A Bioenergética do Músculo Esquelético na Doença
de Alzheimer
Dissertação de mestrado em
BIOLOGIA CLÍNICA LABORATORIAL
Marisa Alexandra Torres de Castro
Orientador Doutor Romeu António Videira
Co-orientadora: Prof. Doutora Maria Manuel Oliveira
Composição do Júri:
__________________________________________
__________________________________________
__________________________________________
Instituição Universidade de Trás-os-Montes e Alto Douro Curso Mestrado em Biologia Clínica Laboratorial
Título “A Bioenergética do músculo esquelético na Doença de Alzheimer”
Autor Marisa Alexandra Torres de Castro Orientador Doutor Romeu António Videira Co-orientador Prof. Doutora Maria Manuel Oliveira
Este trabalho foi suportado pela Fundação para a Ciência e Tecnologia (FCT), FEDER e COMPETE, bolsa de investigação PTDC/SAU-NMC/115865/2009.
“Este trabalho foi expressamente elaborado como dissertação original para o efeito de obtenção do grau de Mestre em Biologia Clínica Laboratorial”
Agradecimentos
Esta dissertação não representa apenas o resultado de extensas horas de estudo, reflexão e trabalho durante as diversas etapas que a constituem. É igualmente o culminar de um objectivo académico a que me propus e que não seria possível sem um número considerável de pessoas.
Ao Doutor Romeu Videira, possibilitando a realização desta dissertação e sugestão de um tema actual e pertinente que me motivou. A sua receptividade e acompanhamento foram relevantes, assim como críticas, correcções e sugestões durante a orientação.
À co-orientadora, Professora Doutora Maria Manuel Oliveira, pela competência científica e orientação dada, bem como pela disponibilidade e amizade.
Ao Professor Doutor Francisco Peixoto, pela cooperação e conhecimentos transmitidos, através do isolamento mitocondrial de músculo-esquelético de murganhos. Criando condições indispensáveis à concretização da parte experimental desta dissertação.
À mestre Vera Cardoso, Joana Faria, pela participação e entreajuda que demostraram ao longo do tempo, não menos importantes para o meu trabalho. Sem esquecer os bolseiros, com quem ao longo desta jornada pude sempre contar e os meus amigos, pela amizade e carinho sempre demonstrado, assim como palavras de incentivo durante esta etapa.
Ao Centro de Ciência Animal e Veterinária (CECAV) e Centro de Química (CQ) pelas condições indispensáveis à concretização da parte experimental desta dissertação, sem esquecer os demais profissionais que contribuíram de alguma forma para este trabalho.
À Fundação para a Ciência e Tecnologia (FCT), FEDER e COMPETE pelo financiamento ao projecto PTDC/SAU-NMC/115865/2009, do qual esta dissertação emergiu.
Ao meu namorado, André Pinto, agradeço o contributo, motivação, apoio, compreensão e carinho que sempre me dedicou ao longo desta etapa.
Por fim, mas não menos importante, agradeço à minha família, nomeadamente aos meus pais, Rui e Regina e à minha irmã, Cláudia e seu amor incondicional essencial para suportar esta etapa.
Trabalhos apresentados com base nesta dissertação
Faria, J., Castro, M., Cardoso, V., Peixoto, F., Oliveira, M.M., Videira, R. A. (2012) Alzheimer's disease beyond the brain: the liver and skeletal muscle dysfunction. XVIII Encontro Luso-Galego de Química. 28-30 Novembro, Vila Real, Portugal.
Joana Faria and Marisa Castro have similar contribution to the work (Comunicação por poster).
Aceite para publicação/apresentação - Castro M, Monteiro-Cardoso V F, Peixoto F, Oliveira MM & Videira RA. Age-dependent skeletal muscle dysfunction in triple-transgenic mice model of Alzheimer’s disease. 47th Annual Scientific Meeting of the European Society for Clinical Investigation, Albufeira, Portugal 17 – 20 April 2013 (Poster). Resumo a ser publicado em: “The European Journal of Clinical Investigation”.
Resumo
A doença de alzheimer (AD) é a doença neurodegenerativa mais comum, que afecta milhões de pessoas em todo mundo. O cérebro de pacientes com AD é caracterizado por neurodegeneração, com perda de sinapses e neurónios, incluindo atrofia e depleção do sistema colinérgico. Considerando a relevância da acetilcolina na regulação da actividade muscular, a disfunção colinérgica cerebral pode estender-se à doença de Alzheimer como disfunção fisiológica no músculo esquelético. Utilizando murganhos triplo-transgénicos (3xTg-AD) com 3, 6 e 12 meses de idade como modelo para a doença de Alzheimer e murganhos Wilde
Type (WT) com idades equivalentes como controlo, analisámos o impacto fisiológico da
progressão da doença de Alzheimer na actividade do músculo esquelético, considerando quatro aspectos: i) os efeitos progressivos sobre a actividade da Acetilcolinesterase no músculo-esquelético; ii) o papel do stresse oxidativo nas células musculares, avaliando as actividades da Superóxido Dismutase e Catalase e o conteúdo total de tióis nas proteínas; iii) as perturbações na actividade bioenergética mitocondrial no músculo esquelético avaliando a actividade das enzimas mitocondriais (complexos I, II e IV e F0F1-ATPase ); iv) os efeitos no perfil de ácidos gordos da membrana mitocondrial, analisados por GC / MS.
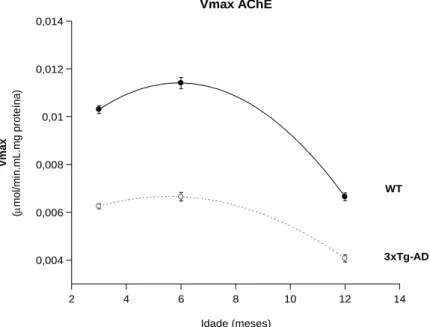
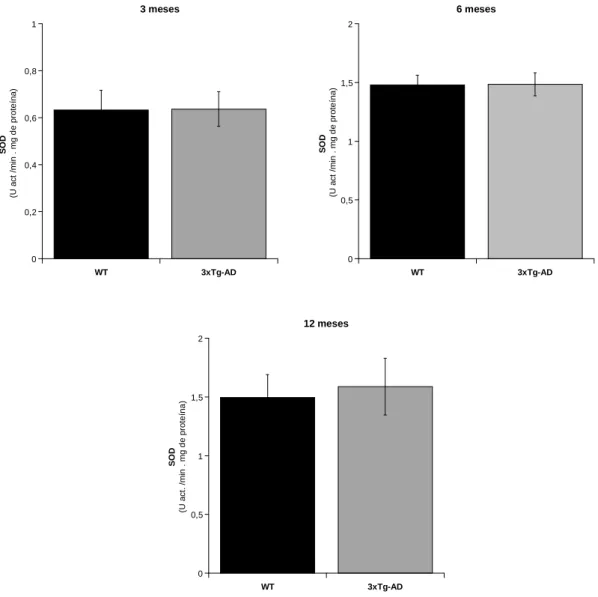
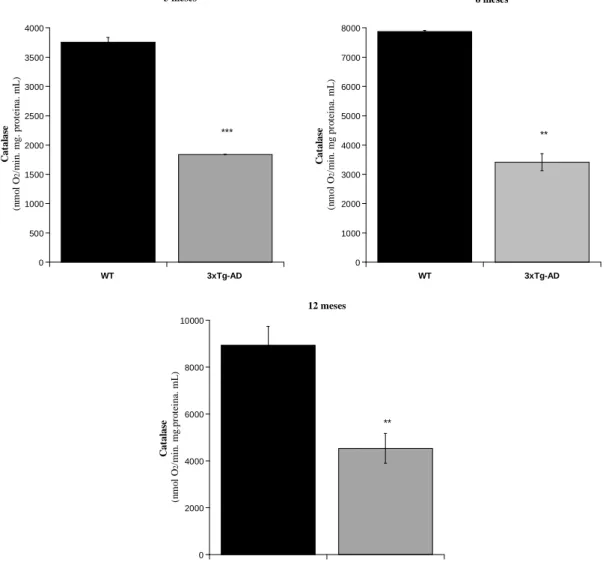
Redução significativa da actividade da Acetilcolinesterase no músculo esquelético é detectada nos modelos 3xTg AD com 3, 6 e 12 meses de idade, sugerindo restrições em estágios iniciais na regulação da actividade muscular. Além disso, a redução significativa da actividade da Catalase, dependente da idade, foi detectada em homogeneizados de músculo esquelético 3xTg-AD, sem alterações na actividade da Superóxido Dismutase e no conteúdo total de tióis nas proteínas. Embora o envelhecimento não induza diferenças na actividade das enzimas mitocondriais de murganhos 3xTg-AD em relação às determinadas nos murganhos WT; aos 6 meses de idade, diferenças significativas no perfil mitocondrial de ácidos gordos 3xTg-AD são detectadas com uma grande acumulação de ácido tri-hidróxidocosahexanóico. Estes dados mostram que o fenótipo da doença de Alzheimer contribui para perturbações na actividade do músculo esquelético, detectado por alterações nas actividades da Acetilcolinesterase e Catalase.
