Agradecimentos
À Prof. Doutora Sílvia Coimbra e ao Prof. Doutor Luís Gustavo Pereira pelo apoio, segurança e motivação transmitidos através da sua orientação e exigência. Por acreditarem desde o início na minha capacidade e competência para realizar este trabalho.
À Doutora Stefanie Sprunck por todo o apoio, pela orientação e exigência, por me ensinar que é preciso muita persistência para se fazer ciência, e pela motivação que me transmitiu.
À Lucija Soljic e à Doutora Stefanie Sprunck pela disponibilização dos resultados das experiências de microarrays, ainda não publicados.
À minha família por todo o apoio, pela paciência, pelo mimo do bom e pela disponibilidade nos momentos mais difíceis e de dúvidas, por sempre me terem dado forças para continuar e me ensinarem a nunca desistir dos meus objectivos.
Aos amigos, por estarem sempre presentes, pela paciência e todo o apoio prestado, pelo carinho e por me aturarem sempre, mesmo quando acabava a falar das minhas plantas, dos meus PCRs azarados ou de transformações com resultados estranhos (ajudaram-me sempre a chegar à conclusão de que não existem resultados estranhos ou azarados, mas sim resultados que nos fazem pensar, tentar compreender, voltar atrás, rever passos, métodos… enfim, fazer ciência).
À Lucija, por todo o apoio que me deu, por tudo o que me ensinou no laboratório, pelas boas conversas, pela boa disposição e o mais importante, pelos crepes com Nutella e gelado que compensavam alguns dias intermináveis no laboratório. E, claro, pela amizade!
A todos os meus colegas e amigos de laboratório, do Porto e de Regensburg, por todo o apoio que me deram, pela paciência para me aturar, mesmo quando queria pôr toda a gente a limpar o laboratório, pelas boas conversas e discussões, por me ajudarem a compreender que para se trabalhar em ciência é preciso saber lidar muito bem com as frustrações (as dos PCRs azarados) e principalmente pela amizade.
Ferramentas para o estudo da expressão de genes de proteínas arabinogalactânicas em diferentes células e tecidos do pistilo de Arabidopsis thaliana
Sumário
Nas plantas com flor a fecundação requer o transporte dos gâmetas masculinos até aos óvulos, que surgem profundamente envolvidos pelos tecidos esporofíticos da flor. Este transporte é feito pelo tubo polínico que responde a sinais de direccionamento e cresce através dos diferentes tecidos do pistilo até ao saco embrionário, conduzindo à dupla fecundação. As proteínas arabinogalactânicas (AGPs) são proteínas altamente glicosiladas que poderão estar envolvidas no direccionamento do tubo polínico até ao saco embrionário. Estudos prévios de imunocitoquímica, feitos com diferentes espécies de plantas, mostraram que estas proteínas estão presentes nas células gametofíticas, nas células do tegumento micropilar e nas células do nucelo micropilar. Estas proteínas são óptimas candidatas a moléculas de atracção para o direccionamento do tubo polínico. Neste trabalho foram construídas ferramentas moleculares que permitirão identificar os epítopos destas proteínas previamente localizados por imunocitoquímica no pistilo de Arabidopsis thaliana, determinando a localização celular das AGPs. Para a selecção das AGPs expressas nos tecidos femininos foram analisados dados de experiências de microarrays relativos à expressão das AGPs nas sinergídeas, na célula central e na oosfera. Foram também analisados dados de microarrays de pólen e gâmetas masculinos. De acordo com o padrão de expressão de cada AGP nas células gametofíticas masculinas e femininas e atendendo à similaridade entre as diferentes proteínas, foram seleccionadas 7 AGPs (AGP1, AGP9, AGP10, AGP12, AGP 15, AGP20 e AGP23). Foram obtidas plantas mutantes que expressam a GFP sob o controlo de sequências promotoras destas AGPs para 6 das AGPs seleccionadas inicialmente: AGP1, AGP9, AGP10, AGP12, AGP 15 e AGP23.
Estas ferramentas permitirão confirmar a informação obtida a partir das experiências de microarrays bem como determinar que AGPs específicas estão presentes ao longo do trajecto seguido pelo tubo polínico até ao saco embrionário, a nível celular. A partir deste estudo será mais fácil analisar e determinar com uma maior precisão a função das AGPs presentes nos tecidos femininos de Arabidopsis thaliana.
Tools for the localization and characterization of AGP genes in Arabidopsis thaliana pistil tissues
Abstract
Successful fertilization in flowering plants requires sperm cells guidance into the ovules, deeply embedded in the female sporophytic tissues. The pollen tube carries the two sperm cells trough the pistil tissues following guidance cues until it reaches the embryo sac to accomplish double fertilization. Arabinogalactan proteins (AGPs) are highly glycosilated proteins that might be related to pollen tube guidance into the embryo sac. Previous immunolocalization studies with different plant species have shown a specific labelling of AGPs carbohydrate epitopes in the gametophytic cells, the filiform apparatus of the synergids, the integument micropylar cells and the micropylar nucellus. These proteins are excellent candidates for chemoattractants that guide pollen tubes growth.
In this study molecular tools were developed, that will allow the specific identification of these proteins epitopes previously identified by immunolocalization studies in Arabidopsis thaliana. Data from microarray experiments using isolated synergids, egg cells and central cells were evaluated in order to identify AGP genes that have the highest expression levels in these cells. These proteins’ expression was also checked in pollen and sperm cells using microarray data. According to the expression pattern of individual AGPs in the female and male gametophytic cells, and to their sequence similarity, 7 AGPs were selected for further analysis (AGP1, AGP9, AGP10, AGP12, AGP15, AGP20 and AGP23). Arabidopsis transgenic plants were generated that express GFP under the control of AGP promoter sequences for 6 (AGP1, AGP9, AGP10, AGP12, AGP15 and AGP23) of the 7 AGPs initially identified.
The analysis of these plants will allow the verification of the data obtained from the microarray experiments and the identification of the AGPs present along the pollen tube pathway, as well as will permit the identification of their specific cellular location. The identification of the AGPs present in the female reproductive tissues is of major importance to initiate future function analysis studies regarding this large family of proteins.
Índice
1. Introdução 1
1.1 Dupla fecundação nas plantas com flor 1
1.2. Direccionamento do tubo polínico para o saco embrionário 2
1.3. Pela estrada fora – interacções pólen–pistilo 4
1.4 AGPs – Proteínas Arabinogalactânicas 9
1.5 AGPs – moléculas de sinalização? 11
1.6 AGPs no trajecto do tubo polínico 12
1.7 Objectivos 13
2. Materiais e Métodos 15
2.1 Material vegetal e condições de crescimento 15
2.2 Alinhamento das sequências proteicas 15
2.3 Estirpes bacterianas e vectores 15
2.3.1 Meios e condições de crescimento 16
2.3.2 Preparação de células de E. coli DH5α competentes 16 2.3.3 Transformação de células de E.coli TOP10 e E. coli DH5 α 17 2.3.4 Preparação de células de A. tumefaciens gv3101::pMP90::pSOUP competentes 17
