w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Original
article
Hemoglobin
A
2
values
in
sickle
cell
disease
patients
quantified
by
high
performance
liquid
chromatography
and
the
influence
of
alpha
thalassemia
Silvana
Fahel
da
Fonseca
a,∗,
Tatiana
Amorim
b,
Antônio
Purificac¸ão
b,
Marilda
Gonc¸alves
c,
Ney
Boa-Sorte
baUniversidadedeBrasília(UnB),Brasília,DF,Brazil
bAssociac¸ãodePaiseAmigosdosExcepcionais(APAE),Salvador,BA,Brazil cFundac¸ãoOswaldoCruz(FIOCRUZ),Salvador,BA,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received10March2013
Accepted11May2015
Availableonline7June2015
Keywords:
HemoglobinA2
Alphathalassemia
Betathalassemia
Sicklecelldisease
Highperformanceliquid
chromatography
a
b
s
t
r
a
c
t
Background:Insicklecelldisease,thequantificationofHbA2isimportantforthedifferential
diagnosisbetweensicklecellanemia(HbSS)andHbS/0-thalassemia.
Objective:TodetermineHbA2levelsasquantifiedbyhighperformanceliquid
chromatogra-phyinpatientswithsicklecellanemia(HbSS)andwiththeSChemoglobinopathy,withor
withoutconcomitantalphathalassemia.
Methods:Thisisaretrospectivestudyof242childrenagedbetweentwoandsixyearswith
diagnosesofHbSSorHbSC.Thehemoglobinwasevaluatedusinghighperformanceliquid
chromatographyandalphathalassemia[3.7kbdeletion(−␣3.7)]wasdetectedbypolymerase
chainreaction.Patientswereclassifiedashomozygous(−␣3.7/−␣3.7),heterozygous(−␣3.7/␣),
orhomozygouswild-type.AnalysisofvariancewasusedtocomparethemeanHbA2values
betweenthealphathalassemiagroups.
Results:Themean(±standarddeviation)HbA2concentrationsintheHbSSgroup(n=135)
was3.68±0.65%.ThemeanvaluesforindividualswithHbSSandheterozygous(n=28)or
homozygousforalphathalassemia(n=3)were3.98±0.64%and4.73±0.25%,respectively.
ThemeanHbA2ofalltheHbSCpatients(n=107)was4.01±0.507with4.29±0.41%and
4.91±0.22%inindividualsheterozygous(n=23)andhomozygous foralphathalassemia
(n=7),respectively.AllpatientshomozygousforalphathalassemiahadHbA2levelsabove
3.5%.However,Hb A2 values above5.2%wereseenin patientswithHbSS andHb SC,
independentlyofalphathalassemia.
Conclusion:HbA2levelsareelevatedinpatientswithHbSorHbC,andaredirectlyinfluenced
bythealphathalassemiagenotypes.
©2015Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.Published
byElsevierEditoraLtda.Allrightsreserved.
∗ Correspondingauthorat:SQN214,BlocoC,Apt214,AsaNorte,70873-030Brasília,DF,Brazil.
E-mailaddress:[email protected](S.F.daFonseca).
http://dx.doi.org/10.1016/j.bjhh.2015.05.005
1516-8484/©2015Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.PublishedbyElsevierEditoraLtda.Allrights
Introduction
Sickle cell disease is one of the most common genetic
pathologiesintheworld.Itischaracterizedbyhomozygous
hemoglobinS(HbS)orHbSassociatedtootherHbvariants.1
Thereisgreatclinicalvariationintheclinicalmanifestations
betweensicklecelldiseasepatients;severalfactorsare
asso-ciatedwiththedifferentpresentations.Somedeterminants
are already well established, such as genetic, clinical and
laboratoryfactors,while others,suchaspsycho-social and
nutritionalfactors,havebeenlesswellstudied.2–5
Ofthe geneticfactors,the importanceofthephenotype
ofthe hemoglobinopathyiswellcharacterizedinthat
indi-vidualsdoublyheterozygousforsicklecellanemiaandthose
withHbS/0-thalassemiahaveamoresevereclinicalprofile.