Palavras-chave: Doença de Alzheimer; Músculo esquelético; Bioenergética mitocondrial; Acetilcolinesterase; Stresse oxidativo.
Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disease, affecting millions of people worldwide. The brains of patients with AD are characterized by neurodegeneration, loss of synapses and neurons, including atrophy and depletion of cholinergic system. Considering the relevance of acetylcholine in the regulation of muscle activity, the cerebral cholinergic dysfunction may extend Alzheimer's disease physiological dysfunction to skeletal muscle.
Using 3, 6 and 12 months-old triple-transgenic (3xTg-AD) and wild-type (WT) mice, we analyzed the physiological impact of Alzheimer disease progression on skeletal muscle activity, considering: i) effects on skeletal muscle Cholinesterase activity; ii) effects on redox state of muscle cells, evaluating Superoxide Dismutase and Catalase activities and protein thiol groups content; iii) perturbations on skeletal muscle mitochondrial bioenergetics activity, evaluating individual mitochondrial enzymes (complexes I, II and IV and F0F1 -ATPase); iv) effects on mitochondrial fatty acid profiles, analysed by GC/MS.
Significant reduction of skeletal muscle Acetylcholinesterase activity is detected in 3, 6 and 12 months-old 3xTg-AD models, suggesting early restraints in the regulation of muscle activity. Additionally, significant and age-dependent reduction of Catalase activity was detected in 3xTg-AD skeletal muscle tissue without alterations in Superoxide Dismutase activity and redox status of protein thiol groups. Although aging does not induce differences in 3xTg-AD mitochondrial enzymes activity relating WT condition; at 6 months-old significant differences are detected in 3xTg-AD mitochondrial fatty acid profile with large accumulation of trihydroxy-docosahexaenoic acid. These data show that Alzheimer´s disease phenotype contributed to skeletal muscle activity disruption, mainly by skeletal muscle acetylcholinesterase and catalase activities changes.
Keywords: Alzheimer Disease; Skeletal muscle; Mitochondrial bioenergetics; Acetylcholinesterase; Oxidative stress.
Índice
geral
Agradecimentos ... vi
Trabalhos apresentados com base nesta dissertação ... vii
Resumo ... viii
Abstract ... ix
Índice geral ... x
Índice de figuras ... xii
Abreviaturas ... xiv
I-Introdução ... 1
Doenças neurodegenerativas ... 2
A Doença de Alzheimer ... 2
Desregulação do cálcio na doença de Alzheimer ... 5
Bioenergética e a doença de Alzheimer ... 7
O músculo esquelético e a doença de Alzheimer ... 9
Modelos para a doença de Alzheimer ... 11
A necessidade de novos marcadores para detectar a progressão da doença de Alzheimer .. 13
II-Objectivos ... 16
III-Material e métodos ... 18
1-Modelo animal para estudar a doença de Alzheimer ... 19
2-Preparação do material biológico ... 19
2.1 Preparação dos homogeneizados de músculo esquelético ... 19
2.2 Isolamento de mitocôndrias de músculo esquelético ... 20
2.3 Determinação da concentração da proteína nas amostras biológicas ... 21
3-Estudos no sobrenadante nos homogeneizados dos músculos esqueléticos ... 22
3.1 Determinação da actividade da Acetilcolinesterase (AChE) ... 22
3.2 Determinação da actividade da Superóxido dismutase (SOD) ... 22
3.3 Determinação da actividade da Catalase ... 23
3.4 Determinação do conteúdo total de tióis nas proteínas ... 23
4-Determinação da actividade das enzimas mitocondriais ... 24
4.1 NADH:ubiquinona oxidoreductase (complexo I) ... 24
4.2 Succinato-coenzima Q redutase (complexo II) ... 25
4.4 F0F1-ATPase ... 27
4.5 Citrato sintase ... 27
5-Lipidómica... 28
5.1 Extracção de lípidos das mitocôndrias do músculo esquelético ... 28
5.2 Quantificação do fosfato dos extractos lipídico ... 28
5.3 Preparação dos ésteres metílicos dos ácidos gordos ... 29
5.4 Identificação e quantificação dos ésteres metílicos dos ácidos gordos por cromatografia gasosa/espectrometria de massa (GC/MS) ... 30
6-Tratamento estatístico... 30
IV-Resultados e Discussão ... 31
1-Estudos no sobrenadante dos homogeneizados dos músculos esqueléticos ... 31
1.1 Efeitos da produção da doença de Alzheimer na actividade da Acetilcolinesterase do músculo esquelético ... 31
1.2 Efeitos da progressão da doença de Alzheimer nas actividades da SOD e Catalase e no conteúdo total de tióis nas proteínas: parâmetros relacionados com stresse oxidativo 36 2-Estudos nas mitocôndrias isoladas dos músculos esqueléticos ... 41
2.1 Efeitos da progressão da doença de Alzheimer na actividade dos sistemas enzimáticos relacionados com a bioenergética mitocondrial ... 41
3-Lipidómica... 48
3.1 Efeitos da progressão da doença de Alzheimer no perfil de ácidos gordos dos fosfolípidos isolados das mitocôndrias do músculo esquelético ... 48
V-Conclusão ... 55
Índice de figuras
Figura 1.1 Comparação entre o cérebro de um indivíduo saudável (A) e o cérebro de um
doente de Alzheimer (B). ... 3
Figura 1.2 Representação esquemática da localização de placas senis e tranças neurofibrilares ... 4
Figura 1.3 Esquema elucidativo da desregulação da homeostase do Ca2+ e proteínas relacionadas com a AD. ... 6
Figura 1.4 Duplo papel da mitocôndria: produção de ATP e produção de radicais livres ... 7
Figura 1.5 Esquema elucidativo da junção neuromuscular.. ... 10
Figura 1.6 Murganho triplo-transgénico (3xTg-AD) ... 13
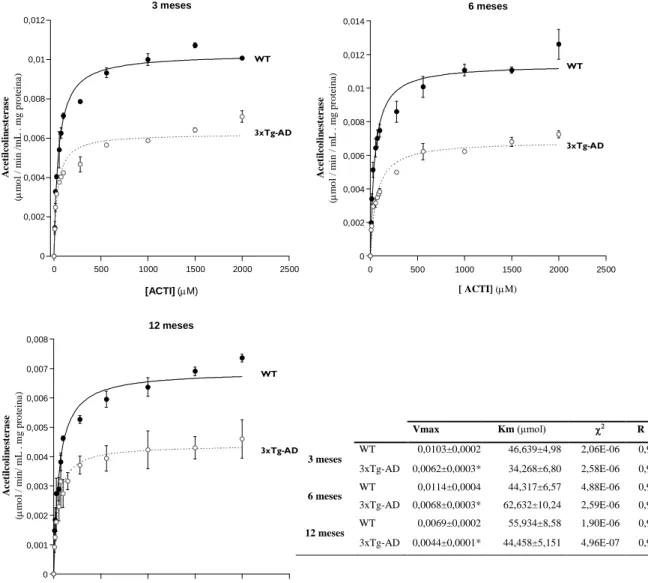
Figura 4.1 Actividade da acetilcolinesterase no músculo esquelético de murganhos WT e 3xTg-AD em função do substrato (ACTI) para 3, 6 e 12 meses de idade. ... 34
Figura 4.2 Velocidades máximas, obtido a partir da cinética de Michaelis-Menten, em função da idade. ... 35
Figura 4.3 Actividade da SOD avaliada em tecido do músculo esquelético de murganhos 3xTg-AD e WT com idades de 3, 6 e 12 meses. ... 37
Figura 4.4 Actividade da catalase avaliada em tecido do músculo de murganhos 3xTg-AD e WT com idades de 3, 6 e 12 meses. ... 38
Figura 4.5 Comparação da actividade da catalase, obtida entre os grupos de estudo (3XTg-AD e WT) em função da idade. ... 39
Figura 4.6 Conteúdo total de tióis nas proteínas avaliado em homogeneizado de tecido do músculo esquelético de murganhos 3xTg-AD e WT com idades de 3, 6 e 12 meses. ... 40
Figura 4.7 Actividade individual dos complexos enzimáticos da cadeia transportadora de electrões I, II, IV e da F0F1-ATPase avaliada em mitocôndrias isoladas de músculo esquelético de murganhos WT e 3xTg-AD com idade de 3 meses, normalizada pela actividade da Citrato sintase. ... 43
Figura 4.8 Actividade individual dos complexos enzimáticos da cadeia transportadora de electrões I, II, IV e da F0F1-ATPase avaliada em mitocôndrias isoladas de músculo esquelético de murganhos WT e 3xTg-AD com idade de 6 meses, normalizada pela actividade da Citrato sintase. ... 44 Figura 4.9 Actividade individual dos complexos enzimáticos da cadeia transportadora de electrões I, II, IV e da F0F1-ATPase avaliada em mitocôndrias isoladas de músculo esquelético de murganhos WT e 3xTg-AD com idade de 12 meses, normalizada pela actividade da Citrato sintase. ... 45 Figura 4.10 Actividade da Citrato sintase avaliada em mitocôndrias isoladas de músculo esquelético de murganhos WT e 3xTg-AD com idade de 3, 6 e 12 meses. ... 47 Figura 4.11 Cromatogramas e a abundancia relativa dos ésteres metílicos dos ácidos gordos obtidos a partir de mitocôndrias do músculo-esquelético dos animais WT e 3xTg-AD com 3 meses de idade. ... 49 Figura 4.12 Cromatogramas e a abundancia relativa dos ésteres metílicos dos ácidos gordos obtidos a partir de mitocôndrias do músculo-esquelético dos animais WT e 3xTg-AD com 6 meses de idade. ... 50 Figura 4.12.1 Cromatograma e espectros de massa relativa dos ésteres metílicos dos ácidos gordos obtidos a partir de mitocôndrias do músculo-esquelético dos animais 3xTg-AD com 6 meses de idade. ... 52 Figura 4.13 Cromatogramas e a abundancia relativa dos ésteres metílicos dos ácidos gordos obtidos a partir de mitocôndrias do músculo-esquelético dos animais WT e 3xTg-AD com 12 meses de idade. ... 54