2.3.5 Transformação de A. tumefaciens 18
2.4 Técnicas de manipulação e análise de DNA 18
2.4.1 Extracção de DNA genómico de A. thaliana 18
2.4.2 Electroforese em gel de agarose 19
2.4.5 RT-PCR em tempo real 23 2.4.6 Recuperação de DNA a partir de géis de agarose 25
2.4.7 Minipreparação de DNA plasmídico 25
2.4.8 Digestão de DNA plasmídico 25
2.4.9 Sequenciação 25
2.5 Produção de linhas de A. thaliana transgénicas 26
2.5.1 Ligação dos fragmentos amplificados no vector pENTR/D-TOPO 26
2.5.2 Linearização dos vectores de entrada 27
2.5.3 Obtenção das construções pAGP::NLS:3xGFP 27
2.5.4 Transformação de A. thaliana pelo método de “floral dip” 28
3. Resultados 30
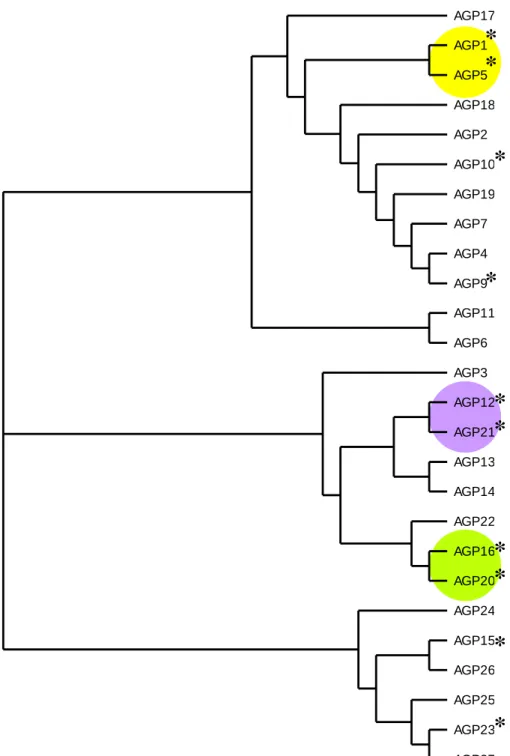
3.1 Selecção das AGPs 30
3.2 Análise dos alinhamentos proteicos 30
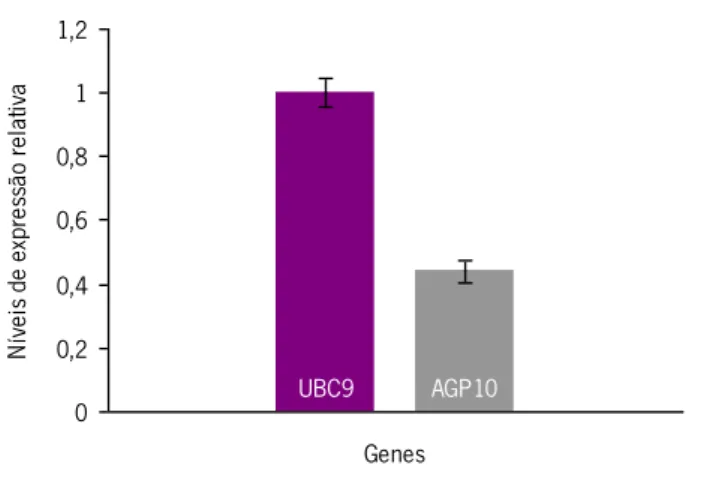
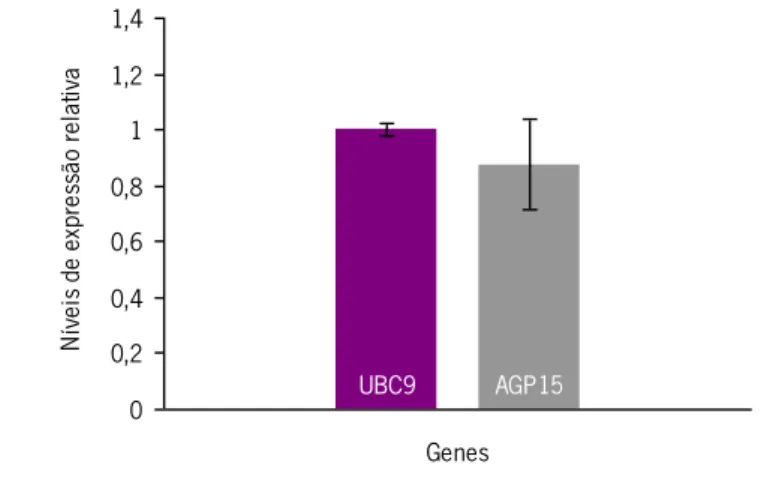
3.3 Expressão de genes de AGPs nos óvulos 32
3.4 Amplificação dos fragmentos das regiões promotoras 36
3.5 Obtenção dos vectores de entrada pENTR/D-TOPO/pAGP 37
3.6 Obtenção das construções pAGP::NLS:3xGFP 39
4. Discussão 43
4.1 Selecção das AGPs 44
4.2 Obtenção das construções pAGP::NLS:3xGFP 45
4.3 Considerações finais 46
5. Referências Bibliográficas 48
Anexos 53
Anexo I – Representação esquemática das reacções de recombinação BP e LR que
Anexo II – Curvas de dissociação dos genes em estudo nas experiências de RT-PCR em
tempo real 54
Anexo III – Curvas padrão dos genes em estudo nas experiências de RT-PCR em tempo
real para determinação da eficiência das reacções 58
Anexo IV – Representação esquemática do vector pENTR/D-TOPO 64 Anexo V – Resultado da sequenciação das construções obtidas após recombinação com o
vector pENTR/D-TOPO 65
Anexo VI – Resultado da sequenciação das construções obtidas após recombinação do vector pENTR/D-TOPO, contendo a sequência promotora de cada AGP, com o vector
1. Introdução
1.1 Dupla fecundação nas plantas com flor
O ciclo de vida das plantas com flor alterna entre um organismo multicelular diplóide, o esporófito, e um organismo multicelular haplóide, o gametófito. Os gametófitos e os esporófitos diferem morfologicamente e funcionalmente. Após a meiose o esporófito origina 2 tipos de esporos sexualmente diferenciados: os microsporos e os megasporos. Estes esporos dividem-se mitoticamente e desenvolvem-se em gametófitos, cuja principal função é a produção de gâmetas. A fusão dos gâmetas masculinos e dos gâmetas femininos (oosfera e célula central) estabelece a nova geração esporofítica, completando assim o ciclo de vida (Drews et al., 1998; Raven et al., 2005). Uma das características que define o desenvolvimento reprodutivo das Angiospérmicas é exactamente esta dupla fecundação (Raghavan, 2003). Este é um processo biológico único, pelo qual um gâmeta masculino se funde com a oosfera para produzir o embrião enquanto o outro gâmeta se funde com a célula central para formar o endosperma, um tecido nutritivo fundamental para o desenvolvimento do embrião (Russell, 1992). Esta forma de reprodução foi descrita pela primeira vez nas Liliáceas Lilium martagon e Fritillaria tenella nos finais do século XIX por S. Nawaschin em 1898. No ano seguinte L. Guinard confirmou o fenómeno observado por Nawaschin, de forma independente, em Lilium martagon e Lilium pyrenaicum (Raghavan, 2003).
A dupla fecundação envolve uma série de interacções complexas entre o gametófito masculino (GM), o esporófito e o gametófito feminino (GF). Os gametófitos feminino e masculino desempenham um papel fundamental na dupla fecundação (Weterings e Russell, 2004).
O GM maduro, também designado por grão de pólen ou microgametófito, desenvolve-se no interior do lóculo da antera e é constituído por duas ou três células haplóides: uma célula vegetativa que transporta no seu interior uma células generativa (grão de pólen bicelular) ou dois gâmetas masculinos (grão de pólen tricelular) (Lord e Russell, 2002; McCormick, 2004). A célula generativa sofre uma mitose originando os dois gâmetas masculinos, esta mitose pode em algumas plantas ocorrer no interior da antera (gramíneas e crucíferas), mas na maioria das plantas ocorre durante o crescimento do tubo polínico (TP). A célula vegetativa é responsável pela formação do TP e pela libertação dos dois gâmetas masculinos no GF (McCormick, 1993; McCormick, 2004).
15 padrões diferentes de desenvolvimento do GF, sendo a forma mais comum designada por GF tipo Polygonum, por ter sido pela primeira vez descrita em Polygonum divaricatum, sendo exibido por espécies tão importantes como Arabidopsis thaliana ou Zea mays. O desenvolvimento do GF tipo Polygonum ocorre em duas fases: megasporogénese (formação do megasporo) e megagametogénese (desenvolvimento do saco embrionário). Na megasporogénese uma célula, o megasporócito sofre uma meiose originando quatro megasporos haplóides. Os três megasporos localizados na região do micrópilo sofrem morte celular programada e o megasporo localizado na região da caláza torna-se o megasporo funcional. Este passa por três ciclos de mitoses originando uma estrutura cenocítica constituída por oito núcleos. Após a 3ª mitose formam-se paredes celulares em torno destes núcleos, e ao mesmo tempo um núcleo de cada pólo (os núcleos polares) migra em direcção ao centro do GF e fundem-se antes ou durante a fecundação originando o núcleo da célula central. Após todos estes eventos o GF maduro do tipo Polygonum é constituído por sete células: três antípodas na região da caláza, uma célula central grande a ocupar toda a região central do saco embrionário, duas sinergídeas e uma oosfera na região do micrópilo (Huang e Russell, 1992; Drews et al., 1998; Yadegari e Drews, 2004).
Todos os processos relacionados com a dupla fecundação ocorrem de forma coordenada conduzindo ao desenvolvimento da semente. Estes processos podem ser organizados por ordem temporal: atracção e crescimento do TP, libertação dos dois gâmetas masculinos numa sinergídea degenerada, migração dos gâmetas masculinos até aos gâmetas femininos, reconhecimento e fusão dos gâmetas, fusão do material genético parental durante a cariogamia e a reiniciação do ciclo celular, transcrição e tradução conduzindo à formação e crescimento do zigoto (Berger, 2008; Berger et al., 2008). O primeiro passo, de atracção e crescimento do TP, é um dos mais estudados actualmente, embora ainda pouco se saiba sobre como ocorre este direccionamento do TP para o saco embrionário.
1.2. Direccionamento do tubo polínico para o saco embrionário
O direccionamento do TP para o saco embrionário precede a dupla fecundação, sendo uma etapa crítica para a sua ocorrência. Durante este direccionamento o TP atravessa diferentes tecidos do pistilo (Figura 1.1a) (Johnson e Preuss, 2002). Após a adesão do grão de pólen à superfície estigmática e sua posterior hidratação (Lord e Russell, 2002), o TP emerge e inicia o seu
transmissão (Hülskamp et al., 1995; Johnson e Preuss, 2002; Lord e Russell, 2002). O tecido de transmissão é um tecido especializado que faz a ligação entre o estigma, o estilete e os óvulos suportando o crescimento do TP através de uma matriz extracelular até ao saco embrionário (Hülskamp et al., 1995; Lennon et al., 1998; Lord, 2000).
