético das dietas
Ingredientes (g/Kg)
AIN-93 SM
G
(Jovens)* (Adultos)* M Jovens Adultos
Amido de milho 529,50 620,70 - -
Sacarose 100,00 100,00 - -
Frutose - - 33,0 34,20
Caseina 200,00 140,00 180,50 180,50
Leite condensado - - 316,00 316,00
Oleo de soja 70,00 40,00 - -
Banha de porco - - 370,00 370,00
Fibra (celulose) 50,00 50,00 50,00 50,00
Mix de minerais (AIN-93G-MX)* 35,00 - 35,00 -
Mix de vitaminas
(AIN-93M-MX)* 35,00 - 35,00
Mix de vitaminas (AIN-93G-VX)* 10,00 10,00 10,00 10,00
DL-Metionina 3,00 1,80 3,00 1,80
Cloridrato de colina 2,50 2,50 2,50 2,50
Macronutrientes (% por
peso)
Carboidratos 62,95 72,07 20,68 20,80
Lipídeos 5,00 4,00 39,53 39,53
Proteinas 20,00 14,00 20,26 20,26
Macronutrientes (% Kcal)
Carboidratos 66,82 75,82 15,92 16,00
Lipídeos 11,95 9,46 68,48 68,42
Proteinas 21,23 14,72 15,60 15,58
Kcal/g 3,77 3,80 5,19 5,20
Kj/g 15,78 15,91 21,73 21,77
Adaptado de De Castro U.G. e cols.; 2013
Tabela I: composição da dieta (g/Kg) padrão AIN-93 e da dieta hiperlipídica utilizada para indução da SM; AIN 93 M = dieta controle de manutenção (Reeves, 1997), G=dieta para gestação, amamentação e crescimento. No presente estudo foi utilizada a dieta AIN93-M como dieta padrão e a dieta hiperlipídica SM de animais jovens para indução de características da síndrome metabólica.
3.3) Calculo da Concentração de Ang-(1-7) na HPβCD/Ang-(1-7)
43
da HPβCD/Ang-(1-7) a ser administrada por gavagem diariamente respeitava-se as seguintes razões:
1. Concentração de Ang-(1-7) a ser administrada por rato– 40μg de Ang-(1-7) por 1000g do rato (40μg/Kg)
2. Concentração de Ang-(1-7) inclusa na HPβCD/Ang-(1-7) – Cada 76μg do composto HPβCD/Ang-(1-7) possui 30μg de Ang-(1-7) inclusa ou aproximadamente 0,3947μg de Ang-(1-7) por μg de HPβCD.
3. Diluição – diluía-se a quantidade calculada da HPβCD/Ang-(1-7) em 200μl de água destilada para cada animal
Observações:
Verificava-se diariamente o peso do rato para poder administrar a dose de 40μg
/Kg de Ang-(1-7). Exemplo: em um rato de 300g, administrava-se 12μg de Ang-(1-7), através da pesagem de 30μg do composto HPβCD/Ang-(1-7);
Diluía-se a quantidade calculada individualmente da HPβCD/Ang-(1-7) em 200μl de água destilada por rato, que era dada via gavagem diariamente;
Nos grupos que recebiam tratamento placebo, era dada a mesma quantidade da ciclodextrina vazia via gavagem diariamente.
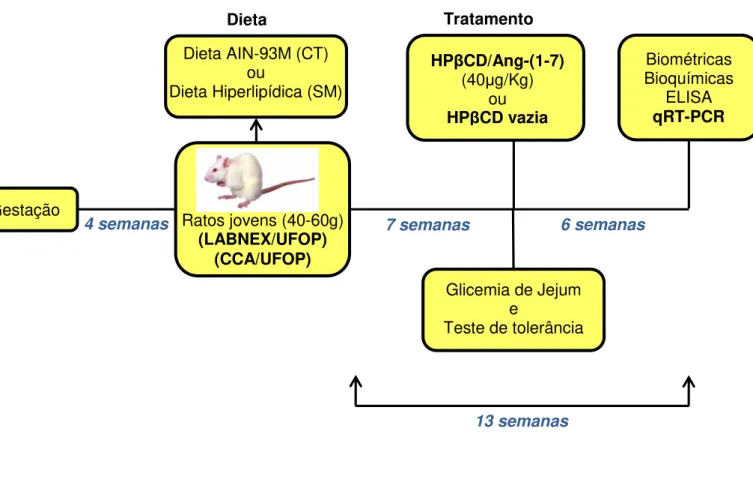
3.4) Protocolo Experimental e Tratamento com HPβCD/Ang-(1-7)
Após sete semanas do consumo das dietas, os animais foram subdivididos em animais que receberam o tratamento diário por gavagem com 40µg/Kg de HPβCD/Ang -(1-7) [CT-Ang--(1-7) ou SM-Ang--(1-7)] ou animais que foram tratados com veículo (V), que consiste na ciclodextrina sem Ang-(1-7) dentro (CT-V ou SM-V) durante as seis semanas finais das dietas. Após esses procedimentos, quatro grupos distintos foram definidos no presente estudo:
1) CT – V: Submetido à dieta padrão (AIN-93M) e que recebeu tratamento placebo (V) com ciclodextrina vazia por gavagem;