Abreviaturas
Acetil-CoA – Acetilcoenzima A AChE – Acetilcolinesterase
ACTI - Iodeto de acetiltiocolina (acetylthiocholine iodide)
AD – Doença de Alzheimer (Alzheimer disease) ADP – Adenosina difosfato
ATP – Adenosina trifosfato
APP – Proteína precursora amilóide (Amyloid precursor protein) BSA – Albumina sérica bovina (Bovin Seric Albumin)
Complexo I – NADH ubiquinona oxireductase Complexo II – Succinato coenzima Q reductase Complexo IV – Citocromo c oxidase
DCIP – 2,6 – diclorofenolindofenol
DNA – Ácido desoxirribonucleico (Deoxyribonucleic acid)
DTNB – Ácido 5,5-ditiobis-(2-nitrobenzóico) (Ellman’s Reagent,
5,5’-dithiobis-(2-nitrobenzoic acid))
EDTA – Ácido etienodiamina tetra-acético (Ethylenediamine tetraacetic acid) EGTA – Ácido etilenoglicol tetra-acético (Ethylene glycol tetraacetic acid) EI – Impacto electrónico
GC/MS – Cromatografia gasosa / espectrometria de massa (Gas chomatography/Mass
spectrometry)
IP3 –1,4,5 - trifosfato inositol
Km – Constante de Michaelis Menten
mtDNA – Ácido desoxirribonucleico mitocondrial (mitochondrial Deoxyribonucleic acid) NADH – Nicotinamida adenina dinucleótido (Nicotinamide adenine dinucliotid)
NBT – Cloreto de azul de nitrotetrazólio (Nitro blue tetrazolium) PI – Padrão interno
PS1 – Presenilina 1 PS2 – Presenilina 2
RE – Retículo Endoplasmático
ROS –Espécies reactivas de Oxigénio (Reactive Oxygen Species) RT – Tempo de retenção
SERCA – ATP, Ca2+ - ATPase
SDS – Dodecilsulfato de sódio (Sodium dodecyl sulfate) SOD – Superóxido dismutase
SNC – Sistema Nervoso Central
TMPD – tetramethyl-p-phenylenediamine Vmáx – Velocidade máxima
“Imaginar é mais importante que saber, pois o conhecimento é limitado enquanto a imaginação abraça o Universo”
Doenças
neurodegenerativas
O largo espectro de doenças neurodegenerativas constitui um dos principais problemas de saúde pública das sociedades ocidentais, pois a sua incidência continua a crescer promovendo a degradação progressiva da qualidade de vida de um número cada vez maior de pessoas apesar dos investimentos crescentes em cuidados de saúde e em investigação que vários governos têm mobilizado para o seu tratamento. Entre os diversos distúrbios neurodegenerativos destacam-se a doença de Alzheimer, a doença de Parkinson, a Esclerose múltipla e a Esclerose lateral amiotrófica pela sua incidência e gravidade (Krieger, 2002). Os mecanismos fisiopatológicos subjacentes ao aparecimento e/ou à progressão destas doenças ainda não estão bem esclarecidos e consequentemente todas elas carecem de tratamento efectivo. A disfunção mitocondrial parece ser um atributo comum aos vários processos neurodegenerativos (Krieger, 2002; Moreira, 2010) apesar da disfunção neuronal e a morte celular subsequente serem, normalmente explicadas por mecanismos diferentes em diferentes neuropatologias (Coskun, 2011).
A Doença de Alzheimer
A doença de Alzheimer (AD), descrita pela primeira vez por Alois Alzheimer em 1906 como uma doença neurológica progressiva, é considerada a forma mais comum de demência. Durante algum tempo foi considerada uma patologia rara, no entanto estudos mais recentes mostram que entre pessoas com idades iguais ou superiores aos 80 anos têm uma a prevalência de cerca de 40%. Actualmente, há cerca de 36 milhões de pessoas em todo o mundo (cerca de 90 000 em Portugal) que sofrem desta doença e, prevê-se que este valor quadruplique até 2050 (Atamna, 2007).
O aumento da idade tem sido reconhecido como o principal factor de risco, apesar de alterações fisiológicas ao nível da vasculatura cerebral serem também, inúmeras vezes, referidas como factores de risco (Alieva, 2004). Modificações neuroquímicas primárias no sistema colinérgico têm sido observadas em cérebros de doentes de Alzheimer, sugerindo que a disfunção colinérgica está envolvida nas alterações de memória, aprendizagem, atenção e de outros processos cognitivos afectados em pacientes. Alterações tanto na densidade de receptores muscarínicos como nos processos de sinalização intracelular induzida por estes receptores têm sido detectadas em diversas regiões cérebro de doentes com Alzheimer
(Francis, 1999). Adicionalmente, concentrações elevadas de uma proteína denominada inibidor endógeno de baixo peso molecular (IEBP) dos receptores muscarínicos foram detectadas por vários investigadores. A IEBP proporciona um antagonismo endógeno aos receptores colinérgicos muscarínicos que parece estar relacionado com as manifestações clínicas observadas na neuropatologia degenerativa (Cummings, 1998).
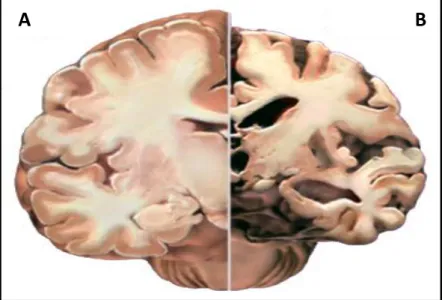
Do ponto de vista clínico, a doença de Alzheimer é caracterizada por perda severa de memória e pela redução das capacidades cognitivas, que nos estágios avançados evolui para uma perda do controlo sobre as funções fisiológicas (Mansur, 2005; Moreira, 2010). Histopatologicamente, a doença de Alzheimer é caracterizada pela diminuição do volume cerebral resultante da perda de neurónios e sinapses (figura 1.1), pela presença extracelular de placas senis, constituídas principalmente pela proteína β-amilóide e por tranças neurofibrilares intracelulares que contêm a proteína Tau hiperfosforilada (Cassano, 2012).
Figura 1.1 Comparação entre o cérebro de um indivíduo saudável (A) e
o cérebro de um doente de Alzheimer (B). A AD afecta as regiões do cérebro envolvidas nos processos de memória e aprendizagem, pelo que em (B) detecta-se uma diminuição significativa do volume dos lóbulos temporais e frontal. Adaptado de Neuroscience News (2012).
As regiões cerebrais envolvidas no processo de memória e aprendizagem, como o hipocampo e o córtex, são as regiões mais afectadas. Dado que muitos dos sintomas apresentados pelos pacientes da doença de Alzheimer (e.g. a perda de memória) são comuns a outras formas de demência, o diagnóstico definitivo só pode ser realizado em análises
Post-mortem, detectando e/ou quantificando a presença de placas senis e de tranças neurofibrilares
por ensaios de imunohistoquímica (Aiyaz, 2012). Cerca de 2% dos casos são familiares, sendo associadas a mutações hereditárias nos genes que codificam a proteína precursora
N ormal
A
B
amilóide (APP) no cromossoma 21, da presenilina 1 (PS1) no cromossoma 14 e da presenilina 2 (PS2) no cromossoma 1 (Moreira, 2010; Cassano, 2012).
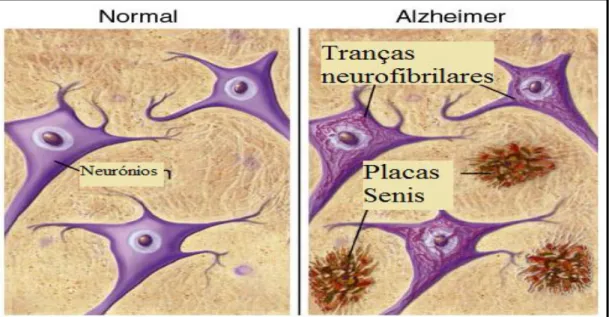
Vários estudos indicam que a produção e acumulação intraneuronal da proteína β-amilóide precede a formação extracelular de placas senis bem como a formação de tranças neurofibrilares intracelulares (figura 1.2) quer nos cérebros dos pacientes com doença de Alzheimer quer em pessoas com síndrome de Down (Gyure, 2001). De facto, a β-amilóide é o maior componente das placas senis, também conhecidas como placas neuríticas, encontradas principalmente em regiões do cérebro responsáveis pela aprendizagem e memória. A β-amilóide é um produto de clivagem da proteína membranar percursora da β-β-amilóide (APP) por proteólises sequenciais da β-secretase e γ-secretase. A clivagem pela γ-secretase pode gerar duas formas distintas de β-amilóide: uma formada por um polipeptídeo com 40 resíduos de aminoácidos e outra maior, composta por 42 resíduos de aminoácidos. O polipeptídeo maior da β-amilóide é considerado tóxico, tendo a capacidade de auto-organização em oligómeros, promovendo a fibrilogenesis e a formação das placas senis. Assim, os níveis de β-amilóide no cérebro de pacientes com Alzheimer são controlados pela sua produção, remoção e degradação (Reddy, 2011).