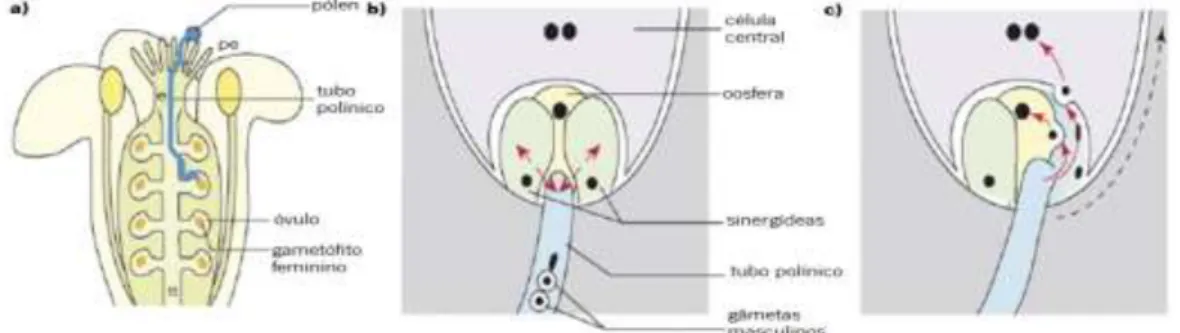
Figura 1.1 – Representação esquemática da dupla fecundação. a) Germinação do TP à superfície do estigma e seu
crescimento ao longo do estilete e tecido de transmissão até atingir o GF. b) Entrada do TP no GF através da região micropilar onde ocorrem interacções (setas vermelhas) entre o TP e as sinergídeas para atrair o TP até ao local de entrada no saco embrionário. c) Libertação dos gâmetas masculinos na sinergídea receptiva, migração dos gâmetas e fusão dos gâmetas masculinos com a oosfera e com a célula central. pe: papilas estigmáticas, e: estilete, tt: tecido de transmissão (Adaptado a partir de Berger et al., 2008)
Em muitas espécies, como em A. thaliana, o tecido de transmissão prolonga-se através do septo que divide o ovário em 2 compartimentos. Consequentemente nestas espécies, o TP tem de atravessar a camada de células da placenta que envolve o tecido de transmissão, e crescer à superfície do septo até atingir o funículo. Na região funicular o TP direcciona o seu crescimento para a região micropilar do óvulo, crescendo à superfície das células do funiculo até entrar pelo micrópilo e atingir o saco embrionário (Johnson e Preuss, 2002; Yadegari e Drews, 2004). Normalmente, apenas um TP cresce em direcção a cada saco embrionário (Kandasamy et al., 1994). Uma vez na região micropilar o TP entra no GF, mais especificamente numa das sinergídeas, entrando junto ao aparelho filiforme (estrutura constituída por expansões da parede celular) num local de menor resistência, rompe e aí liberta o seu conteúdo (Figura 1.1b e 1.1c). Esta sinergídea sofre morte celular programada pouco antes da entrada do TP ou mesmo aquando a sua entrada (Faure et al., 2002). Forma-se então uma pequena abertura na extremidade do TP ou na sua proximidade, e o seu conteúdo é libertado para o interior do citoplasma da sinergídea degenerada de uma forma mais ou menos explosiva (Russell, 1992). Daqui, os gâmetas masculinos migram de forma a ocorrer fusão de um deles com a célula central, e do outro com a oosfera (Figura 1.1c), iniciando
assim o desenvolvimento do endosperma e do zigoto, respectivamente (Johnson e Preuss, 2002; Weterings e Russell, 2004; Yadegari e Drews, 2004).
Todo o trajecto seguido pelo TP desde do estigma até ao saco embrionário é suportado por uma rede molecular complexa responsável pela adesão e atracção do TP até ao seu alvo final, o saco embrionário. Muitos destes mecanismos moleculares têm sido desvendados ao longo da última década, permanecendo ainda várias questões por explicar (Higashiyama e Hamamura, 2008). A maioria das questões que permanece por responder diz respeito às moléculas responsáveis pela comunicação entre o GF e o GM na região funicular e micropilar, bem como a comunicação com os tecidos esporofíticos envolventes.
1.3. Pela estrada fora – interacções pólen–pistilo
De uma forma geral, e de acordo com os mecanismos moleculares que regulam o crescimento e a atracção do TP, todo o processo de direccionamento do TP pode ser dividido em duas fases: uma fase esporofítica, em que ocorre crescimento do TP inicialmente à superfície das papilas estigmáticas e, posteriormente, crescimento intercelular ao longo do estilete, de um modo independente do GF, e uma fase gametofítica, dependente de sinais produzidos pelo GF para conduzir o TP até ao saco embrionário (Ray et al., 1997; Higashiyama et al., 2003).
Os dois gâmetas masculinos têm de ser transportados desde o estigma até ao saco embrionário, que se encontra profundamente envolvido pelo nucelo e pelos tegumentos interno e externo do óvulo, o qual, por sua vez, se encontra rodeado pelo tecido da placenta no interior do ovário (Weterings e Russell, 2004). O TP cresce através dos diferentes tecidos do pistilo sem nunca perder a orientação do seu crescimento e atingindo a região micropilar do saco embrionário de forma bastante precisa (Higashyiama e Hamamura, 2008). Todo o processo de direccionamento do TP é controlado por múltiplas interacções celulares que envolvem vários mecanismos de sinalização quer para o reconhecimento do grão de pólen pelos tecidos da flor quer para o direccionamento do crescimento do TP no interior do pistilo até ao GF (Sanchez et al., 2004; Dresselhaus, 2006). Estes mecanismos têm sido, nos últimos anos, dos principais processos estudados no que diz respeito à reprodução nas plantas com flor.
Foram estudadas mutações em Arabidopsis em que o saco embrionário e o tecido esporofítico são afectados (bell, bel1 e short integuments1, sin1) e mutações que afectam apenas o
direccionamento do tubo polínico até ao óvulo, não afectando, no entanto, a fase anterior de crescimento do tubo polínico, até à superfície da placenta, a fase esporofítica. Em todos estes mutantes, após crescimento dos TPs no septo, estes não cresciam mais através do funículo em direcção ao micrópilo do óvulo, crescendo ao acaso sobre qualquer superfície, como por exemplo as paredes externas dos óvulos. Estes estudos indicam que diferentes sinais controlam a migração dos tubos polínicos através do tecido de transmissão e a sua emergência deste tecido em direcção aos óvulos (Hülskamp et al., 1995). Estudos semi in-vitro efectuados em Torenia fournieri utilizando estiletes polinizados cortados e co-cultivados com óvulos, permitiram verificar que, quando alguns dos óvulos eram danificados pelo calor, os tubos eram atraídos para outros óvulos viáveis (Higashiyama et al., 1998). Neste estudo foram também feitas experiências apenas com TPs co-cultivados com óvulos, verificando-se que os TPs apenas se direccionam para os óvulos quando atravessam o estigma e o estilete. Isto demonstra a importância da passagem do TP por estes tecidos de modo a adquirir competência para depois responder aos sinais de direccionamento para o GF. Este estudo sugere que o GF não é necessário para o direccionamento do TP desde o estigma até à base do estilete.
Assume-se que existem moléculas responsáveis pela atracção e crescimento do TP em todo o seu trajecto até ao saco embrionário, tendo sido efectuados vários estudos para identificar estas moléculas (Higashiyama e Hamamura, 2008). Inicialmente o direccionamento é controlado apenas pelo tecido esporofítico do pistilo, sinais moleculares presentes no estigma e no estilete serão os responsáveis por este direccionamento (McCormick e Yang, 2005).
Em Nicotiana tabacum certas proteínas arabinogalactânicas (AGPs) TTS (transmitting tissue-specific), presentes na matriz extracelular do tecido de transmissão, foram relacionadas com o direccionamento do tubo polínico para os óvulos. Estas proteínas estimulam o crescimento do tubo polínico in vitro, atraem os tubos polínicos em sistemas de cultura semi-in vivo e são necessárias para o crescimento óptimo do tubo polínico in vivo, funcionando como uma fonte de nutrientes e moléculas de adesão para os tubos polínicos em crescimento (Cheung et al. 1995; Wu et al., 2001). Outro estudo feito com Arabidopsis mostra que em mutantes no transmitting tract (ntt) que apresentam um desenvolvimento anormal do tecido de trasmissão, este tecido é fundamental para estimular o crescimento do TP. Os TP neste pistilo mutante crescem menos e mais devagar do que nos pistilos da variedade selvagem, apresentando a planta mutante uma taxa de fecundação muito reduzida (Crawford et al., 2007). Estes estudos comprovam que o tecido de transmissão é fundamental para um rápido crescimento do TP ao longo do pistilo.
O GABA (ácido γ-aminobutírico) foi identificado como possível molécula de atracção produzida pelos tecidos esporofíticos do pistilo de Arabidopsis. Esta molécula apresenta um gradiente de concentração ao longo destes tecidos, com concentrações mais baixas no estigma e concentrações mais elevadas no tegumento interno dos óvulos (Palanivelu et al., 2003). No mesmo estudo foi verificado que o gene POP2 (pollen-pistil interaction) codifica uma transaminase que degrada o GABA, contribuindo para o estabelecimento do gradiente de concentração desde o estigma até aos tegumentos do óvulo. No entanto, a actividade do GABA como molécula de atracção do tubo polínico nunca foi reproduzida in vitro, sugerindo que outras moléculas serão necessárias para actuar em conjunto com o GABA. Na verdade, pensa-se que exista não uma molécula sinal, mas sim várias moléculas componentes de um sistema de sinalização bastante complexo (McCormick e Yang, 2005). Outras moléculas de atracção produzidas pelos tecidos esporofítico têm sido descobertas tais como a quimiocianina, uma pequena proteína produzida pelos estigmas de Lilium longiflorum (Kim et al., 2003). Glicoproteínas ricas em hidroxiprolina (HRGPs – hidroxyproline rich glycoproteins) como as proteínas arabinogalactânicas (AGPs – arabinogalactan proteins) presentes no pistilo foram também relacionadas com o processo de direccionamento do tubo polínico até ao GF (Sanchez et al., 2004).
Nos estudos efectuados por Hülskamp (1995), referidos acima, os TPs não crescem em direcção ao saco embrionário nos mutantes sin1 e 47H4, em que não se desenvolve saco embrionário, indicando que o gametófito feminino poderá ser a fonte do sinal de atracção responsável pelo seu direccionamento. Estudos efectuados com os mutantes magatama (maa1 e maa3) de A. thaliana que apresentam um atraso no desenvolvimento do GF, mostram que os TPs são direccionados para o GF, mas perdem a sua orientação imediatamente antes de entrarem no micrópilo (Shimizu e Okada, 2000). Estes trabalhos mostram que a fase gametofítica, última fase de direccionamento do TP, pode ser dividida em duas fases: uma fase de direccionamento funicular e uma fase de direccionamento micropilar (Figura 1.2) (Shimizu e Okada, 2000; Higashiyama e Hamamura, 2008).
