Aplicação de funcionais não locais da densidade a sólidos, superfícies e agregados
125
0
0
Texto
Imagem
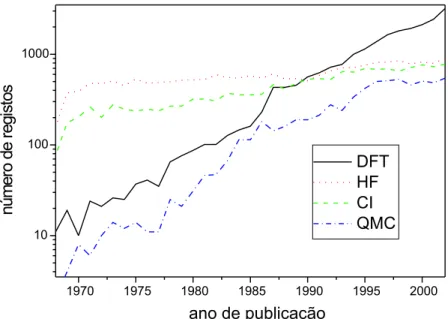



+7
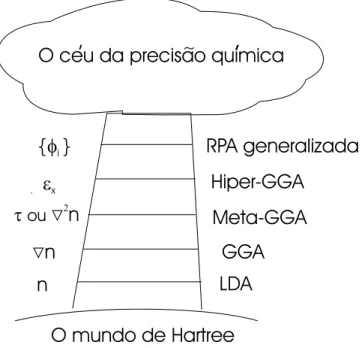
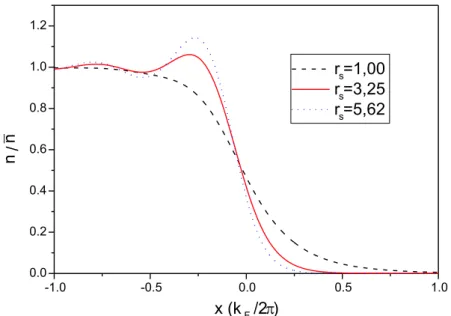
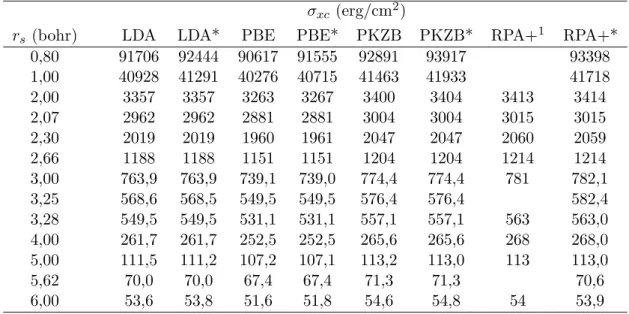


Documentos relacionados