Ontheotherhand,carriersofHbSCtogetherwithHbS/+
-thalassemiahavebetteroutcomes,whichmakesthecorrect
diagnosisofthesesyndromesanissueofgreatimportancefor
abetterunderstandingandadequateclinicalandtherapeutic
managementofpatients.4–6
Inthediagnosisofsicklecelldisease,quantificationofHb
A2, aswell asacomplete bloodcount(CBC),familyhistory
andclinicaldata,helptoestablishthedifferentialdiagnosis
betweensicklecellanemia(HbSS)andHbS/0-thalassemia.7,8
ThechoiceofthemethodologytoaccuratelymeasureHb
A2hasbeenthesubjectofmanydiscussionsinthemedical
lit-erature,andhighperformanceliquidchromatography(HPLC)
hasbeenregardedthemethodofchoiceformanyyears.The
referencevalueofHbA2inhealthyadultswhodonothave
thalassemiaisusuallybetween2.0and3.3%.5,9–12
In1996,Suhetal.13reportedthatHbA
2valuesobtained
byHPLCincreasedsignificantlyinsamplescontainingHbS
andsuggestedthepossibilitythatHbSSindividualsmayhave
beenincorrectlydiagnosed withHbS/0-thalassemia.They
alsosuggestedthatincreasesinHbA2couldbeexplainedby
thepresenceofsmallercomponentsofHbSthatco-eluted
withHbA2.
In 2000, Shokrani et al.14 proposed that blood samples
withHb valuesofup to 5.9%and up to5.2% ofHb A2 as
analyzedbyHPLCfrom Hb SS or HbSC patients and
indi-vidualswiththesicklecelltrait(HbAS),respectivelycanbe
considerednormal.In2004,Headetal.7confirmedthe
find-ingsofShokranietal.andexplainedthatthefalselyelevated
Hb A2 values were due to the presence of Hb S that had
sufferedpost-translationalmodifications,andconsequently
had the same retention time as Hb A2. They also
demon-stratedthatthedegreeofuncertaintydirectlycorrelated to
theconcentrationofHbS,whichishigherinHbSSpatient
samplesthaninsamplesfromindividualswithHbAS.
Addi-tionally,theyconcludedthatthepercentageofHbA2ishigher
in Hb AS individuals with alpha thalassemia (AT) than in
thosewithoutAT;andsuggestedthatthisoccursbecausethe
deltachainshavegreateraffinitytoalphachainsthantheS
chains.7
Morerecently,studiesusingmassspectrometryhave
pro-posedthatelevatedHbA2valuesinsampleswithHbScanbea
consequenceoftheelutionofsmallerHbcomponentsformed
byaSchainassociatedwithanalpha-globinchainthatwas
modifiedbycarbamylationaftertranslation.15
TheinterferenceofanomalousHbinthequantificationof
HbA2isnotlimitedtothepresenceofHbSandHbC,butcan
alsoberelatedtothepresenceofother betachainvariants
suchasHbE,HbDandHbLepore.16,17
In2007,Ondeietal.,18 inanefforttoestablishreference
valuesfortheBrazilianpopulation,usedHPLCtoanalyze136
sampleswiththeHbASphenotype,106withATandHbAS,
18withHbSSandninewithHbSC,andconcludedthatHb
A2valuesof2.9–5.2%forHbAS,2.8–5.2%forATtogetherwith
HbAS,0.8–5.6%forHbSSand3.8–5.7%forHbSCshouldbe
considerednormal.
Considering the widespread use of HPLC inthe
labora-torypracticeandtheimportanceofanaccuratediagnosisto
definethetypeofsicklecelldiseaseinordertofacilitatebetter
clinical andlaboratoryfollow-upofthesepatients,the
cur-rentstudysoughttoverifytheHbA2valuesasquantifiedby
HPLC,insamplesofpatientswithHbSandHbC,inthe
pres-enceorabsenceofthe␣2-thalassemia3.7kbdeletionasboth
hemoglobinopathies maybeprevalent inthesame
popula-tion.
Methods
Studydesignandpopulation
Thisis aretrospective study conductedbetweenJune2008
andJune2009withaninitialsamplecomposedof287
chil-dren.Forty-fivewereexcluded:eightforpresentingadiagnosis
ofHb S/-thalassemia, elevenforhavingbeen analyzedby
HPLC after the transfusion of blood components, and 26
duetoirondeficiency.Thisleftatotalof242children
diag-nosedwithsicklecellanemiaorHbSCscreenedandfollowed
at the Neonatal Screening Referral Service (SRTN) of the
Associac¸ão de Pais eAmigos dos Excepcionaisin Salvador
(APAE/Salvador), Bahia, Brazil. The SRTN-APAE/Salvador is
registeredwiththeBrazilianMinistryofHealth(Ordinance822
–06/06/2001)asareferralserviceinthestateofBahia,Brazil.