O mecanismo de direccionamento funicular não é ainda conhecido, algumas moléculas de atracção poderão ser libertadas directamente pelo GF maduro ou em desenvolvimento ou um sinal do GF poderá conduzir, indirectamente, à produção de um sinal de atracção pelas células dos tecidos esporofíticos do óvulo (Higashiyama e Hamamura, 2008).
este adquire a competência para atrair o TP, sendo por isso provavelmente a fonte do sinal de atracção para o TP.
Figura 1.2 – Fase gametofítica de direccionamento do TP em A. thaliana. As linhas azuis representam TPs a crescer a partir dos grãos de pólen em direcção ao GF. Nas regiões rodeados por círculos a tracejado alguns TPs perdem a sua orientação quando o GF não apresenta um desenvolvimento correcto, sugerindo que existem sinais produzidos pelo GF responsáveis pelo direccionamento do TP nesta região. Nesta fase o direccionamento pode ser dividido numa fase funicular e numa fase micropilar. GM: gametófito masculino, GF: fametófito feminino. (Adaptado a partir de Higashiyama
et. al., 2003).
Higashiyama e colaboradores (1998) fizeram estudos em Torenia fournieri, sugerindo que as sinergídeas são as células responsáveis por esta atracção. Na maioria das espécies, o saco embrionário está envolvido por várias camadas de tecido esporofítico, o que não facilita o acesso ao saco embrionário, no entanto, Torenia apresenta um saco embrionário nu, que quando maduro, é projectado para o exterior do óvulo através do micrópilo, permitindo assim o fácil acesso às células do saco embrionário. Num sistema in vitro, estes autores observaram o crescimento dos TPs em direcção a ovários dissecados, verificando que os tubos polínicos são atraídos directamente para o saco embrionário nu, sendo que, uma vez atraídos, não mais abandonam a região micropilar do saco embrionário. Verificaram ainda que, quando alguns dos óvulos eram danificados pelo calor, os tubos eram atraídos para outros gametófitos femininos viáveis. Estes resultados sugerem fortemente que o saco embrionário produz sinais de atracção para o TP. Seguidamente, os mesmos autores (Higashiyama et al., 2001) efectuaram trabalhos de ablação com laser das diferentes
verificando que a ablação da célula central e da oosfera não afectava o direccionamento normal do TP, mas que a ablação das sinergídeas alterava este direccionamento. Quando as duas sinergídeas eram eliminadas, nenhum saco embrionário atraía tubos polínicos. Se apenas uma sinergídea era eliminada, a atracção diminuía mas não cessava, sugerindo que as duas sinergídeas são capazes de atrair o TP e que uma sinergídea é suficiente para gerar um sinal de atracção, embora este seja mais forte quando as duas células estão presentes. As sinergídeas foram assim identificadas como sendo a fonte do sinal de atracção a curta distância para os tubos polínicos. O mesmo foi provado em A. thaliana mas utilizando uma abordagem diferente. Kasahara e colaboradores (2005) efectuaram estudos com o gene MYB98, que no óvulo é expresso exclusivamente nas sinergídeas. O mutante myb98 apresenta um GF e sinergídeas aparentemente normais, no entanto, a estrutura do aparelho filiforme é anormal, não possuindo as expansões da parede celular típicas desta estrutura. Neste mutante os TPs crescem desde a placenta até ao funículo mas não daqui até ao micrópilo. Tal como na Torenia fournieri em Arabidopsis o direccionamento micropilar do TP parece ser controlado pelas sinegídeas (Kasahara et al., 2005; Punwani e Drews, 2008).
Até à data, os processos moleculares que regulam o desenvolvimento e as funções das sinergídeas, tais como os sinais de atracção e factores que controlam o direccionamento do tubo polínico até ao saco embrionário, e a libertação dos gâmetas nas sinergídeas, não são ainda conhecidos (Punwani e Drews, 2008).
Uma das questões fundamentais acerca da reprodução nas plantas com flor é determinar qual a base molecular para o direccionamento do tubo polínico até ao saco embrionário. Recentemente Márton e colaboradores (2005) identificaram uma pequena proteína em Zea mays, a ZmEA1 (Zea mays EGG APPARATUS1), produzida pelas sinergídeas e pela oosfera, necessária para a atracção do tubo polínico até ao gametófito feminino. O gene ZmEA1 é expresso exclusivamente nestas células antes da fecundação, após a qual a sua expressão diminui progressivamente até desaparecer por completo nos embriões mais desenvolvidos, já com 10 dias. Esta proteína possui um domínio transmembranar, e para determinar a sua localização foi utilizada uma proteína de fusão ZmEA1:GFP C-terminal, observando-se expressão da ZmEA1 não só nas sinergídeas e oosfera mas também no aparelho filiforme e nas células do nucelo micropilar, sugerindo estes resultados que a região C-terminal do domínio transmembranar da proteína é clivada para funcionar como molécula sinal nos tecidos circundantes. A ZmEA1 é uma proteína candidata para o sinal de atracção produzido pelas sinergídeas em Zea mays.
Recentemente Okuda e colaboradores (2009) identificaram em Torenia fournieri polipéptdos ricos em cisteína (CRPs - cysteine-rich polypeptides) pertencentes a um sub-grupo das proteínas tipo defensinas: LURES, como moléculas de atracção produzidas pelas sinegídeas. Neste estudo foram isoladas sinergídeas de T. fornieri e identificados transcritos que codificam proteínas candidatas a moléculas de atracção. Foram encontradas duas CRPs abundantemente expressas nas sinergídeas, e que são secretadas para a superfície do aparelho filiforme. Verificaram também que estas moléculas são capazes de atrair TPs in vitro. Foi ainda demonstrado pela utilização de oligomeros antisense para os LUREs que quando estes oligomeros são injectados no GF há uma diminuição da atracção dos TPs para o GF. Este estudo sugere fortemente que as LUREs são as moléculas de atracção produzidas pelas sinergídeas em T. fornieri.
Não se sabe no entanto, se existe apenas uma molécula de atracção produzida pelas sinergídeas ou várias moléculas que poderão funcionar de forma redundante ou em conjunto (Higashiyama e Hamamura, 2008). Será mais provável que existam várias moléculas, ou uma complexa rede de sinalização uma vez que não há nenhum estudo em que a mutação de um gene ou diminuição da sua expressão tenha conduzido a um bloqueio total do crescimento dos TPs para o saco embrionário.
Até à data, nenhuma molécula sinal para o direccionamento do TP foi ainda identificada em A. thaliana. Vários estudos apontam as proteínas arabinogalactânicas (AGPs) como candidatas a moléculas de sinalização para o direccionamento do tubo polínico até ao saco embrionário (Coimbra et al., 2007).
1.4 AGPs – Proteínas Arabinogalactânicas
As AGPs encontram-se amplamente distribuídas no reino das plantas, desde as Briófitas até às Angiospérmicas, sendo particularmente abundantes nas paredes celulares, membranas plasmáticas e secreções extracelulares (Majewska-Sawka e Nothnagel, 2000). As AGPs são uma classe de glicoproteínas ricas em hidroxiprolina e altamente glicosiladas, estruturalmente bastante complexas (Showalter, 2001; Johnson et al., 2003). Foram já identificados, em Arabidopsis, 47 genes que codificam as estruturas proteicas de AGPs (Schultz et al., 2000, 2002). Em Arabidopsis, as AGPs podem ser divididas em 4 sub-famílias, de acordo com as características do seu núcleo proteico: as AGPs clássicas, os péptidos AG, as AGPs ricas em lisina e as AGPs tipo fasciclina, FLAs
um núcleo proteico rico em Pro, Ala, Ser e Thr, possuem uma sequência N-terminal, que é removida da proteína madura, um domínio rico em resíduos de Prolina/Hidroxiprolina e um domínio hidrofóbico C-terminal contendo uma sequência sinal para uma âncora lipídica glicosilfosfotidilinositol (GPI). Os péptidos AG possuem um núcleo proteico curto, com cerca de 10 a 15 resíduos de aminoácidos. As AGPs ricas em lisina apresentam um pequeno domínio rico em lisina, e as FLAs são AGPs que possuem um ou dois domínios de fasciclina, podendo ou não apresentar uma âncora GPI (Schultz et al., 2000, 2002; Sun et al., 2005; Seifert e Roberts, 2007).
Figura 1.3 – Representação esquemática da estrutura das AGPs. a) AGPs clássicas. b) AGPs ricas em lisina. c)
Péptidos AG. d) FLAs. Lys: domínio rico em lisina.
Diversos estudos mostram que as AGPs desempenham funções importantes em diferentes aspectos do desenvolvimento e crescimento das plantas. Estão envolvidas na proliferação celular, na expansão celular, na diferenciação celular, no reconhecimento entre células, na embriogénese somática, no crescimento do tubo polínico e na morte celular programada (Majewska-Sawka e Nothnagel, 2000).
comunidade científica mostra que as AGPs são alvo de uma regulação muito refinada e são diferencialmente expressas durante o desenvolvimento da planta, mais precisamente durante a fase de reprodução sexuada da planta (Coimbra e Salema, 1997; Coimbra e Duarte, 2003; Pereira et al., 2006; Coimbra et al., 2007)
No entanto, os resultados obtidos recorrendo à utilização de MAbs dirigidos para a porção glicosídica destas proteínas, identifica grupos de AGPs com o mesmo tipo de epítopos glicosídicos, não permitindo identificar quais as AGPs específicas que se encontram presentes num determinado tecido.
1.5 AGPs – moléculas de sinalização?
As AGPs foram sempre consideradas fortes candidatas a mediadoras de interacções celulares e reguladoras do crescimento celular. No entanto, o mecanismo exacto, através do qual as AGPs poderão mediar a sinalização entre células, permanece ainda desconhecido. Um possível mecanismo que pode explicar o funcionamento das AGPs como moléculas de sinalização poderá estar associado à clivagem da âncora GPI.