Figura 1.2 Representação esquemática da localização de placas senis e tranças neurofibrilares,
adaptado de American Health Assistance Foundation (2012)
Outra importante característica fisiopatológica encontrada no tecido cerebral de pacientes com doença de Alzheimer são as tranças neurofibrilares intracelulares, compostas principalmente pela proteína Tau hiperfosforilada. A proteína Tau, em condições normais, é específica dos neurónios e desempenha um papel importante na estabilização dos
microtúbulos, os quais são responsáveis pela manutenção do funcionamento e da estrutura neuronal. Adicionalmente, ela também tem um papel importante na regulação da actividade motora dos microtúbulos, na transdução de sinais para o citoesqueleto celular e em outros processos dependentes da activação da fosfolipase C (Jenkins, 1999; Brandt, 2005). A proteína Tau tem mais de 30 zonas (grupos) susceptíveis de serem fosforiladas. Os processos de fosforilação/desfosforilação regulam a actividade desta proteína e consequentemente a montagem e dinâmica dos microtúbulos (Wang, 2008). Foi demostrado que a capacidade da Tau se ligar e estabilizar os microtúbulos é drasticamente reduzida quando hiperfosforilada (Lindwall, 1984), preferindo, nesta situação, formar filamentos helicoidais pareados que se agregam em emaranhados neurofibrilares como os detectados no cérebro de pacientes com doença de Alzheimer (Reddy, 2011). De facto, a densidade dos emaranhados neurofibrilares nos cérebros de pacientes com Alzheimer está relacionada com a gravidade da doença (Mohs, 2002). No entanto, os emaranhados neurofibrilares não são específicos da doença de Alzheimer dado que também são detectados noutras desordens neurodegenerativas, incluindo a doença de Parkinson pós-encefalítica e encefalites esclorosantes subagudas (Selkoe, 2004). Assim, outros factores como stresse oxidativo, processos inflamatórios, disfunção mitocondrial e a desregulação dos fluxos de cálcio ao nível dos neurónios têm sido também implicados na perda progressiva de sinapses e de neurónios característica da doença de Alzheimer (Mistur, 2009).
Desregulação do cálcio na doença de Alzheimer
A desregulação da homeostase iónica tem sido implicada na patologia da doença de Alzheimer, constituindo a base de uma popular hipótese formulada em 1994 para explicar a doença (Khachaturian, 1994). De acordo com esta hipótese, a acumulação progressiva de alterações na homeostase do cálcio seria uma causa proximal nas doenças neurodegenerativas, incluindo a doença de Alzheimer (Stutzmann, 2005).
O movimento do Ca2+ no retículo endoplasmático (RE) é controlado por várias proteínas localizadas no seu interior em resposta a estímulos ambientais. O gradiente de Ca2+ é mantido por uma bomba dependente de ATP, Ca2+- ATPase (SERCA) localizada na membrana do RE. É necessário uma activação eléctrica, sináptica ou mediada por receptor para que o cálcio seja libertado do RE via dois tipos de canais (receptor IP3 e de rianodina) localizados na membrana que permitem a libertação rápida do ião (Mattson, 2000).
A descoberta da existência de um metabolismo associado à APP levou a uma incessante procura do efeito dos seus fragmentos. Estudos mostram que agregados de β-amilóide alteram a sinalização do cálcio e quando adicionados a células em meios de cultura, os níveis de Ca2+ no citosol são alterados (Goodman, 1994). Uma alteração no metabolismo do Ca2+, normalmente regulado, pode causar uma variedade de efeitos secundários como a activação de enzimas celulares, a activação da apoptose e a modificação do citoesqueleto. Foi também referido que a β-amilóide desencadeia a libertação de Ca2+
através do RE, mediado tanto por IP3 como por rianodina (Ferreiro, 2004). A β-amilóide diminui a actividade da enzima Ca2+- ATPase, que desempenha um papel importante no controlo de cálcio no citosol (Berrocal, 2009).
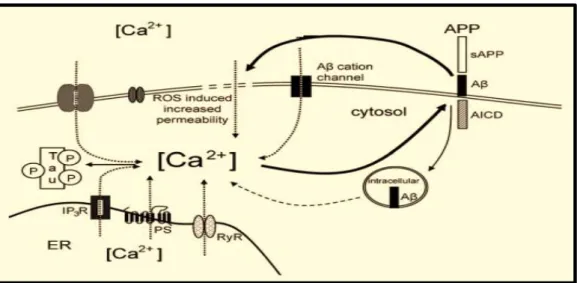
Figura 1.3 Esquema elucidativo da desregulação da homeostase do Ca2+ e proteínas relacionadas com a AD. O processamento da APP resulta não fluxo de iões Ca2+ para dentro da célula através da formação de canais selectivos a catiões, aumento da permeabilidade da membrana induzido por ROS. Elevada concentração do ião aumenta fosforilação da Tau (Bojarski, 2008).
Outro mecanismo pelo qual a β-amilóide interactua com a homeostase intracelular do Ca2+ relaciona-se com a sua capacidade para a formação de ROS, que podem induzir peroxidação lipídica na membrana (Hensley, 1994), promovendo alterações nas propriedades membranares, o que modifica as funções de transportadores e canais iónicos que conduzem ao aumento dos níveis do ião Ca2+, como ilustra a figura 1.3(Keller, 1997).
Os estudos relativos à alteração da homeostase do Ca2+ não permitem afirmar se as suas alterações são a chave para o desencadear da doença de Alzheimer, apesar de não existirem dúvidas que os distúrbios na homeostase do Ca2+ ocorrem no princípio da patologia, antes dos primeiros sintomas clínicos. Estratégias terapêuticas, destinadas à correcção da
desregulação da concentração do ião Ca2+ podem reduzir a progressão da doença (Bojarski, 2008).
Bioenergética e a doença de Alzheimer
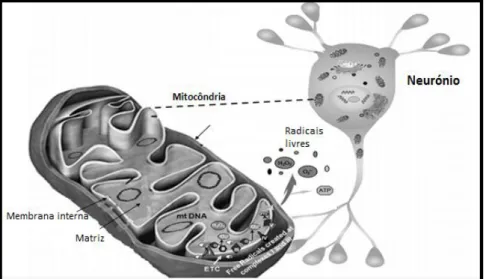
A doença de Alzheimer é acompanhada por um decréscimo na expressão e na actividade de várias enzimas mitocondriais, com consequente comprometimento do funcionamento da cadeia transportadora de electrões e da síntese de ATP (Yao, 2009). A mitocôndria é essencial para a função neuronal, pois a limitada capacidade glicolítica destas células torna-as altamente dependentes da fosforilação oxidativa aeróbia para satisfazer as suas necessidades energéticas. Contudo, a fosforilação oxidativa é também a maior fonte endógena de radicais livres (Moreira, 2010).
Figura 1.4 Duplo papel da mitocôndria: produção de ATP e produção de
radicais livres, adaptado de Krieger (2002)
O sistema nervoso central (SNC) é particularmente vulnerável ao ataque por radicais livres por várias razões, nomeadamente: i) está permanentemente exposto a concentrações relativamente elevadas de oxigénio, consumindo cerca de 20% da energia metabólica do organismo; ii) as membranas das suas células possuem um elevado conteúdo de ácidos gordos polinsaturados, os quais são muito susceptíveis ao ataque por radicais livres; iii) apresenta um sistema antioxidante menos activo do que outros tecidos, evidenciado pela baixa actividade da catalase e por uma actividade moderada da glutationa peroxidase e da superóxido dismutase; iv) algumas regiões do SNC apresentam elevadas concentrações de ferro e ascorbato, os quais tem a capacidade de promover a formação de radicais livres através da reacção de
Fenton/Haber Weiss (Moreira, 2010). Quando a geração de radicais livres supera a capacidade de supressão desses radicais pelo sistema celular de defesas antioxidante ocorre dano em múltiplas biomoléculas da célula, denominado genericamente por stresse oxidativo. Nos pacientes com a doença de Alzheimer, os danos celulares promovidos pelo stresse oxidativo estão associados à desregulação da homeostase do cálcio e à morte de populações vulneráveis de neurónios por apoptose (Yao, 2009).