Thisservicediagnosesandfollowsupallnewbornswithsickle
cellanemiainBahia,thestatewiththehighestincidenceof
thishemoglobinopathyinBrazil(onein601newborns/year).19
Theobjectivesandproceduresinvolvedinthestudywere
explained,andparentswhoagreedtoparticipatesignedan
informedconsentform.Allparentsofparticipatingchildren
wereasymptomatic,andweresubjectedtoaCBC,Hbprofiling,
andmeasurementofserumferritin.Allpresentedalaboratory
profilecompatiblewithheterozygosityforHbSorHbCand
normalironreserves.
Analysesandlaboratorytechniques
Venousbloodsampleswerecollectedin3.0-mLtubes
contain-ing 3.6mgK2-ethylenediaminetetraacetic acid (EDTA) as an
anticoagulationagentandinatubewithagelseparator.The
qualitativeandquantitativeHbprofileswereinvestigatedby
automatedHPLCusingtheVariantExpressapparatusandthe
-thalassemiaShortProgramkit(Bio-Rad,California,USA).
Cation-exchangeHPLCisaprocessinwhichamixtureof
molecules(suchasnormalandvariantHbs)withanet
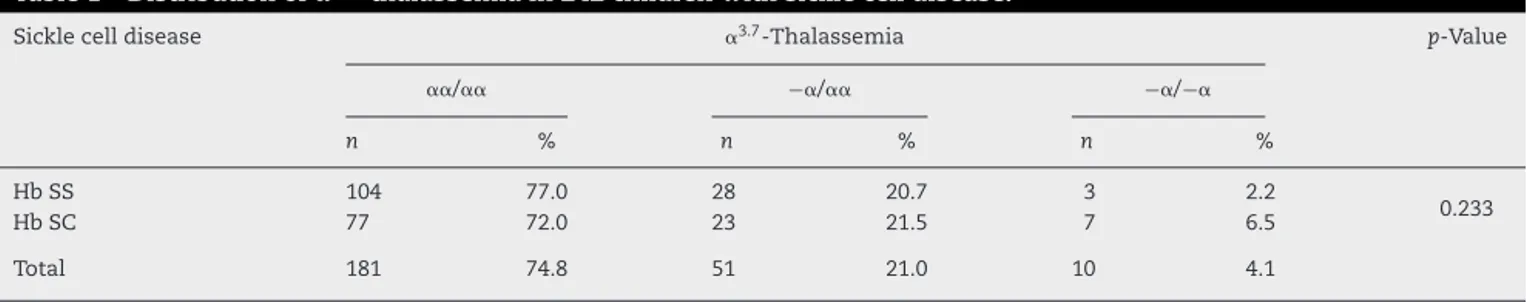
Table1–Distributionof␣3.7-thalassemiain242childrenwithsicklecelldisease.
Sicklecelldisease ␣3.7-Thalassemia p-Value
␣␣/␣␣ −␣/␣␣ −␣/−␣
n % n % n %
HbSS 104 77.0 28 20.7 3 2.2
0.233
HbSC 77 72.0 23 21.5 7 6.5
Total 181 74.8 51 21.0 10 4.1
HbSS:sicklecellanemia;HbSC:SChemoglobinopathy.
ofmoleculesinanegatively chargedstationaryphasein a chromatographycolumn,followedbytheirelutioninamobile phase.Inthemobilephasealiquidwithanincreased con-centrationofcationsflowsthroughthecolumn;thecations inthemobilephasecompetewiththeadsorbedproteinsfor anionicbindingsites.Thus,theadsorbedpositivelycharged Hb molecules are eluted from the column into the liquid phaseataraterelatedtotheiraffinityinthestationaryphase. Thedifferent Hbs elutedfrom the columnare represented graphicallyandautomaticallyquantified.Theresultisa chro-matogramwiththepercentageandretentiontimeofeachHb fraction.Thetimeofelution(retentiontime)ofanynormalor variantHbpresentiscomparedwiththatofknownHbs pro-vidingquantificationofnormalHbs(HbA,HbF,andHbA2) andmanyvariants.ThemostpositivelychargedHbs(e.g.HbS andHbC)havealongerretentiontime.20
Patientsampleswere consideredtocontainHbSand/or
HbCwhenthechromatogramspresentedvariantswith
reten-tiontimesequivalenttotheseHbs.Assuch,thediagnosisof
sicklecellanemiawasmadeonlyifHbS,HbFandHbA2were
present,hemametric indicesof the CBCwere normal, and
theparentspresentednormalCBCswithHbprofileconsistent
withsicklecelltrait.Theserumferritinlevelwasdetermined
bychemoluminescence(ADVIACentaurCPSystem,Siemens).