Pensa-se que a maioria das AGPs estejam ancoradas à membrana plasmática por âncoras lipídicas glicosilfosfatidilinositol (GPI) (Borner et al., 2002; Schultz et al., 2004). A presença desta âncora proporciona um potencial mecanismo pelo qual as AGPs poderão estar envolvidas em processos de sinalização e atracção polarizada, muito importante para muitas das funções sugeridas para estas proteínas (Gaspar et al., 2001). Esta âncora poderá fornecer um mecanismo de libertação das AGPs a partir da membrana citoplasmática, eventualmente por acção de enzimas como as fosfolipases (C ou D), para o meio extracelular (Gaspar et al., 2001; Showalter, 2001). Para além desta hipótese, vários outros modelos existem para explicar a possível função das AGPs como moléculas sinal. Num outro modelo, o facto de as AGPs serem proteínas muito ricas em carbohidratos e de muitos açúcares estarem envolvidos em diversas vias de transdução de sinal nas plantas, sugere que as AGPs poderão ser processadas enzimaticamente para libertar oligossacarídeos que funcionarão como sinais, que se ligam a receptores da membrana citoplasmática relacionados com um sistema de transducção de sinal (Showalter, 2001). No entanto, estudos mais aprofundados são necessários para compreender melhor como poderão estas moléculas funcionar como sinais nas complexas interacções celulares. Em 2006, Lamport e
GPI seria clivada, permitindo que as AGPs passassem da membrana plasmática para o espaço periplasmático, deste espaço para a parede celular e, por fim, para o espaço extracelular.
1.6 AGPs no trajecto do tubo polínico
Vários estudos identificam e sugerem o envolvimento de AGPs específicas na reprodução sexuada das plantas. Estudos recentes de Acosta-García e Vielle-Calzada (2004) revelaram que uma AGP clássica, a AGP18, é essencial para a iniciação da gametogénese feminina em A. thaliana. A AGP18 é expressa não só nas células do tecido reprodutor feminino (célula mãe do megásporo, células do nucelo adjacente, megásporo funcional e gametófito feminino), mas também no embrião em desenvolvimento e no grão de pólen maduro. Em mutantes AGP18-RNAi, não ocorre crescimento nem divisão mitótica do megásporo, não havendo formação do gametófito feminino, indicando que esta AGP é fundamental para a iniciação da gametogénese feminina.
Estudos de RT-PCR mostraram que os genes de duas outras AGPs clássicas, AGP6 e AGP11, são expressos especificamente nos grãos de pólen e tubos polínicos (Pereira et al., 2006), tendo sido posteriormente verificado que de facto estão envolvidas no desenvolvimento do grão de pólen (Coimbra et al., 2009) e no crescimento do tubo polínico em A. thaliana (Coimbra et al., 2009).
Em Nicotiana tabacum foram identificadas AGPs específicas do tecido de transmissão, as proteínas TTS (transmitting tissue-specific), e relacionadas com o direccionamento do tubo polínico para os óvulos. Este estudo mostrou que estas proteínas estimulam o crescimento do tubo polínico in vitro, atraem os tubos polínicos em sistemas de cultura semi-in vivo e são necessárias para o crescimento óptimo do tubo polínico in vivo, funcionando como uma fonte de nutrientes e moléculas de adesão para os tubos polínicos em crescimento (Cheung et al., 1995).
Recentemente, Coimbra e colaboradores (2007), mostraram que as AGPs funcionam também como marcadores moleculares durante o desenvolvimento do pistilo de Arabidopsis. A marcação para estas proteínas surge nas células da região central do estigma e do tecido de transmissão do estilete, ou seja, no trajecto seguido pelo tubo polínico na fase inicial do seu crescimento. O mesmo tipo de marcação tinha sido anteriormente observado noutras espécies como Amaranthus hypocondriacus (L.), Actinidia deliciosa (A. Chev.) (Coimbra e Duarte, 2003), Nicotiana alata (Wu et al., 2001) e Nicotiana tabacum (Cheung et al., 1995). Ainda no mesmo estudo, Coimbra e
aparelho filiforme de Arabidopsis. Outros trabalhos de imunolocalização de AGPs realizados anteriormente em A. hypochondriacus (Coimbra e Salema, 1997) e A. deliciosa (Coimbra e Duarte, 2003) revelaram um padrão de marcação dos epítopos das AGPs semelhante, ou seja, um padrão de marcação que corresponde precisamente ao trajecto seguido pelo tubo polínico, na fase final do seu processo de direccionamento até atingir o saco embrionário. Estes dados sugerem o possível envolvimento das AGPs no processo de atracção e direccionamento do tubo polínico.
Em 2001, Higashiama e colaboradores identificaram as sinergídeas como fonte de moléculas de atracção para o tubo polínico, mas nenhum estudo, até agora, identificou a natureza bioquímica desta molécula de atracção em A. thaliana. Os resultados obtidos em Arabidospsis e noutras espécies, e a possível função das AGPs como moléculas sinal, permitem levantar a hipótese do seu envolvimento no processo de direccionamento do tubo polínico até ao saco embrionário. A forte marcação, muito específica, com MAbs, da parede do saco embrionário e das sinergídeas, mais precisamente do aparelho filiforme, é uma forte indicação de que as AGPs poderão ter alguma função relacionada com as secreções destas células, através do micrópilo. A marcação dos tegumentos que envolvem o micrópilo sugere que estas moléculas, ou os resíduos de açúcares por elas libertados, poderão estar relacionados com o fenómeno de atracção do tubo polínico em crescimento até ao saco embrionário. O mecanismo molecular de acção das AGPs é ainda desconhecido, principalmente devido às dificuldades impostas pela estrutura bastante complexa destas glicoproteínas. Mas, por outro lado, a estrutura complexa da sua porção glicosídica torna as AGPs uma potencial fonte de pequenas moléculas de sinalização, como oligossacarídeos biologicamente activos (Coimbra et al., 2007).
Será de enorme interesse determinar que sinais são percepcionados pelo tubo polínico durante o seu direccionamento até ao saco embrionário, bem como determinar se estes sinais são específicos para as diferentes espécies, e determinar se as AGPs ou fragmentos de AGPs estarão entre os sinais responsáveis por este direccionamento, conforme os dados de imunocitoquímica sugerem.
1.7 Objectivos
O objectivo central do presente trabalho é produzir ferramentas que permitam identificar os genes de AGPs de A. thaliana que podem estar envolvidos nos mecanismos de sinalização que
ocorrem durante o processo de dupla fecundação, mais precisamente durante o direccionamento do tubo polínico para o saco embrionário. Para tal:
(i) seleccionar-se-ão as AGPs expressas nos tecidos femininos por análise de dados de experiências de microarrays relativos à expressão das AGPs nos carpelos, nas sinergídeas, na célula central, na oosfera, no pólen e em gâmetas masculinos e atendendo-se à similaridade entre as diferentes proteínas;
(ii) isolar-se-ão sequências da região promotora de 7 genes de AGPs previamente seleccionadas em (i) (AGP1, AGP9, AGP10, AGP12, AGP 15, AGP20 e AGP23) e estas serão usadas para a obtenção de construções com GFP;
(iii) por último, plantas que expressam a GFP sob o controlo de sequências dos promotores endógenos das AGPs seleccionadas serão obtidas para futura localização destas proteínas nos tecidos do pistilo.
2. Materiais e métodos
2.1 Material vegetal e condições de crescimento
Foram usadas plantas de Arabidopsis thaliana (L.) variedade selvagem (ecótipo Columbia 0 [Col 0]) (NASC, The European Arabidopsis Stock Center). As sementes foram colocadas directamente em vasos com terra. Estes permaneceram na obscuridade, a 4ºC durante 3 dias, após os quais foram transferidos para uma câmara de crescimento com um fotoperíodo de dias curtos (9h de luz, 22ºC, 70% humidade relativa). Após 3 a 4 semanas, as plantas foram transferidas para um fotoperíodo de dias longos (16h de luz, 22ºC, 70% humidade relativa).
2.2 Alinhamento de sequências proteicas
Todas as sequências proteicas foram obtidas a partir do NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/). Foi feito o alinhamento múltiplo das sequências proteicas das diferentes AGPs de Arabidopsis thaliana, e obteve-se uma àrvore filogenética utilizando o T-Coffee (http://www.igs.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi, Notredame et al., 2000). Os alinhamentos daqui resultantes foram tratados utilizando o software GeneDoc, versão 2.6 (http://www.nrbsc.org/downloads, Nicholas et al., 1997). A árvore filogenética obtida no T-Coffe foi desenhada utilizando o software TreeView, versão 1.4 (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html, Page, 1996).
2.3 Estirpes bacterianas e vectores
Neste estudo foram utilizadas as seguintes estirpes bacterianas: Escherichia coli estirpe DH5α (Hanahan, 1983), Escherichia coli estirpe TOP10 (Invitrogen) e Agrobacterium tumefaciens estirpe GV3101::pMP90::pSOUP (Koncz e Shell, 1986), e os seguintes vectores: pENTR/D-TOPO (Invitrogen) e pNLS3xGFPnost-pGII (Shinobu Takada, Universidade de Tübingen, modificado por Milly Ron, Universidade da California, Berkeley).
2.3.1 Meios e condições de crescimento
Para as culturas de células de E. coli foi utilizado o meio de Luria Bertani (LB) (bacto-triptona 1% [p/v], extracto de levedura 0,5% [p/v] e NaCl 1% [p/v] para os meios líquidos e com agar 1,5 % [p/v] para os meios sólidos). Todas as culturas de E. coli foram incubadas a 37ºC, durante 12 - 16 h . Para as culturas de células de A. tumefaciens foi utilizado o meio YEP (bacto-peptona 1% [p/v], extracto de levedura 1% e NaCl 0,5% [p/v] para os meios líquidos e com agar 1,5 % [p/v] para os meios sólidos). Todas as culturas de A. tumefaciens foram incubadas a 28ºC, durante 2 dias. Todos os meios de cultura foram esterilizados durante 20 min a 120ºC num autoclave. Os meios sólidos foram armazenados a 4ºC e os meios líquidos à temperatura ambiente. Em todos os meios suplementados com antibióticos, estes foram adicionados após esterilização do meio, e estes conservados a 4ºC. Para a conservação das bactérias contendo os plasmídeos de interesse, as culturas bacterianas foram armazenadas em stocks de glicerol 40% a -80ºC.