Problemas ao nível do funcionamento de várias enzimas mitocondriais como o complexo piruvato desidrogenase, citocromo c oxidase e complexo α-cetoglutarato desidrogenase têm sido associados à doença de Alzheimer. Por exemplo, no córtex cerebral de pacientes com doença de Alzheimer, a actividade da enzima citocromo c oxidase (complexo IV da cadeia respiratória) apresenta menor actividade do que na população controlo (Gibson, 1998). Anomalias na estrutura e função da mitocôndria, incluindo danos no mtDNA, foram detectadas não só nas células do sistema nervoso central mas também nas células de vários tecidos periféricos de pacientes com a doença de Alzheimer (Schapira, 2012). Todavia, a sua relevância para o aparecimento e/ou progressão da doença permanece controversa e os resultados nem sempre são reprodutíveis (Schapira, 2012). Outras descobertas, como a que mostra uma relação positiva entre o risco de contrair a doença de Alzheimer e o polimorfismo do gene TOMM40 (2 kilobases de distancia do gene APOE 4 no cromossoma 4), também apoiam o envolvimento da disfunção mitocondrial na doença de Alzheimer. TOMM40 é uma proteína que se localiza na membrana externa da mitocôndria e que está envolvida no poro que permite a importação de proteínas citoplasmáticas para a mitocôndria, o qual permite também a acumulação da β-amilóide (Devi, 2006). As PS1, PS2 e γ-secretase encontram-se também associadas à membrana mitocondrial, em particular nas regiões de junção entre o retículo endoplasmático e a mitocôndria (Area-Gomez, 2009). Esta região membranar inter-organelos também possui um papel importante no metabolismo de lípidos, incluindo na síntese e transporte de fosfatidiletanolaminas, classe de fosfolípidos maioritária nas mitocôndrias que desempenha também um papel importante na fosforilação da Tau. Adicionalmente contem a enzima acil-CoA colesterol aciltransferase, uma enzima fundamental no metabolismo do colesterol, o qual ajuda a regular a composição e organização das membranas, incluindo a formação das jangadas lipídicas (rafts) nas membranas plasmáticas envolvidas na formação da β-amilóide (Puglielli, 2001).
Assim, a contribuição da disfunção mitocondrial para o aparecimento e progressão da doença de Alzheimer continua em investigação, considerando várias vertentes, incluindo o
papel de mutações e/ou polimorfismos ao nível do mtDNA, o papel de alterações nos processos bioquímicos subjacentes à produção de energia, de radicais livres ou do controlo da homeostase do ião cálcio.
O músculo esquelético e a doença de Alzheimer
O músculo esquelético representa cerca 40% do tecido do corpo humano enquanto o músculo liso e cardíaco, no seu conjunto, representam 10%. O músculo esquelético é um componente essencial do sistema locomotor. Dado que as funções locomotivas requerem a manutenção e a renovação de proteínas do tecido contráctil bem como um suplemento contínuo de energia na forma de ATP, então a funcionalidade deste tecido requer a produção contínua de ATP (Barazzoni, 2011). O papel relevante da mitocôndria na organização e actividade do músculo esquelético emerge das diferenças na densidade deste organelo entre os diferentes tipos de músculos detectadas por microscopia. As fibras musculares do tipo I, também designadas por fibras “lentas”, apresentam uma grande densidade de mitocôndrias permitindo ao músculo esquelético uma actividade aeróbia prolongada e a resistência à fadiga (Saunders, 2004). Por outro lado, as fibras do tipo II, com menor densidade de mitocôndrias, dependem fortemente da glicólise (fermentação) para suprimir as suas necessidades energéticas, pelo que só conseguem sustentar a contracção muscular por curtos períodos de tempo devido à fadiga precoce (Saunders, 2004). O músculo humano é constituído por estes dois tipos de fibras, mas a prevalência de um dado tipo de fibra tem um impacto importante nas características do músculo em termos de função e utilização, bem como potenciais implicações clínicas. Assim, a actividade da mitocôndria, responsável pelo consumo de oxigénio e pela produção de ATP, está directamente relacionada com a capacidade do músculo esquelético sustentar o trabalho contráctil e inversamente relacionada com a fadiga do músculo (Rasmussen, 2004).
As células musculares, com forma cilíndrica, são multinucleadas e preenchidas por feixes longitudinais de miofibrilas. Cada miofibrila é composta por dois tipos de filamentos proteicos: filamentos grossos, compostos por agregados de miosina e filamentos finos, formados por agregados de actina. Estas proteínas estão organizadas em unidades contracteis denominadas de sarcómeros. É curioso que durante a contracção muscular, em que a miofibrila (sarcómero) encurta, estes filamentos mantém constante o seu cumprimento, pois o encurtamento do sarcómero ocorre por interpenetração dos filamentos de miosina e actina.
Assim, a contracção muscular resulta da formação reversível e cíclica de pontes transversais entre as extensões das moléculas de miosina e os filamentos de actina que promovem o deslize dos filamentos de actina em relação aos filamentos de miosina, isto é, o encurtamento do sarcómero ou interpenetração dos filamentos. A formação reversível e cíclica das pontes transversais entre os filamentos de miosina e actina, que promove a contracção muscular requer ATP e é iniciada pelo aumento da concentração de Ca2+ no citoplasma (Huxley, 1974). No entanto, a contracção do músculo esquelético é controlada pela actividade do SNC que envia sinais, na forma de potenciais de acção, através dos axónios dos neurónios motores que inervam as fibras musculares. O terminal do axónio segrega a acetilcolina (neurotransmissor) que se liga e activa os receptores nicotínicos da acetilcolina (canal de sódio/potássio) localizados na membrana plasmática das células musculares na região da junção neuromuscular (figura 1.4), induzindo uma despolarização na região da placa motora que desencadeia um potencial de acção nas regiões adjacentes da membrana. Este potencial de acção propaga-se ao longo da membrana da fibra muscular, do mesmo modo que se desloca nas membranas das fibras nervosas, actuando indirectamente sobre o retículo sarcoplasmático promovendo a abertura dos canais de cálcio e, consequentemente, a saída do Ca2+ armazenado no retículo sarcoplasmático para o citoplasma. O aumento da concentração ião cálcio no citoplasma desencadeia o processo de contracção muscular. A contracção muscular cessa pela remoção dos iões cálcio do citoplasma por acção de bomba de cálcio dependente de ATP, que os transporta, contra o gradiente de concentração, de volta para o retículo sarcoplasmático. O cálcio permanece aí armazenado até à chegada de um novo potencial de acção que inicia um novo ciclo de contracção (Berchtold, 2000; Brooks, 2003).
Figura 1.5 Esquema elucidativo da junção neuromuscular. Adaptado de Amabis e Martho
(Amabis, 2004).
Considerando que a doença de Alzheimer é caracterizada desde os seus estágios iniciais por perturbações nos processos subjacentes à actividade da acetilcolina (Cummings,
1998; Fotiou, 2009) e que este neurotransmissor também controla a actividade dos músculos esqueléticos, então perturbações na actividade destes músculos são esperadas como consequência do aparecimento e/ou progressão da doença. De facto, os pacientes com doença de Alzheimer, para além de apresentarem um comprometimento progressivo da actividade intelectual, apresentam também uma diminuição progressiva da força e da actividade muscular associada a uma redução de massa muscular (Sica, 1997). Adicionalmente, o primeiro fármaco a ser aprovado para tratamento da doença de Alzheimer (Tacrina e donepezil) é um inibidor da acetilcolinesterase, enzima que na fenda sináptica degrada o neurotransmissor acetilcolina, controlando o intervalo de tempo em que o neurotransmissor exerce acção sobre as células pós-sinápticas (Al-Jafari, 1997). Actualmente, a tacrina não é muito utilizada devido aos efeitos hepatotóxicos (Patocka, 2008). No entanto, os fármacos actualmente mais utilizados ( rivastigmina, galantamina) também são moduladores da actividade colinérgica (Eschenbach, 2007). Pelo exposto, a associação da progressão da doença de Alzheimer com alterações na actividade do músculo-esquelético, a qual é controlada pela acetilcolina libertada pelos neurónios e depende da actividade da mitocôndria e do controlo dos fluxos de cálcio no citoplasma, está subjacente à hipótese que suporta a presente investigação.
Modelos para estudar a doença de Alzheimer
O desenvolvimento de modelos biológicos adequados para estudar as doenças humanas é um desafio permanente que a comunidade científica da área das ciências médicas enfrenta. Os estudos em modelos biológicos são fundamentais para desvendar os mecanismos moleculares subjacentes ao aparecimento e à progressão das doenças, cujo conhecimento é fundamental para conceber agentes terapêuticos específicos e para identificar biomarcadores capazes de identificar as diferentes etapas da doença. Adicionalmente, os modelos biológicos são também fundamentais para a avaliação da eficiência terapêutica e da toxicidade dos fármacos desenvolvidos antes da sua utilização em humanos. Apesar de vários modelos biológicos terem sido propostos e utilizados para estudar a doença de Alzheimer, nenhum manifesta cabalmente a sequência de eventos detectados durante a progressão da doença em humanos.