Ferritinlevels<12ng/mLwereconsideredasirondeficient.21
The deletion of 3.7kb in AT was investigated by
allele-specific polymerase chain reaction (PCR)22 and the
participants were identified as heterozygotes (−␣3.7/␣␣),
homozygotes(−␣3.7/−␣3.7)orwildtype(␣␣/␣␣).
Statisticalanalysis
HbA2concentrationsweredescribedaccordingtothetypeof
sicklecelldisease(HbSCorHbSS)together withATstatus
(wild-type,heterozygousorhomozygous)usingcentral
ten-dencyanalysisanddispersion.TheKolmogorov–Smirnovtest
wasusedforanalysisofnormaldistribution.One-wayanalysis
ofvariance(ANOVA)wasusedtoanalyzedifferencesbetween
groups,andtwo-wayANOVAwasusedtoanalyzethe
influ-enceoftheinteractionandconfoundingfactorsbetweentype
ofsicklecellanemiaandpresenceofAT(independent
vari-ables)onHbA2levels(dependentvariable).Posthocanalysis
wasperformedusingTukey’sHonestlySignificantDifference
(HSD)testandtheGames–Howelltesttoidentifysituationsin
whichequalityofvariancewasverified.Duetothereduced
sizeofthegroupwithhomozygousmutations,statistical
sig-nificancewassetatap-value<0.01.
Hb A2 valueswerecategorizedinthreegroupsbasedon
standard reference values: (1) below 2.0%; (2) between 2.0
and3.5%;and(3)above3.5%.Thesecategorieswereanalyzed
accordingtotheATdatausingthechi-squaredtendencytest.
AlldatawereanalyzedusingEPIINFOforWindows(version
3.5.1)andtheStatisticalPackagefortheSocialSciences(SPSS®
–version13.0).
Ethicalconsiderations
The present study was approved by the Ethics
Commit-tee for Research in Human Beings of the Gonc¸alo Moniz
Research Centerfrom the OswaldoCruz Foundation, Bahia
(CEP-CPqGM/FIOCRUZ:protocol#112/2006)and followedthe
researchethicsguidelinesasdefinedinresolution196/96of
theHelsinkideclarationof2008.
Results
Thefinalcohortanalyzedconsistedof242patients(107HbSC
and135HbSS)agedfromtwotosixyearswithanaverageage
of3.05years(±1.01),andmedian(p25–p75)of4(3.0–5.0)years.
Thedistributionof␣3.7-thalassemiaisdescribedinTable1.
There was no significant difference between the
distribu-tion of heterozygous or homozygous AT between the two
typesofhemoglobinopathies (HbSS andHb SC)(2=2.917;
p-value=0.233).
ThemeanHbA2levelinHbSSsamplesindependentlyof
thepresenceofAT(n=135)was3.68%(±0.65);inHbSSsamples
withheterozygousAT(n=28)itwas3.98%(±0.64)andinHbSS
sampleswithhomozygousAT(n=3)itwas4.73%(±0.25).The
meanHbA2inallHbSCsamples(n=107)was4.01%(±0.50);
inHbSCsampleswithheterozygousAT(n=23)itwas4.29%
(±0.41),andinHbSCsampleswithhomozygousAT(n=7)it
was4.91%(±0.22).
WithregardtothemeanHbA2valuesandthesicklecell
diseasetype(HbSSorHbSC),significantlyhigheroverall
val-ues were observedinpatients withHbSC comparedtoHb
SS(p-value<0.001)asshowninTable2.Whenthedatawere
stratifiedaccordingtotheATstatus(wildtype,heterozygous
or homozygous),asignificantdifferencewas observedonly
whensampleswithoutATwerecompared.Valueswerenot
significantforheterozygous(HbSC:4.29±0.41%versusHbSS:
3.98±0.64%;p-value=0.074),orforhomozygousATmutation
(HbSC:4.91±0.22%versusHbSS:4.73±0.25%;p-value=0.267
Table2–Mean,standarddeviation,maximumandminimumvaluesforHbA2in242childrenaccordingtothepresence of␣3.7-thalassemia,stratifiedbytypeofsicklecelldisease.