2.3.2 Preparação de células de E. coli DH5α competentes
Para a preparação de células de E. coli DH5α competentes foi utilizado um método descrito por Inoue et al. (1990), com pequenas alterações. Foi inoculada uma colónia isolada de E. coli em 5 mL de meio LB líquido e esta cultura foi incubada a 37ºC, com agitação a 250 rpm, durante 12 - 16 h. Esta cultura foi adicionada a 250 mL de SOB (triptona 2% [p/v], extracto de levedura 0,5% [p/v], NaCl 10mM e KCl 2,5 mM aos quais foram adicionados, após esterilização, 20 mL de SOC 50x [glucose 1M, MgCl2 0,5 M e MgSO4 0,5 M]) e foi incubada a 18ºC, com agitação a 250 rpm, até que o valor de absorvância a 600 nm estivesse próximo de 0,6. Atingido este valor de absorvância a cultura foi arrefecida em gelo, com agitação durante 10 min. A partir deste ponto todos os passos seguintes foram efectuados em gelo. As células foram depois recolhidas por centrifugação a 3000 rpm (SORVALL Evolution, rotor SLA-1500) durante 15 min a 4ºC. O sobrenadante foi removido e o sedimento ressuspendido em 80 mL de tampão TB a 4ºC (PIPES 10 mM, MnCl2 55 mM, CaCl2 15 mM e KCl 250 mM, pH 6,7 com KOH, armazenado a 4ºC). Esta suspensão celular foi mantida em gelo durante 10 min após os quais foi centrifugada a 3000 rpm durante 15 min a 4ºC. As células foram ressuspendidas em 20 mL de tampão TB a 4ºC suplementado com DMSO a uma
concentração final de 7% (v/v). As células foram divididas por alíquotas de 150 µL, congeladas imediatamente em azoto líquido e armazenadas a -80ºC.
2.3.3 Transformação de células de E.coli TOP10 e E.coli DH5α
A transformação de células de E. coli foi efectuada utilizando o método de choque térmico. As células de E. coli foram descongeladas em gelo e foram adicionados 10 – 100 ng de DNA (1 - 5 µL) a uma alíquota de E. coli One Shot TOP10 (Invitrogen) ou a 150 µL de células de E. coli DH5α competentes. Estas foram incubadas em gelo durante 30 min. De seguida foi provocado um choque térmico transferindo as células rapidamente para um banho a 42ºC durante 30 segundos. Após o choque térmico as células foram mantidas durante 5 minutos em gelo. Foram depois adicionados 250 µL de SOC (Invitrogen) e incubados a 37ºC durante 1 h com agitação a 150 – 200 rpm. As células foram depois espalhadas em placas com meio LB sólido suplementado com canamicina 50 µg/mL e incubadas a 37ºC, durante 12 - 16 h.
2.3.4 Preparação de células de A. tumefaciens gv3101::pMP90::pSOUP competentes
Para a preparação de células de A. tumefaciens competentes foi inoculada uma colónia de A. tumefaciens isolada em 5 mL de meio YEP líquido suplementado com rifampicina 50 µg/mL (resistência cromossómica), gentamicina 25 µg/mL (resistência do plasmídeo Ti) e tetraciclina 2 µg/mL (resistência do pSOUP) e incubada a 28ºC durante 2 dias, com agitação a 150 rpm. Estes 5 mL foram inoculados em 200 mL de meio YEP líquido suplementado com rifampicina 10 µg/mL, gentamicina 15 µg/mL e tetraciclina 2 µg/mL e incubados a 28ºC com agitação a 150 rpm até que a absorvância a 600 nm se encontrasse entre 0,5 – 1. Após atingir a absorvância desejada a cultura foi arrefecida em gelo durante 10 min. Esta cultura foi depois centrifugada a 5300 rpm (rotor SLA 1500, SORVALL Evolution), durante 20 min a 4ºC. A partir deste passo as células foram mantidas sempre em gelo. O sobrenadante foi descartado e as células foram ressuspendidas em 200 mL de tampão TE (Tris-HCl 100mM e EDTA 10 mM, pH8), previamente arrefecido a 4ºC. As células foram novamente centrifugadas a 5300 rpm (rotor SLA 1500, SORVALL Evolution), durante 20 min a 4ºC. O sobrenadante foi descartado e as células ressuspendidas em 15 mL de meio LB.
Estas foram divididas por alíquotas de 200 µL, congeladas imediatamente em azoto líquido e posteriormente armazenadas a -80ºC.
2.3.5 Transformação de A. tumefaciens
A transformação de células de A. tumefaciens foi efectuada utilizando o método de freeze– thaw (Weigel e Glazebrook, 2002). As células foram descongeladas em gelo e foram adicionados 0,5 – 10 µg (10 – 30 µL). Estas células foram mantidas 5 min em gelo e depois transferidas para azoto líquido durante 5 min. Foram depois submetidas a choque térmico por incubação num banho a 37ºC durante 5 minutos. Após este choque térmico foram adicionados 300 µL de meio YEP líquido às células e estes foram incubados a 28ºC, durante 4 h com agitação a 150 rpm. De seguida, a suspensão bacteriana foi espalhada sobre meio YEP sólido suplementado com rifampicina 10 µg/mL, gentamicina 15 µg/mL, tetraciclina 2 µg/mL e canamicina (50 µg/mL). As placas com as bactérias foram depois incubadas a 28ºC, durante 2 dias.
2.4 Técnicas de manipulação e análise de DNA
2.4.1 Extracção de DNA genómico de A. thaliana
O DNA genómico foi extraído de acordo com o método de Li e Chory (1998) com algumas alterações. Cerca de 300 mg de tecido foliar foram congelados em azoto líquido e macerados com um pilão e um almofariz, pré-arrefecidos (-80ºC). O material vegetal foi transferido para um tubo tipo Falcon e após evaporação do azoto líquido foram adicionados 5 mL de tampão de extracção (Tris–HCl 100 mM pH 7.5, NaCl 500 mM, EDTA 50 mM, β-mercaptoetanol 10 mM e SDS 1% [v/v]) pré-aquecido a 65ºC. Após agitação vigorosa esta suspensão foi incubada a 65ºC durante 10 min, com agitação ocasional. De seguida foram adicionados 1,67 mL de acetato de potássio 5 M, e após agitação vigorosa a suspensão foi incubada em gelo durante 20 min. O material foi depois centrifugado a 30000g (SORVALL Evolution, rotor SLA-1500) durante 20 min, a 4ºC. O sobrenadante foi transferido para um novo tubo e adicionou-se um volume de isopropanol
Para recuperação dos ácidos nucleicos a mistura foi novamente centrifugada a 20000g (SORVALL Evolution, rotor SLA-1500) durante 15 min, a 4ºC. O sobrenadante foi removido e o DNA seco com os tubos invertidos sobre papel absorvente durante c. de 40 min. O DNA foi ressuspendido em 500 µL de tampão TE (Tris-HCL 50 mM pH 8 e EDTA 10 mM pH8), transferido para um tubo Eppendorf e centrifugado durante 10 min a 4500 rpm. O sobrenadante foi transferido para um novo tubo ao qual se adicionou RNAse 0,1 mg/mL, tendo esta mistura sido incubada a 37ºC durante 1 h. Foram adicionados 500 µL de fenol:clorofórmio (1:1) e toda a solução foi centrifugada durante 10 min a 4500 rpm. O sobrenadante foi transferido para um novo tubo Eppendorf e o passo de extracção com fenol:clorofórmio foi repetido. A fase aquosa foi transferida para um novo tubo e foram adicionados 50 µL de acetato de sódio 3 M, pH 4,8 e 350µL de isopropanol. Esta mistura foi incubada a -20ºC durante 12 – 16 h. No dia seguinte o DNA foi centrifugado durante 10 min a 4500 rpm. Após remoção do sobrenadante foram adicionados 500 µL de etanol 75% a -20ºC. O etanol foi removido e o DNA foi seco num Speed-Vac durante 5 min e ressuspendido em 100 µL de tampão TE, sendo armazenado a 4ºC.
2.4.2 Electroforese em gel de agarose
Os géis de agarose (0,8 - 1% [p/v] em tampão TAE 1x [Trizma-base 40 mM, ácido acético glacial 10% e EDTA 10 mM]) continham brometo de etídio 250 ng/mL. A separação electroforética foi feita em tampão TAE 1x. Após electroforese os fragmentos de DNA foram visualizados utilizando um transiluminador de UV e fotografados com o Gene Genius Bio Imaging System (Syngene). Como marcadores de tamanhos moleculares foram utilizados o GeneRuler 1 kb Plus DNA Ladder (Fermentas) e o Lambda DNA/Eco 471 (AvaII) - Marker13 (Fermentas).