O conhecimento do genoma humano e o de várias outras espécies mostrou que um grande número de genes foi conservado durante a evolução da vida e, consequentemente
vários processos biológicos, dependentes da expressão desses genes, foram também conservados e são regulados por mecanismos similares independentemente da espécie. Assim, não é de estranhar que organismos como Saccharomyces cervisiae, Drosophila melanogaster e Caenohabiditis elegans sejam modelos adequados para estudar alguns aspectos de várias doenças humanas (Culetto, 2000; Moloney, 2010). Por exemplo, em Drosophila
melanogaster foram encontrados genes envolvidos em vários processos neurológicos cujo
mau funcionamento provoca doenças “tipicamente” humanas, incluindo os da via de sinalização de Notch, com relevância na arteriopatia cerebral autossómica dominante (síndrome de Cadasil) e os genes da APP e presenilina com relevância na doença de Alzheimer (Rubin, 2000). Assim, a Drosophila melanogaster foi usada como modelo para estudar o metabolismo da β-amilóide e sua toxicidade a nível genético (Finelli, 2004), para mostrar que a APP é relevante para a formação da memória de longa duração sem relevância nos processos de aprendizagem (Goguel, 2011) e também para evidenciar os benefícios da restrição calórica nos processos de envelhecimento e no desenvolvimento da doença de Alzheimer (Kerr, 2011).
Porém, o modelo biológico mais usado para estudar a doença de Alzheimer são os murganhos geneticamente modificados. Entre os modelos transgénicos encontram-se os modelos com modificações genéticas apenas no gene da APP, os que apresentam modificação simultânea nos genes APP e PS1 (Olcese, 2009). Contudo, os modelos mais recentes e actualmente mais utilizados possuem mutações em três genes (3xTg-AD). Este modelo desenvolvido por Oddo e colaboradores possui mutações nos genes APPSWE, TauP301L e PS1M146V (Oddo, 2003), os quais estão directamente associados à doença de Alzheimer, pelo que os animais apresentam um fenótipo neuropatológico relacionado com a idade, incluindo a deposição da β-amilóide e a hiperfosforilação da Tau (Resende, 2008; Yao, 2009). Os animais 3xTg-AD são obtidos pela microinjecção de dois transgenes numa única célula de embriões PS1 (M146V) homozigóticos de ratinhos knockin, gerando assim animais com a mesma origem genética capazes de se reproduzir entre si, preservando as modificações ao longo de várias gerações (Oddo, 2003). Portanto, este modelo permite seleccionar para os estudos quer populações de animais de um só género (masculino ou feminino) como populações mistas mediante o objectivo pretendido (Yao, 2009). Murganhos 3xTg-AD do género masculino foram também seleccionados para os estudos efectuados no presente trabalho.
Figura 1.6 Murganho triplo-transgénico (3xTg-AD) utilizado no
presente trabalho.
Várias linhas celulares têm sido usadas para procurar desvendar os mecanismos subjacentes ao aparecimento e à progressão da doença de Alzheimer (Neill, 1994; Swerdlow, 1997). Um exemplo são as células cibridas, ou híbridos citoplasmáticos, que combinam o genoma nuclear de uma célula com o genoma mitocondrial de outra célula, permitindo dissociar a contribuição genética do genoma mitocondrial do genoma nuclear (Trimmer, 2004). Cibridos preparados pela fusão do citoplasma (contendo o DNA mitocondrial) de células dum paciente clinicamente afectado com doença de Alzheimer com neurónios saudáveis mostraram que a citocromo c oxidase (complexo IV da cadeia respiratória) está afectada, promovendo a formação de quantidade anormais de ROS e o consequente dano celular por stresse oxidativo (Swerdlow, 1997), concluindo os autores que estes aspectos são relevantes para a doença de Alzheimer.
A necessidade de novos marcadores para detectar a progressão da
doença de Alzheimer
Os biomarcadores são parâmetros biológicos mensuráveis (fisiológicos, bioquímicos, histológicos ou anatómicos) que permitem detectar no organismo a ocorrência de uma determinada função patológica e avaliar a resposta do organismo a um dado agente farmacológico ou toxicológico (Growdon, 1999). Adicionalmente, os biomarcadores podem também desempenhar um papel importante na identificação de pacientes pré-sintomáticos ou de subgrupos específicos de pacientes para uma dada patologia. No campo da investigação médica, os biomarcadores mais relevantes são os de natureza bioquímica que podem ser
avaliados nos fluidos corporais (e.g. sangue, urina), os quais são obtidos com facilidade por técnicas não invasivas e sem risco para a saúde humana.
O desenvolvimento e validação de marcadores bioquímicos capazes de detectar a doença de Alzheimer nos estágios iniciais e de distinguir as diferentes fases durante a sua progressão é uma necessidade premente e um dos maiores desafios da investigação na área, pelas seguintes razões: i) actualmente não existe qualquer tratamento efectivo para doença; ii) algumas terapias que são benéficas nos estágios iniciais da doença não o são nos estágios mais avançados; iii) as actuais técnicas de diagnóstico, baseados essencialmente na avaliação das funções cognitivas usando testes como o do Mini Exame do Estado Mental (Folstein, 1975), não detectam a doença nos estágios precoces, têm dificuldade em distinguir a doença de Alzheimer dos outros tipos de demência e apresentam uma grande margem de erro, pois os resultados dependem muito do nível cultural e de escolaridade dos pacientes; iv) o diagnóstico definitivo requer análises Post-mortem aos cérebros dos pacientes para detectar os marcadores histopatológicos (Aiyaz, 2012), não tendo por isso utilidade terapêutica para o respectivo paciente.
De acordo com Di Luca e colaboradores (Di Luca, 2005), um bom biomarcador para a doença de Alzheimer deve ser sensível para detectar precocemente as alterações cognitivas e biológicas e, ser capaz de diferenciar esta doença do envelhecimento normal, de pseudo-demências e de outras doenças neurodegenerativas. Adicionalmente deve ser uma ferramenta confiável, simples e facilmente aplicável que permita avaliar, em tempo útil, o desempenho das diversas opções terapêuticas e que permita ainda o desenvolvimento de novas opções terapêuticas, dado que deverá ajudar a compreender os mecanismos moleculares subjacentes à doença (Vanderstichele, 2012).
As alterações bioquímicas subjacentes à doença de Alzheimer mais bem caracterizadas são resultado de processos celulares patológicos que ocorrem ao nível do sistema nervoso central como o metabolismo da proteína percursora amilóide (APP) que promove a formação das placas senis, a hiperfosforilação da proteína Tau que leva à formação dos emaranhados neurofibrilares, processos inflamatórios associados à produção de citocinas e stresse oxidativo ligado à oxidação de lípidos, proteínas e DNA. Assim, os marcadores bioquímicos propostos para a doença pretendem detectar algumas ou todas estas alterações (Irizarry, 2004).
Dado que as alterações bioquímicas no cérebro são reflectidas na composição do líquido cefalorraquidiano, então a detecção/quantificação da proteína Tau e do fragmento 1-42 β-amilóide no líquido cefalorraquidiano têm sido as principais ferramentas no diagnóstico
da doença de Alzheimer. Alguns investigadores mostraram que a combinação destes dois biomarcadores tem grande sensibilidade permitindo detectar com especificidade a doença de Alzheimer em mais de 85% dos casos analisados (Blennow, 2003). Outros sugerem que a presença da proteína Tau fosforilada na treonina 181 no líquido cefalorraquidiano é um marcador específico para a doença de Alzheimer, permitindo descriminar esta doença de outras neuropatologias (Kohnken, 2000; Hampel, 2004). De facto, estudos com autópsias mostraram que o diagnóstico adquire elevada precisão (superior a 80%) quando são detectados no líquido cefalorraquidiano os três marcadores em simultâneo (Vanderstichele, 2012). Apesar destes marcadores permitirem diagnosticar, com grande confiança, que o paciente sofre de doença de Alzheimer não fornecem qualquer informação do estágio clínico da doença, tendo por isso utilidade limitada na definição das estratégias terapêuticas mais adequadas (Vanderstichele, 2012). Adicionalmente, a recolha do líquido cefalorraquidiano não é uma análise de rotina e requer equipas especializadas, representando sempre um risco para o doente. Por outro lado, a detecção e quantificação da β-amilóide no plasma têm-se revelado pouco significativa para o diagnóstico, apesar de proporcionar informações importantes para avaliar a resposta a um dado tratamento. Aparentemente, a β-amilóide aumenta de modo progressivo e significativo no plasma dos pacientes de Alzheimer apenas durante os estágios avançados da doença, restringindo assim a sua utilidade como biomarcador (Vanderstichele, 2012). Do exposto, novos biomarcadores para detectar os diferentes estágios da doença de Alzheimer são uma necessidade premente e fundamental para travar a progressão desta patologia que cada vez afecta mais pessoas.