␣3.7-Thalassemia n Average SD Minimum–maximum p-Value
<0.001a
␣␣/␣␣ 181 3.68 0.56 1.50–5.20
0.001b
HbSS 104 3.57 0.61 1.50–5.20
HbSC 77 3.84 0.42 2.90–4.80
−␣/␣␣ 51 4.12 0.57 2.30–4.90
0.074b
HbSS 28 3.98 0.64 2.30–4.90
HbSC 23 4.29 0.41 3.30–4.90
−␣/−␣ 10 4.86 0.23 4.50–5.20
0.267b
HbSS 3 4.73 0.25 4.50–5.00
HbSC 7 4.91 0.22 4.60–5.20
Total 242 3.82 0.62 1.50–5.20
<0.001c
HbSS 135 3.68 0.65 1.50–5.20
HbSC 107 4.01 0.51 2.90–5.20
SD:standarddeviation;HbSS:sicklecellanemia;HbSC:SChemoglobinopathy.
a ComparisonofaverageHbA2valuesaccordingtopresenceof␣3.7-thalassemia(wild-type,heterozygousandhomozygous)independentlyof typeofhemoglobinopathy–non-parametricKruskal–Wallistest.
b ComparisonofaverageHbA2valuesbetweenthetypesofhemoglobinopathy(HbSSorHbSC)foreachalphathalassemiaclassification
(wild-type,heterozygousandhomozygous)–non-parametricMann–Whitneytest.
c ComparisonofaverageHbA2valuesbetweenthetypesofhemoglobinopathy(HbSSorHbSC)independentlyofalphathalassemia
classifi-cation(wild-type,heterozygousandhomozygous)–non-parametricMann–Whitneytest.
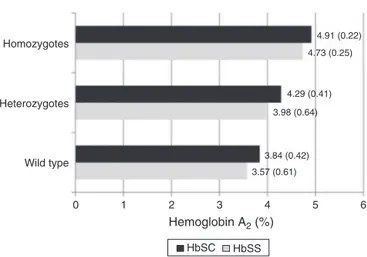
0 1 2 3
HbSC HbSS 4 3.57 (0.61) Wild type
Heterozygotes Homozygotes
Hemoglobin A2 (%) 3.84 (0.42)
3.98 (0.64) 4.29 (0.41)
4.73 (0.25) 4.91 (0.22)
5 6
Figure1–HbA2values(%)accordingto␣3.7-thalassemia
genotypeandsicklecelltype(HbSS/HbSC)in242children.
WhencomparinggroupsaccordingtoATstatusonly
(wild-type,heterozygousorhomozygous),highermeanvalueswere
observed for Hb A2 in patients with the −␣/−␣ genotype
(F=30.985;p-value<0.001–Table2).
Theanalysisoftheinteractionbetweenthetypeofsickle
celldiseaseandATgenotypewascarriedoutafterthe
differ-encebetweentheaverageHbA2valuesforHbSSandHbSC
wasfoundtobesignificant.Therewasnosignificant
inter-action(F=0.583; p-value=0.559) orinfluence ofthe typeof
hemoglobinopathy(F=2.156;p-value=0.43)onthemeanHb
A2values.OnlytheATgenotypeinfluencedthevariationof
themeanHbA2values(F=28.676;p-value<0.001).Theposthoc
analysisindicatedasignificantdifferencebetweenthethree
ATgenotypegroups,showingaprogressiveincreaseinHbA2
levels(Figure1).
UsingthereferenceHbA2valuesdescribedbyOndeietal.in
2007,18itwasobservedthatallhomozygousmutantsamples
had elevatedHb A2 levels (above3.5%),and atendencyfor
increasesinthepercentageofchildrenwithHbA2levelsabove
3.5%fromthewild-type(59.7%)totheheterozygous(88.2%)
andthehomozygous(100%)samples(2lineartendency=18.871;
p-valuetendency<0.001),asshowninTable3.
Discussion
Hbdisordersarerecognizedasoneofthemostcommon
inher-ited diseases worldwide. Among the hemoglobinopathies,
sicklecelldiseaseand-thalassemiahavethegreatestimpact
onmorbidityandmortality,affectingmillionsofindividuals
worldwide.5
Sicklecelldiseaseshouldbeconsideredasbotha
qualita-tiveandquantitativegeneticdisorderinthatitiscausedby
thepresenceofanabnormalHbvariant–HbS.Homozygosity
forHbSorsicklecellanemiaisthemostcommongenotype;
theother causativegenotypesincludecompound
heterozy-gousstatesofHbSwithHbC(HbSC)or-thalassemiavariants
(HbS/0-thalassemiaandHbS/+-thalassemia).5
Diagnosisofspecificsicklecelldiseasesisaccomplishedby
integratingclinicalandhematologicalparametersalongwith
laboratory Hbanalysis. Combiningtheseelements to
prop-erlydiagnose Hbdisorders isessential forthetreatmentof
anemia,primarypreventionandgeneticcounselingfor
under-lyingdisorders.Inthemajorityofpatients,thepresenceofa
hemoglobinopathycanbediagnosedwithsufficientaccuracy
forclinicalpurposesfromknowledgeofthepatient’sethnical
backgroundandclinicalhistory(includingfamilyhistory)and
theresultsofthephysicalexaminationcombinedwith
rela-tivelysimplebloodtests.Initialinvestigationsshouldinclude
Table3–ProportionofHbA2above3.5%foundin242accordingtothepresenceof␣3.7-thalassemia.