2.4.3 Reacção da polimerase em cadeia – PCR
Todos os PCRs foram efectuados num UNO-Thermoblock Thermal Cycler ou no TGradient Thermal Cycler (ambos da Biometra), quer para as reacções de PCR com Taq DNA polymerase (Fermentas) quer para as reacções de PCR com Phusion High Fidelity DNA polymerase (Finnzymes). Todos os testes de oligonucleótidos de iniciação e confirmação de bactérias que
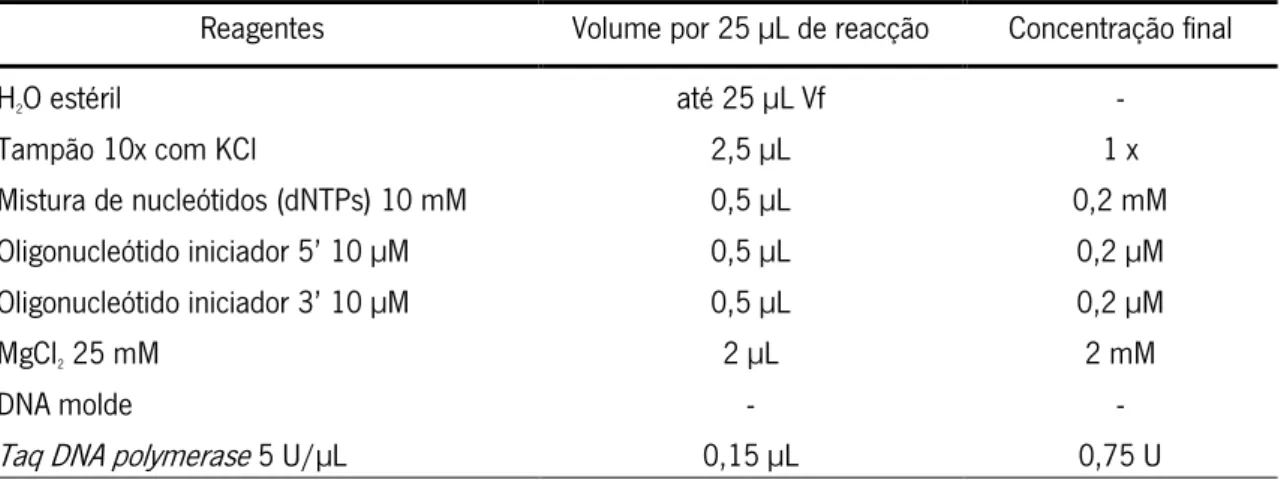
Polymerase. Para a amplificação dos fragmentos de regiões promotoras das diferentes AGPs foi utilizada a Phusion High Fidelity DNA Polymerase, de forma a minimizar a incorporação de erros nas sequências amplificadas. O gene da actina3 (ACT3) foi utilizado como gene de referência para todos os PCRs. Os programas de PCR para a Taq DNA polymerase consistiram num primeiro passo de desnaturação (2 min, 95ºC) seguido por 30 a 35 ciclos de desnaturação (45 segundo, 95ºC), de emparelhamento (45 segundos, Ta do par de oligonucleótidos iniciadores) e de extensão (1 min por 1kb de amplificado, 72ºC) seguidos por um passo de extensão final (5 - 10 min, 72ºC) e um passo de arrefecimento (4ºC). Foram utilizadas misturas de reacção de 25 µL cujos componentes estão descritos na tabela 2.1.
Tabela 2.1: Componentes das reacções de PCR utilizadas para testes de oligonucleótidos de iniciação e
PCR de colónias de bactérias.
Reagentes Volume por 25 µL de reacção Concentração final
H2O estéril até 25 µL Vf - Tampão 10x com KCl 2,5 µL 1 x Mistura de nucleótidos (dNTPs) 10 mM 0,5 µL 0,2 mM Oligonucleótido iniciador 5’ 10 µM 0,5 µL 0,2 µM Oligonucleótido iniciador 3’ 10 µM 0,5 µL 0,2 µM MgCl2 25 mM 2 µL 2 mM DNA molde - -
Taq DNA polymerase 5 U/µL 0,15 µL 0,75 U
Vf – volume final
Para os testes de oligonucleótidos de iniciação foram usados c. de 0,5 - 1 µL de DNA molde que correspondem a cerca de 50-100 ng de DNA. Para as reacções de PCR de colónias foi utilizada uma pequena porção da colónia a testar. A temperatura de emparelhamento ou annealing (Ta) para todos os oligonucleótidos de iniciação foi determinada por PCR com gradiente de temperatura. A temperatura de cada par de oligonucleótidos de iniciação foi calculada com base na equação 2.1, e em todos os PCR com gradiente de temperatura foram utilizadas temperaturas 5ºC acima e 5ºC abaixo da Tm de cada oligonucleótido de iniciação:
G C
A T
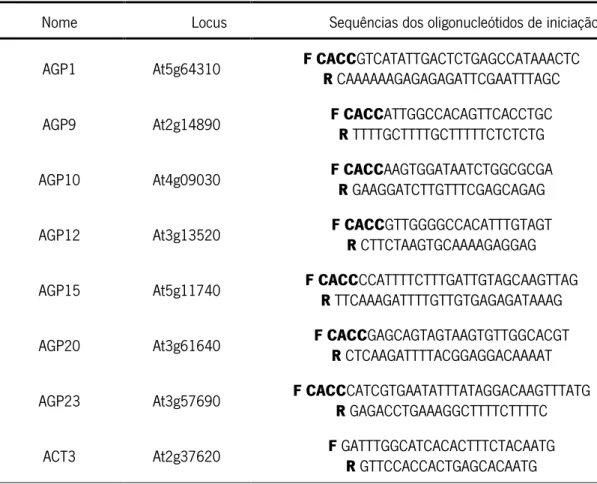
Foram amplificados fragmentos das regiões promotoras de 7 AGPs: da AGP1, da AGP9, da AGP10, da AGP12, da AGP15, da AGP20 e da AGP23 utilizando os oligonucleótidos de iniciação descritos na tabela 2.2, a partir de DNA genómico extraído de folhas de A. thaliana (2.4.1). As sequências promotoras foram isoladas, sempre que possível, desde o final da UTR do gene mais próximo situado a montante, até ao codão de iniciação do gene de interesse. Para os genes que apresentavam regiões promotoras com mais do que cerca de 3000 bp foram isoladas sequências desde 3000 – 3200 bp a montante do codão de iniciação da tradução do gene de interesse até ao respectivo codão de iniciação. A todos os oligonucleótidos de iniciação à direita foi adicionada uma sequência CACC (ver oligonucleótidos representados a negrito na tabela 2.2) à extremidade 5’ de modo a permitir a clonagem direccional destas sequências de DNA no vector pENTR/D-TOPO (Invitrogen).
Tabela 2.2: Sequências dos oligonucleótidos de iniciação utilizados para amplificação dos fragmentos
das regiões promotoras das AGPs (representados no sentido 5’→3’).
Nome Locus Sequências dos oligonucleótidos de iniciação
AGP1 At5g64310 F CACCGTCATATTGACTCTGAGCCATAAACTC R CAAAAAAGAGAGAGATTCGAATTTAGC AGP9 At2g14890 F CACCATTGGCCACAGTTCACCTGC R TTTTGCTTTTGCTTTTTCTCTCTG AGP10 At4g09030 F CACCAAGTGGATAATCTGGCGCGA R GAAGGATCTTGTTTCGAGCAGAG AGP12 At3g13520 F CACCGTTGGGGCCACATTTGTAGT R CTTCTAAGTGCAAAAGAGGAG AGP15 At5g11740 F CACCCCATTTTCTTTGATTGTAGCAAGTTAG R TTCAAAGATTTTGTTGTGAGAGATAAAG AGP20 At3g61640 F CACCGAGCAGTAGTAAGTGTTGGCACGT R CTCAAGATTTTACGGAGGACAAAAT AGP23 At3g57690 F CACCCATCGTGAATATTTATAGGACAAGTTTATG R GAGACCTGAAAGGCTTTTCTTTTC
ACT3 At2g37620 F GATTTGGCATCACACTTTCTACAATG R GTTCCACCACTGAGCACAATG F – oligonucleótido de iniciação direito; R – oligonucleótido de iniciação esquerdo.
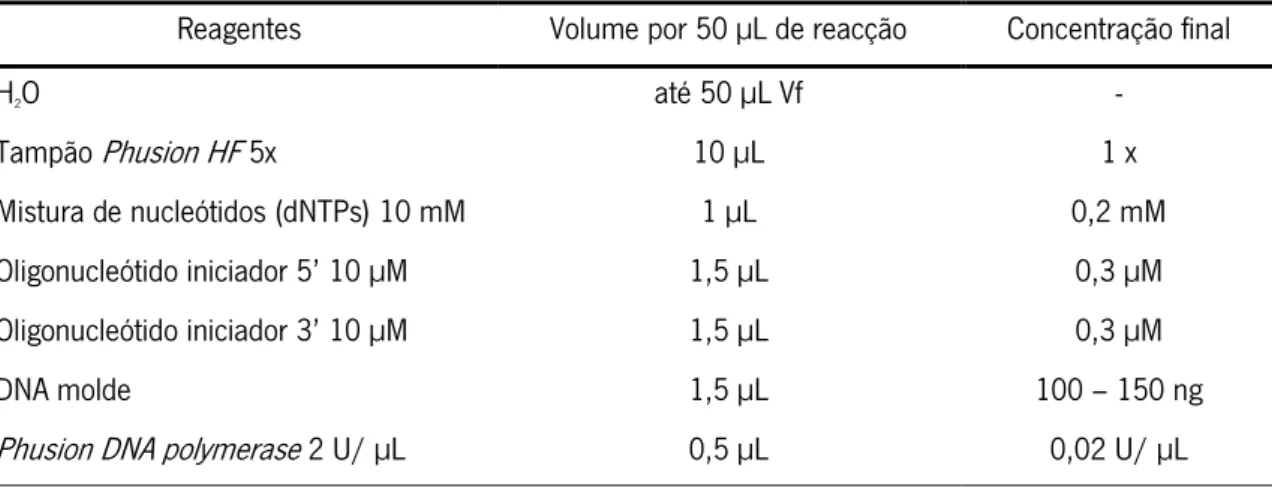
Os programas de PCR para a Phusion High Fidelity DNA Polymerase consistiram num primeiro passo de desnaturação (30 segundos, 98ºC) seguido por 30 a 35 ciclos de desnaturação (5 - 10 segundos, 98ºC), de emparelhamento (30 segundos, Ta do par de oligonucleótideos iniciadores) e de extensão (15 - 30 segundos por 1kb de amplificado, 72ºC) seguidos por um passo de extensão final (5 - 10 min, 72ºC) e um passo de arrefecimento (4ºC). Foram utilizadas misturas de reacção de 50 µL cujos componentes estão descritos na tabela 2.3.