Objectivos
No presente projecto formulou-se a hipótese de que a doença de Alzheimer, caracterizada desde os estágios iniciais por perturbações nos processos subjacentes à actividade da acetilcolina ao nível do sistema nervoso central, promove perturbações progressivas na actividade dos músculos esqueléticos, dado que a actividade destes músculos é controla por este neurotransmissor. Assim, utilizaram-se murganhos triplo-transgénicos (3xTg-AD) com 3, 6 e 12 meses de idade como modelo para a doença de Alzheimer e murganhos “wild type” com idades equivalentes como controlo com o objectivo geral de investigar os mecanismos moleculares subjacentes à relação entre o processo neurodegenerativo e actividade muscular, identificando assim novos biomarcadores para a doença. Os objectivos específicos são:
1. Caracterizar a influência da idade na actividade da acetilcolinesterase do músculo esquelético para avaliar a sua relação com a progressão da doença de Alzheimer;
2. Caracterizar como é que o conteúdo total de tióis nas proteínas e as actividades da Superóxido Dismutase e da Catalase se alteram com idade para determinar o papel do stresse oxidativo ao nível do músculo esquelético na progressão da doença de Alzheimer;
3. Estudar, em mitocôndrias isoladas dos músculos esqueléticos, a influência da idade na actividade das enzimas relacionadas com a bioenergética mitocondrial (Complexos I, II, IV e F0F1-ATPsintase) para avaliar se a progressão da doença de Alzheimer está relacionada com uma menor capacidade da célula muscular em sintetizar ATP por processos aeróbios;
4. Avaliar como é que a composição lipídica das membranas das mitocôndrias do músculo esquelético se alteram com a idade para identificar se as esperadas perturbações no metabolismo da célula muscular durante a progressão da doença de Alzheimer se relacionam com a actividade bioenergética e o stresse oxidativo.
III.1-Modelo animal para estudar a doença de Alzheimer
No presente trabalho foram usados murganhos triplo-transgénicos, com mutações ao nível dos genes Tau, APP e PS1, como modelo da doença de Alzheimer humana (3xTg-AD) e os animais da estirpe selvagem (WT) como controlos. Os animais foram obtidos no Centro de Neurociências e Biologia Celular da Universidade de Coimbra (CNC/UC) que mantém uma colónia em contínuo e mantidos no biotério da UTAD em condições padrão (temperatura de 24 ± 2 ºC, humidade relativa de 55 ± 5% e fotoperíodo de 12 h), em gaiolas de policarbonato até atingirem a idade pretendida para os estudos. Os ratinhos foram alimentados ad libitum com água e dieta padronizada. Nos estudos foram utilizados animais do género masculino com 3, 6 e 12 meses de idade.
III.2-Preparação do material biológico
2.1.Preparação dos homogeneizados de músculo esquelético
Para estudar a possível relação entre a doença de Alzheimer e a disfunção muscular relacionada com a actividade da acetilcolinesterase e o stresse oxidativo preparam-se homogeneizados de músculo esquelético. O animal foi morto por deslocamento cervical e imediatamente decapitado. Os músculos esqueléticos dos membros superiores e inferiores e da região intercostal foram extraídos cuidadosamente e a sua massa determinada numa balança analítica. De seguida, o tecido muscular foi transferido para um copo e o volume de tampão de homogeneização (Sacarose 160 mM, Tris-HCl 10 mM, pH = 7,2), previamente arrefecido a 4 ºC, necessário para se atingir a proporção massa de tecido: volume de 1:10 foi adicionado, isto é, 10 mL de tampão por cada 1 grama de tecido muscular. Todo o material necessário foi mantido em gelo granulado, de forma a manter a temperatura entre 0 e 4 ºC. O tecido em suspensão foi cortado em pequenas fracções e homogeneizado num homogeneizador de vidro tipo “Potter Elvjhem” rodando o pistão de teflon a cerca de 500 rotações por minuto, utilizando um agitador IKA EUROSTAR power basic. De seguida, o homogeneizado foi submetido a ultrassons, quatro ciclos de 5 segundos com intervalos de 10
segundos entre ciclos para aumentar a eficiência de lise das células e novamente homogeneizado no homogeneizador de vidro do tipo “Potter Elvjhem”. O homogeneizado foi transferido para tubos de centrífuga e centrifugado a 16000 x g durante 15 minutos numa centrífuga refrigerada Sigma 2 – 16 K, à temperatura de 4 ºC. O sobrenadante recolhido com uma pipeta foi armazenado à temperatura de -20 ºC para posterior determinação das actividades enzimáticas
.
2.2. Isolamento de mitocôndrias de músculo esquelético
As mitocôndrias foram isoladas a partir do músculo esquelético de animais 3xTg-AD e WT por centrifugação diferencial de acordo com o método previamente descrito (Skalska, 2008), com algumas alterações. O animal foi morto por deslocamento cervical e imediatamente decapitado. Os músculos esqueléticos dos membros superiores e inferiores e da região intercostal foram extraídos cuidadosamente e colocados em meio de homogeneização (sacarose 250 mM, Hepes 5 mM, EGTA 0,2 mM e EDTA 0,1 mM, pH = 7,4) previamente arrefecido a cerca de 4 ºC. Todo o material necessário foi inserido em gelo granulado, para manter a temperatura entre os 0 e 4 ºC. O músculo foi cortado em pequenos pedaços e abundantemente lavado com meio de homogeneização para remover excesso de sangue e alguma gordura. O tecido cortado foi incubado em meio de homogeneização contendo uma protéase (Tripsina do pâncreas bovino, Type I Sigma 8003) 0.15 mg/mL, para facilitar a libertação das células musculares da matriz externa, durante 2 minutos à temperatura ambiente. De seguida, a mistura foi transferida para um homogeneizador de vidro tipo “Potter Elvjhem” e homogeneizado rodando o pistão de teflon a cerca de 500 rotações por minuto, utilizando um agitador IKA EUROSTAR power basic. Nesta operação, o tecido foi totalmente desintegrado e a maior parte das células rompidas, obtendo-se uma suspensão de membranas, organelos intracelulares e algumas células intactas e eritrócitos. O homogeneizado foi centrifugado a 12000 x g, durante 10 minutos numa centrífuga refrigerada Sigma 2 – 16 K, à temperatura de 2 ºC. Esta centrifugação tem como objectivo remover a protéase para evitar a sua acção sobre as membranas dos organelos, nomeadamente sobre as mitocôndrias. O sedimento foi ressuspendido em meio de homogeneização enriquecido com BSA 0,2% e homogeneizado no homogeneizador de vidro tipo “Potter Elvjhem” rodando o pistão a cerca de 400 rpm, para rompimento de tecido que ainda permaneça intacto. O
homogeneizado foi centrifugado a 800 x g durante 10 minutos, para remover os constituintes mais densos da suspensão como por exemplo células intactas e núcleos. O sobrenadante foi cuidadosamente filtrado através de gaze e novamente centrifugado a 12000 x g durante 10 minutos, para obter um sedimento que corresponde à fracção mitocondrial. O sedimento foi ressuspenso num pequeno volume de meio de lavagem (sacarose 250 mM, Hepes 5 mM, ATP 1 mM, pH = 7,4). As ressuspensões, diluídas em 40 mL de meio de lavagem, foram transferidas para novos tubos de centrífuga e centrifugadas novamente a 12000 x g durante 10 minutos. Esta etapa foi repetida e o sedimento (fracção mitocondrial purificada) obtido foi ressuspenso num volume mínimo de meio de lavagem sem ATP. Cada fracção mitocondrial foi transferida para um pequeno tubo cónico (eppendorf), o qual foi mantido a uma temperatura de 4 ºC até à quantificação da proteína mitocondrial. A fracção mitocondrial restante foi congelada em azoto líquido e guardada à temperatura de -20 ºC para posterior utilização.
2.3. Determinação da concentração da proteína nas amostras biológicas
A concentração da proteína foi determinada pelo método colorimétrico do biureto (Gornall, 1949), que se baseia na reacção do CuSO4 em meio alcalino (reagente de biureto) com as ligações peptídicas das proteínas, formando-se um complexo de cor violeta, o qual pode ser quantificado por espectrofotometria.
As amostras da suspensão mitocondrial (30 µL) e do sobrenadante obtido com os homogeneizados do tecido muscular (100 µL) foram solubilizadas pela adição de 100 µL de dodecilsulfato de sódio (SDS) a 10%. A esta mistura adicionou-se água desmineralizada até perfazer um volume de 0,5 mL, em seguida, 2 mL de reagente de biureto (CuSO4.5H2O 0,15%, NaKC4H4O6.4H2O 0,6%, NaOH 3% e KI 0,1%). Prepararam-se padrões de BSA (0 - 2,4 mg) nas mesmas condições das amostras, com excepção das amostras biológicas que foram substituídas por igual volume da correspondente solução tampão. Ao fim de 15 minutos de incubação à temperatura ambiente, determinou-se a absorvância das amostras e dos padrões a 540 nm, num espectrofotómetro UV-Vis Cary 50, contra um branco que continha todos os reagentes excepto a proteína.