HbA2
␣3.7-Thalassemia <2.0%
n(%)
2.0–3.5% n(%)
>3.5% n(%)
p-Value
␣␣/␣␣ (n=181) 1(0.6) 72(39.8) 108(59.7)
<0.001a
−␣/␣␣(n=51) – 6(11.8) 45(88.2)
−␣/−␣(n=10) – – 10(100.0)
Total 1(0.4) 78(32.2) 163(67.4)
a Chi-squaredlineartendencytest.
detailedexaminationofawell-stainedbloodfilmshouldbe
carriedout.OtherimportantbasictestsareHb
electrophore-sisorchromatography,andthemeasurementofHbA2and
HbF.23AutomatedHPLC,byanalyzinglargenumbersof
sam-ples,isincreasinglyreplacingHbelectrophoresisastheinitial
investigativeprocedureinlaboratories.18,23
WhileHPLCisconsideredamethodthatpermitsrapidand
precisedetectionofHbvariants,aswellassensitive
quantifi-cationofHbA2,9 overthelasttwodecadesvariousauthors
have reported the occurrence of falsely elevated Hb A2 in
screeningforHbvariants(HbS,HbC,HbD,HbEandHb
Lep-ore)andforAT.Someoftheseauthorshavecautionedabout
theriskofinterpretationerrorsandthefalsediagnosisofHb
S/0-thalassemia.7,10,13,14,16–18,24–26
Accuratediagnosis ofHb disorders isessential insickle
cellsyndromes.ConsideringtheclinicalimportanceofHbA1
andHbA2and/orHbFtogetherwithclinicalandlaboratory
dataforthediagnosisofHbS/-thalassemia,thisstudy
ana-lyzedHbA2valuesbyHPLCinconjunctionwithHbSandHb
C,andAT,inapopulationwith alowestimatedfrequency
of-thalassemia.27,28Similartoresultsobservedinprevious
studies,7,10,13,14,16–18,24–26 theHbA
2valuesobtainedbyHPLC
wereincreasedinHbSSandHbSCpatientsamples.
␣2-Thalassemia was diagnosed in23.4% of the children
withsickle cell disease,a resultthat agrees withprevious
studies performed inthe population ofthe state ofBahia,
Brazil.29,30Thus,asSuhetal.13andOndeietal.18stated,the
presenceofAThasanimpactonthevalueofHbA2as
quanti-fiedbyHPLCandadditionally,inthepresentstudy,significant
differencesinthemeanlevelsofHbA2wereobserved
accord-ingtothepresenceof␣3.7-thalassemia,withagradualincrease
inthisHbfractionbetweenthethreegroups:wildtypeto
het-erozygoustohomozygous.
Severalstudiesinrecentyears9–12havesoughttocompare
differentmethodologiesandsystemsinordertoidentify
pro-cessesinwhichthepresenceofanomalousHbshavetheleast
effectonthemeasurementofHbA2inordertoreachamore
accuratediagnosis.However,studiesseekingtoexplainhow
thepresenceoftheseHbsandATmayresultinfalseHbA2
levelsareextremelyscarce.Eventhemostrecentpublications,
suchasthatofGreeneetal.in20155seemtoagreewithSuh
etal.from199613inattributingtheapparentincreaseinHb
A2asquantifiedbyHPLCinsamplescontainingHbstructural
variantstothepresenceofglycosylatedfractionsof
anoma-lousHb(suchasHbS1c)thatco-elutewithHbA2.10,14,17,23
OncomparingtheimpactofthetypeofHbvariantonthe
measurementofHbA2,thegreatestimpactwascausedbythe
presenceofAT,andnotbyHbSorHbC.Headetal.7defended
thatthe increasedHb A2 levelsobservedintheassociation
ofsicklecelldiseasewithATareexplainableinthatwitha
lowernumberofalphachainsbeingproduced,thesepositively
chargedmoleculescombinewithotherchainsforwhichthey
havegreataffinity,namelythedeltachains.
