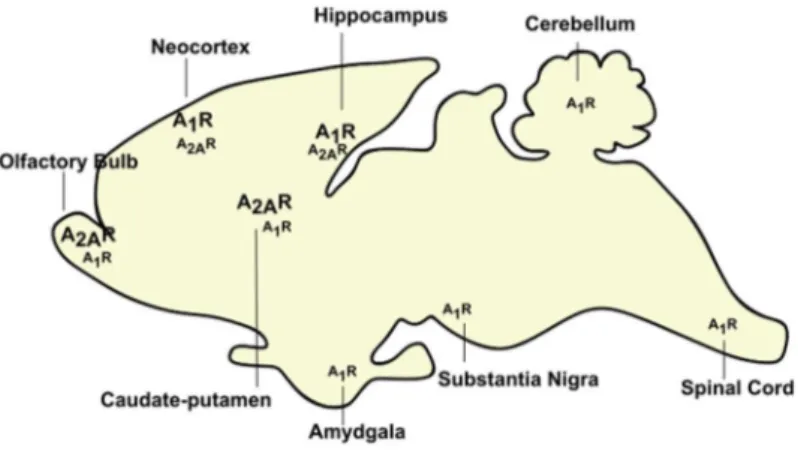
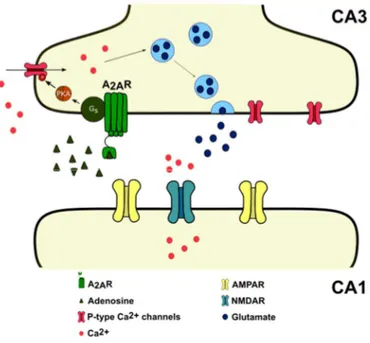
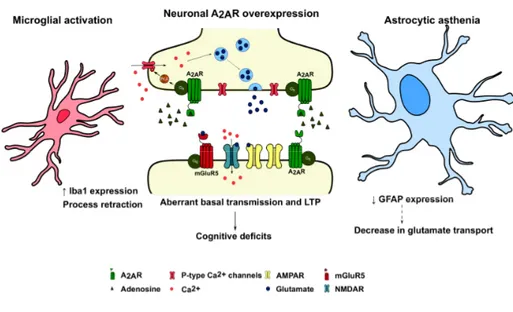
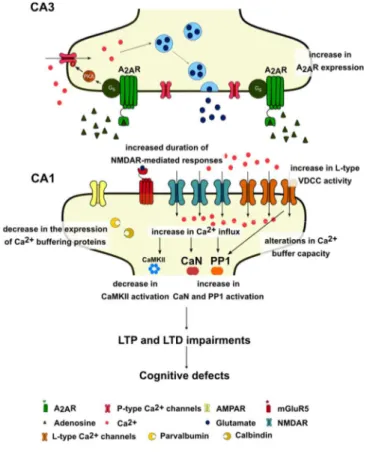
The role of A2A receptors in cognitive decline : decoding the molecular shift towards neurodegeneration
235
0
0
Texto
Imagem
+7
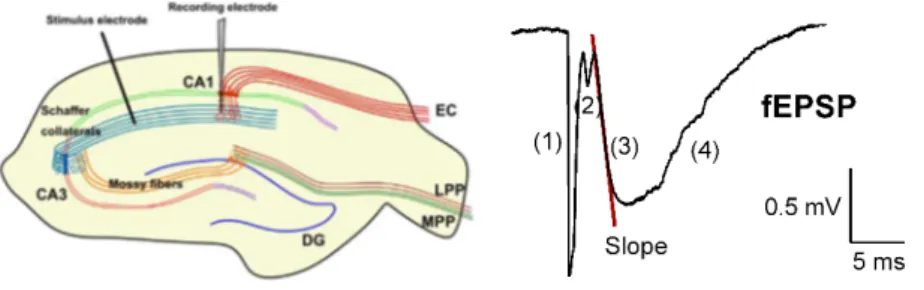
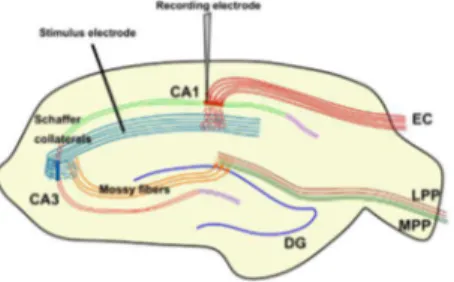
Documentos relacionados