III.3-Estudos no sobrenadante dos homogeneizados dos músculos
esqueléticos
3.1. Determinação da actividade da Acetilcolinesterase (AChE)
A acetilcolinesterase (E.C.3.1.1.7.) é uma enzima que cliva o neurotransmissor acetilcolina em ácido acético e colina na região das sinapses colinérgicas, incluindo na região das junções neuromusculares (Ferreira, 2012; Grubič, 1999). A actividade AChE nos sobrenadantes obtidos com os homegeneizados do músculo esquelético foi avaliada pelo método descrito por Ellman e colaboradores com pequenas alterações (Ellman, 1961), usando a acetiltiocolina (ACTI) como substrato. A actividade da AChE foi avaliada na presença de etopropazina (um inibidor selectivo da butirilcolinesterase) com uma concentração final de 0,1 mM. 30 µL de homogeneizado de músculo foram adicionados a uma cuvete de quartzo contendo 2 mL de tampão fosfato (KH2PO4 0,1 M pH = 7,4) suplementado com 0,1 mM etopropazina e com 10 µL de reagente de Ellman (DTNB 0,15 mM em tampão fosfato 100 mM, pH = 7,4). Após 2 min de incubação, à temperatura de 30 ºC, a reacção enzimática foi iniciada pela adição de ACTI (8 - 2000 µM). A actividade catalítica da enzima foi medida, monitorizando o aumento da absorvância a 412 nm (isto é, a formação do anião de cor amarela, 5-tio-2-nitrobenzoato produzido pela reacção da tiocolina com DTNB), num espectrofotómetro UV-Vis Cary 50, durante 4 minutos. A actividade da AChE foi avaliada em função da concentração do substrato, usando 11 concentrações diferentes de substrato, para determinar os parâmetros cinéticos de Michaelis-Menten, Km (constante de Michaelis) e Vmax (velocidade máxima).
3.2. Determinação da actividade da Superóxido dismutase (SOD)
Superóxido dismutase (E.C. 1.15.1.1.) é uma enzima com um papel relevante na defesa dos organismos contra o stresse oxidativo, dado que protege a célula da actividade do anião superóxido (O2*-), catalisando a sua dismutação em oxigénio molecular (O2) e peróxido de hidrogénio (H2O2). Nos organismos eucariotas, foram identificados três isoformas desta metaloproteína: a Mn-SOD, específica da mitocôndria; a CuZn-SOD citoplasmática e outra extracelular (Ec-SOD) que é secretada pelas células (Rajkumar, 2008). Assim, no presente
trabalho avaliou-se, principalmente, a actividade da CuZn-SOD citoplasmática dado que as amostras biológicas analisadas correspondem ao sobrenadante da centrifugação dos homogeneizados a 16000 x g que exclui as mitocôndrias.
A actividade da SOD foi determinada por uma análise espectrofotométrica de acordo com o método de McCord e colaboradores (McCord, 1969), com ligeiras alterações. Recorrendo ao sistema xantina - xantina oxidase na presença do cromóforo cloreto de azul de nitrotetrazolio (NBT). O volume de homogeneizado de músculo correspondente a 0,596 mg de proteína foi adicionado a uma cuvete contendo tampão fosfato (KH2PO4 50 mM, EDTA 1 mM a pH = 7,4) suplementado com NBT 0,1 mM e hipoxantina 0,1 mM. Após 2 minutos de incubação, à temperatura de 30 ºC, a reacção foi iniciada com 5 µL xantina oxidase (5 U/mL) e monitorizada durante 3 minutos a 560 nm, num espectrofotómetro UV-Vis Cary 50. O aumento na absorvância, graficamente expresso por uma recta cujo declive, determinado usando o software associado ao espectrofotómetro, permitiu a determinação da actividade desta enzima. Os resultados são expressos em U / min . mg de proteína.
3.3. Determinação da actividade da Catalase
A catalase (E.C.1.11.1.6) é uma hemoproteína que catalisa a decomposição de peróxido de hidrogénio (H2O2) a oxigénio molecular (O2) e água (H2O). A catalase que nos animais está presente em todos os tecidos, é essencialmente uma enzima dos peroxissomas (Williams, 2012). A actividade da catalase foi avaliada polarograficamente, numa camara termostatizada a 30 ºC, com um eléctrodo de oxigénio do tipo Clark (Hansatech,) acoplado a um computador. Este método é baseado na determinação da velocidade inicial da produção de oxigénio resultante da decomposição do peróxido de hidrogénio (H2O2) pela catalase (Del Rio, 1977). O volume de homogeneizado de músculo correspondente a 75 µg de proteína foi ressuspendido em 1 mL de tampão fosfato 50 mM a pH = 7. Após 2 minutos de incubação, a reacção foi iniciada com adição de H2O2 12 mM e a produção de oxigénio monitorizado durante dois minutos. A actividade da enzima foi calculada pelo declive da recta e expressa em nmol O2 / min . mg proteína.
3.4. Determinação do conteúdo total de tióis nas proteínas
O conteúdo total de tióis foi determinado usando o método colorimétrico de (Ellman, 1959). De acordo com este método, os grupos sulfidrilo (-SH) das proteínas reagem com o 5,5’-ditiobis-(2-ácido nitrobenzóico) (DTNB) originando um derivado de coloração amarela, com um máximo de absorção a 412 nm, o qual pode ser quantificado por espectrofotometria. Ressuspenderam-se 100 µL de homogeneizado de músculo em tampão fosfato (100 mM a pH = 8) suplementado com DTNB 0,5 mM, perfazendo um volume final de 2 mL. Após 15 minutos de incubação à temperatura ambiente, as amostras foram lidas num espectrofotómetro UV-Vis Cary 50 a 412 nm, utilizando-se água como branco. As misturas foram homogeneizadas em agitação em vórtice e incubadas durante 15 minutos, no escuro, à temperatura ambiente. Em simultâneo, preparou-se uma série de padrões de cisteína (0 a 100 µM) utilizando as mesmas condições das amostras. Após a incubação, as absorvâncias das amostras e dos padrões foram avaliadas, a 412 nm, num espectrofotómetro Varian cary 50 Bio. O conteúdo total de tióis de cada amostra foi determinado a partir da curva padrão de cisteína e expresso em equivalentes de cisteína µmoles / mg proteína.
III.4-Determinação da actividade das enzimas mitocondriais
4.1. NADH:ubiquinona oxidoredutase (complexo I)
A NADH desidrogenase (E.C.1.6.5.3.), também conhecida por NADH-ubiquinona oxidoredutase ou vulgarmente complexo I, é uma enzima localizada na membrana interna da mitocôndria que catalisa a oxidação do NADH com a redução da ubiquinona a ubiquinol, promovendo assim a transferência de electrões do NADH para a coenzima Q (Dykens, 2008). A actividade da NADH desidrogenase foi medida espectrofluorimetricamente pela monitorização do decaimento da florescência a 450 nm, resultante da oxidação do NADH (Melo, 2012). Antes da realização dos ensaios, cada fracção foi submetida a 3 ciclos de congelamento/descongelamento para aumentar o acesso dos substratos ao local activo da respectiva enzima.
O volume da fracção mitocondrial correspondente a 0,3 mg de proteína foi adicionado a uma cuvete de quartzo contendo 2,5 mL de tampão (KH2PO4 25 mM, MgCl2 10 mM a pH = 7,4) suplementado com KCN 1 mM. Após 2 minutos de incubação, à temperatura de 30 ºC, a
reacção foi iniciada pela adição de NADH 40 µM. Após estabilização da intensidade de fluorescência resultante da adição do NADH, adicionaram-se 13 µL de dodecilubiquinona 25 mM, com a consequente queda na intensidade de fluorescência, graficamente expresso por uma recta cujo declive possibilita a determinação da actividade do complexo I. Dois minutos após a adição da dodecilubiquinona, adicionaram-se 3 µL de rotenona 1,5 mM (um inibidor específico do complexo I) para a garantir que as taxas de decaimento de fluorescência resultam da actividade do complexo I. A intensidade da emissão da florescência do NADH foi medida a 450 nm fixando a excitação de 366 nm, num espectrofluorímetro Varian Cary Eclipse com um suporte de células termostatizado e agitação constante. A actividade do complexo I foi calculada pela diferença entre os declives das rectas antes e depois da adição do inibidor, usando o software do espectrofluorímetro. O resultado é expresso em unidades arbitrárias.
A actividade do complexo I de cada extracto mitocondrial, expressa em unidades arbitrárias/min . mg proteína, foi normalizada pela actividade da citrato sintase determinada nesse mesmo extracto para evitar que as diferenças de actividade resultem de diferentes graus de pureza das fracções mitocondriais isoladas.
4.2. Succinato-coenzima Q redutase (complexo II)
Succinato-coenzima Q redutase (E.C.1.3.5.1.), também conhecida por succinato desidrogenase ou complexo II, é uma enzima localizada no folheto interno da membrana interna da mitocôndria. Esta enzima catalisa a oxidação do succinato a fumarato com a redução da ubiquinona a ubiquinol. O fumarato permanece no ciclo de Krebs, enquanto o ubiquinol difunde pela membrana lipídica sendo oxidado pelo complexo III mitocondrial. Assim, o complexo II é uma enzima do ciclo de Krebs que funciona como subsidiária da cadeia transportadora de electrões (Dykens, 2008).
A actividade da enzima succinato desidrogenase foi medida por espectrofotometria, através da monitorização da perda de cor do corante azul DCIP (2,6-diclorofenolindofenol), resultante da redução, a 600 nm. Antes da realização dos ensaios, cada fracção foi submetida a 3 ciclos de congelamento/descongelamento para aumentar o acesso dos substratos ao centro activo da respectiva enzima. O volume da fracção mitocondrial correspondente a 0,2 mg de proteína foi adicionado a uma cuvete contendo 2,2 mL de tampão fosfato 25 mM a pH = 7,4, Dodecilubiquinona 0,1 mM, DCIP 6 µM e inibidores dos complexos mitocondriais I, III e IV