Theprevalenceof-thalassemiaisverylowinthestudied
population,27,28andacarefulhematologicalstudyofthe
par-entsshowednormalerythrogramsandthepresenceofHbS
andHbAconsistentwithheterozygosityforHbS.Giventhese
facts,weconsiderthattheHbA2valuesseeninthepatients
werefalselyelevatedduetointerferenceofthemethodology
usedinthepresenceofstructuralHbvariantsandAT,andas
suchthepatientscanbediagnosedashavingsicklecell
ane-mia,andnotHbS/0-thalassemia,eventhoughwerecognizea
limitationbythefactthatwedidnotperformmolecular
anal-ysestoconclusivelyexcludethediagnosisof-thalassemia.
WesuggestthatincaseswithapossiblediagnosisofHbS/0
thalassemia,theHbprofileshouldnotbeperformedbyHPLC,
orthatifused,thismethodshouldbecomplementedbya
sec-ondconfirmatorytest.Furthermore,whendoubtsstillremain,
amolecularanalysisshouldbecarriedouttoidentifydeletions
orothermutations,inordertoconfirmtheclinicaldiagnosis
andguidegeneticcounseling.5
Conclusion
WeconcludethattheHbA2levelsinsamplescontainingHb
Sand/orHbCcanbeoverestimatedwhenanalyzedbyHPLC,
especiallyinthepresenceofAT,andthatinthesesituations
the referencevaluesofup to3.5%shouldnotbeused.We
reinforcetheimportanceofmulti-centerstudieswhen
estab-lishingpatterns,theneedforindividualevaluationsofcases
inordertoreachadifferentialdiagnosisbetweensicklecell
syndromes,especiallyinregionswithahighprevalenceofthe
differenttypesofhemoglobinopathies,andtheuseof
molec-ularbiologystudiestoclarifyanydoubts.
Funding
CentroNacionaldeDesenvolvimentoCientíficoeTecnológico
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
r
e
f
e
r
e
n
c
e
s
1. WeatherallDJ.Theinheriteddiseasesofhemoglobinarean emergingglobalhealthburden.Blood.2010;115(22):4331–6.
2. NogueiraZD,Boa-SorteN,LeiteME,KiyaMM,AmorimT, FonsecaSF.Breastfeedingandtheanthropometricprofileof childrenwithsicklecellanaemiareceivingfollow-upina newbornscreeningreferenceservice.RevPaulPediatr. 2015;33(2):154–9.
3. RakyanVK,DownTA,BaldingDJ,BeckS.Epigenome-wide associationstudiesforcommonhumandiseases.NatRev Genet.2011;12(8):529–41.
4. TheinSL.Geneticassociationstudiesin
-hemoglobinopathies.HematolAmSocHematolEduc
Program.2013;2013:354–61.
5. GreeneDN,VaughnCP,CrewsBO,AgarwalAM.Advancesin
detectionofhemoglobinopathies.ClinChimActa. 2015;439:50–7.
6. SteinbergMH,ForgetBG,HiggsDR,WeatherallDJ.Disorders ofhemoglobin:genetics,pathophysiologyandclinical
management.2nded.Cambridge:CambridgeUniversity
Press;2009.
7. HeadCE,ConroyM,JarvisM,PhelanL,BainBJ.Some observationsonthemeasurementofhaemoglobinA2andS percentagesbyhighperformanceliquidchromatographyin thepresenceandabsenceof␣thalassaemia.JClinPathol. 2004;57(3):276–80.
8. ClarkeGM,HigginsTN.Laboratoryinvestigationfor hemoglobinopathiesandthalassemias:reviewandupdate. ClinChem.2000;468(Pt2):1284–90.
9. GreeneDN,PyleAL,ChangJS,HokeC,LoreyT.Comparisonof SebiaCapillarysFlexcapillaryelectrophoresiswiththe BioRadVariantIIhighpressureliquidchromatographyinthe evaluationofhemoglobinopathies.ClinChimActa.
2012;413(15–16):1232–8.
10.PaleariR,GulbisB,CottonF,MoscaA.Interlaboratory comparisonofcurrenthigh-performancemethodsforHbA2. IntJLabHematol.2012;34(4):362–8.
11.WeatherallDJ,CleggJB.Inheritedhaemoglobindisorders:an increasingglobalhealthproblem.BullWorldHealthOrgan. 2001;79(8):704–12.
12.StephensAD,AngastiniotisM,BaysalE,ChanV,FucharoenS, GiordanoPC,etal.ICSHrecommendationsforthe
measurementofhaemoglobinA2.IntJLabHematol. 2012;34(1):1–13.
13.SuhDD,KraussJS,BuresK.InfluenceofhemoglobinS adductsonhemoglobinA2quantificationbyHPLC.Clin Chem.1996;42(7):1113–4.
14.ShokraniM,TerrelF,TurnerEA,AguinagaMD.
ChromatographicmeasurementsofhemoglobinA2inblood
samplesthatcontainsicklehemoglobin.AnnClinLabSci. 2000;30(2):191–4.
15.ZurbriggenK,SchmuggeM,SchmidM,DurkaS,KleinertP, KusterT,etal.Analysisofminorhemoglobinsby
matrix-assistedlaserdesorptiontime-of-flightmass spectrometry.ClinChem.2005;51(6):989–96.
16.HigginsTN,KhajuriaA,MackM.QuantificationofHbA(2)in patientswithandwithoutbeta-thalassemiaandinthe
presenceofHbS,HbC,HbE,andHbDPunjabhemoglobin
variants:comparisonoftwosystems.AmJClinPathol. 2009;131(3):357–62.
17.MoscaA,PaleariR,IvaldiG,GalanelloR,GiordanoPC.Therole ofhemoglobinA2testinginthediagnosisofthalassemiaand relatedhemoglobinopathies.JClinPathol.2009;62(1):13–7.
18.OndeiLS,ZamaroPJA,MangonaroPH,ValêncioCR,
Bonini-DomingosCR.HPLCdeterminationofhemoglobinsto
establishreferencevalueswiththeaidofstatisticsand informatics.GenetMolRes.2007;6(2):453–60.
19.AmorimTA,PimentelH,FontesMI,Purificac¸ãoA,LessaP, Boa-SorteN.Evaluationofaneonatalscreeningprogramof Bahiafrom2007to2009lessonsofhemoglobinopathies.Gaz MédBahia.2010;80(3):10–3.
20.BainBJ.Haemoglobinopathydiagnosis.2nded.Oxford: BlackwellPublishingLtd.;2008.
21.WorldHealthOrganization.Centersfordiseasecontroland prevention.DepartmentofNutritionforHealthand Development.Assessingtheironstatusofpopulations.2nd ed;2007.
22.DodéC,KrishnamoorthyR,LambJ,RochetteJ.Rapidanalysis of−alpha3.7thalassaemiaandalphaalphaalphaanti3.7 triplicationbyenzymaticamplificationanalysis.BrJ Haematol.1993;83(1):105–11.
23.LewisSM,BainBJ,BatesI,DacieandLewis.Practical
Haematology.In:ChurchillLivingstone.10thed.Philadelphia: Elsevier;2006.
24.KalleasC,TentesI,MargaritisD,AnagnostopoulosK,ToliA, PendilasD,etal.EffectofHbSinthedeterminationofHbA(2)
withtheTOSOHHLC-723G7analyzerandtheHELENA
Beta-ThalQuikcolumnkit.ClinBiochem.2007;40(3–4):242–7.
25.KalleasC,TentesI,MargaritisD,AnagnostopoulosK,ToliA, PendilasD,etal.EffectofHbSinthedeterminationofHbA2 withtheBioradVariantIIanalyzer.ClinBiochem.
2007;40(9–10):744–6.
26.AnagnostopoulosK,TentesI,KalleasC,MargaritisD,ToliA, PendilasD,etal.EffectofHbSinthedeterminationofHbA2 withtheMenariniHA-8160analyzerandcomparisonwith otherinstruments.IntJLabHematol.2009;31(6):665–72.
27.FonsecaSF,MouraNetoJP,Gonc¸alvesMS.Prevalenceand molecularcharacterizationof-thalassemiainthestateof BahiaBrazil:firstidentificationofmutationHBB:c.135delCin Brazil.Hemoglobin.2013;37(3):285–90.
28.AdornoEV,CoutoFD,MouraNetoJP,MenezesJF,RêgoM,Reis MG,etal.HemoglobinopathiesinnewbornsfromSalvador, BahiaNortheastBrazil.CadSaudePublica.2005;21(1): 292–8.
29.FonsecaSF,AmorimT,RibeiroR,etal.Incidenceofalpha thalassemia,FactorVLieden,Prothrombingenemutation anddeterminationofhaplotypesofbetaglobingenein childrenwithsicklecelldiseasediagnosedbyneonatal screening,Bahia,Brazil.ClinChem.2009;55Suppl.6:A75.