Tabela 2.3: Componentes das reacções de PCR utilizadas para a amplificação dos fragmentos de
regiões promotoras das diferentes AGPs.
Reagentes Volume por 50 µL de reacção Concentração final
H2O até 50 µL Vf - Tampão Phusion HF 5x 10 µL 1 x Mistura de nucleótidos (dNTPs) 10 mM 1 µL 0,2 mM Oligonucleótido iniciador 5’ 10 µM 1,5 µL 0,3 µM Oligonucleótido iniciador 3’ 10 µM 1,5 µL 0,3 µM DNA molde 1,5 µL 100 – 150 ng
Phusion DNA polymerase 2 U/ µL 0,5 µL 0,02 U/ µL
Vf – volume final
Todos os produtos de PCR foram analisados por electroforese em gel de agarose (2.4.2). No caso das reacções para a síntese dos fragmentos de DNA correspondentes às regiões promotoras, todo o volume de reacção (50 µL) foi carregado no gel e as bandas de interesse foram excisadas o mais rápido possível de modo a evitar a danificação dos fragmentos de DNA (2.4.6).
2.4.4 Extracção de RNA de óvulos de A. thaliana
Os óvulos foram isolados a partir de flores nos estados de desenvolvimento 12 e 13 (Smyth et al., 1990). As flores foram cortadas pelo receptáculo e as sépalas, pétalas e estames foram removidos. Os óvulos foram extraídos utilizando agulhas hipodérmicas e colocados imediatamente em tampão de lise RLT do RNeasy Plant Mini Kit (Qiagen), em gelo. Em cada novo processo de extracção o material vegetal foi colocado em novo tampão de lise RLT. No final de cada extracção
do RNA. O RNA de todas as amostras foi extraído utilizando o RNeasy Plant Mini Kit (QIAGEN) seguindo as instruções do fabricante. As amostras de RNA foram tratadas com DNaseI (Promega) para evitar a contaminação com DNA. Para analisar a quantidade e a qualidade do RNA extraído uma alíquota desta amostra, 5 µL, foi separada por electroforese em gel de agarose 1,5% (2.4.2).
2.4.5 RT-PCR em tempo real
As reacções de transcrição reversa foram feitas com o Reverse Transcription System (Promega), a 42ºC (30 min), usando como oligonucleótido de iniciação oligo(dT)15. O cDNA dos óvulos de A. thaliana foi amplificado utilizando o iQ™ SYBR® Green Supermix no termociclador iQ™ 5 Real-Time PCR Detection System (Bio-Rad). A expressão do gene da ubiquitin-conjugating enzyme (UBC9) foi utilizada como controlo e para a normalização dos resultados. Foram efectuados duplicados de todas as reacções de RT-PCR em tempo real para todos os genes. As misturas de reacção foram feitas para 25 µL de volume total em placas de polipropileno para 96 reações (Microseal Semi-Skirted 96-well PCR Plates, Bio-Rad) fechadas com adesivos para microplacas (Optical Sealing Tape, Bio-RAd), contendo os componentes descritos na tabela 2.4. Os oligonucleótidos de iniciação utilizados nesta experiência estão listados na tabela 2.5.
Tabela 2.4: Componentes das reacções de RT-PCR em tempo real utilizadas para amplificação
do cDNA de óvulos de A. thaliana
Reagentes Volume por 25 µL de reacção Concentração final
H2O estéril 10,5 µL -
iQ™ SYBR® Green Supermix 2x 12,5 µL 1 x
Oligonucleótido iniciador 5’ 10 µM 0,5 µL 0,2 µM
Oligonucleótido iniciador 3’ 10 µM 0,5 µL 0,2 µM
cDNA molde 1 µL -
Vf – volume final
Os programas de PCR utilizados consistiram num primeiro passo de desnaturação. Os programas de PCR utilizados consistiram num primeiro passo (4 min, 94ºC), seguido por 40 ciclos de desnaturação (30 segundos, 94ºC), de emparelhamento (30 segundos, Ta do par de oligonucleótidos iniciadores) e de extensão (30 segundos, 72ºC). Obtiveram-se as curvas de
dissociação para cada gene, aquecendo as amostras de 55ºC até 95ºC para verificar a especificidade da amplificação de forma a confirmar a ausência de formação de dímeros de oligonucleótidos de iniciação ou de qualquer outro produto inespecífico. Foram obtidas curvas padrão utilizando uma série de diluições (10-1, 10-2, 10-3) de cDNA de óvulos para todos os genes, em duplicados, de modo a determinar a eficiência dos oligonucleótidos de iniciação. Foram obtidas as curvas de amplificação e valores de CT (threshold cycle), número de ciclos em que se acumula uma quantidade de produto amplificado suficiente para gerar um sinal de fluorescência detectável) para cada gene. Os dados foram analisados utilizando o programa iQ5 2.0, Standard Edition Optical System Software versão 2.0 (Biorad). Todos os valores de CT foram exportados para o MS Excel (Microsoft) e analisados utilizando o método CT ( Livak e Schmittgen, 2001).
Tabela 2.5: Sequências dos oligonucleótidos de iniciação utilizados para amplificação do cDNA
de óvulos de A. thaliana (representados no sentido 5’→3’).
Nome Locus Sequências dos oligonucleótidos de iniciação
AGP1 At5g64310 R CTTCAGTCGGAGAATCGG F AATCCTCTGCTTCACCAC AGP9 At2g14890 F ATCTGTATCGTCCTCATCG R ATGTTGTGACTGGTGGTG AGP10 At4g09030 R CGCTAACTCAATTCTTAATACAAC F CTCGCTGCTGGATCTTTAG AGP12 At3g13520 F CACAACTCATCATTCGCACCAAAG R CCACAATAGCCACCATCAACACC AGP15 At5g11740 R ACGAAATCACCATCACAGTAAC F CACCAGCACCAAGTCCTAC AGP16 At2g46330 R ACCACCATTAGCAAATACGC F TCATCATTTTCGTCGGATCA AGP20 At3g61640 R AGCGAATAAAGCGATTACGG F GGTTTTTGGTTGTCCGTGTC AGP21 At1g55330 F TCATCACAACACAAATCAAAACA R TGGTACAAACATGGCAGCAT
UBC9 At4g27960 R AAATCCCACGATCAAATTCC F GTTTCACCACCCTTTCTTC
2.4.6 Recuperação de DNA a partir de géis de agarose
Todas as bandas contendo DNA de interesse foram rapidamente excisadas do gel de agarose de forma a minimizar os danos causados pela exposição aos raios UV. O DNA foi recuperado através do QIAquick Gel Extraction Kit (Quiagen), seguindo as normas do fabricante.
2.4.7 Minipreparação de DNA plasmídico
Todas as extracções de DNA plasmídico para rastreio de clones positivos e utilização nos processos de sub-clonagem foram realizadas recorrendo ao High-Speed Plasmid Mini kit (Avegene) seguindo as instruções do fabricante.
2.4.8 Digestão de DNA plasmídico
A digestão de DNA plasmídico para rastreio após clonagem e para clonagem no vector de destino foram realizadas de acordo com as instruções do fabricante das enzimas de restrição utilizadas (Fermentas e New England BioLabs). Os resultados das restrições foram analisadas por electroforese em gel de agarose (2.3.2), e no caso das clonagens, a banda que continha o fragmento de DNA de interesse foi excisada e purificada conforme descrito acima (2.3.6).
2.4.9 Sequenciação
Todas as sequenciações foram efectuadas pela empresa 4 Base Lab (Alemanha). Todos os dados provenientes das sequenciações foram analisados utilizando os programas Chromas versão 1.45 (Conor McCarthy, Griffith University) e o Clustal w (http://www.ebi.ac.uk/Tools/clustalw2/index.html, Larkin et al., 2007).
2.5 Produção das linhas de A. thaliana transgénicas
2.5.1 Ligação dos fragmentos amplificados no vector pENTR/D-TOPO
As construções proAGP::NLS:3xGFP utilizadas para a produção de linhas de A. thaliana que expressam a GFP sob o controlo dos promotores endógenos das diferentes AGPs foram obtidas utilizando o sistema de clonagem Gateway (Invitrogen). A tecnologia Gateway é um sistema universal de clonagem e sub-clonagem de sequências de DNA. Uma vez inseridas neste sistema as sequências de DNA podem ser facilmente transferidas entre diferentes vectores utilizando locais específicos de recombinação. O vector pENTR/D-TOPO permite uma clonagem fácil e rápida de fragmentos de DNA com extremidades cegas, constituindo uma forma de entrada no sistema Gateway. Estes vectores possuem locais de recombinação específicos attL para recombinação do vector de entrada com um vector de destino, um local de clonagem direccional que permite a ligação de produtos de PCR de extremidades cegas e possuem o gene de resistência à canamicina para selecção em E. coli (ver anexo I para representação esquemática das reacções de recombinação). As sequências das regiões promotoras das diferentes AGPs foram obtidas por PCR (2.4.3) e purificadas a partir de géis de agarose (2.4.6). Estas sequências foram clonadas no vector de entrada pENTR/D-TOPO (Gateway, Invitrogen) utilizando o pENTR Directional TOPO Cloning Kit (Gateway, Invitrogen), seguindo as instruções do fabricante, com algumas alterações. As reacções de ligação constituídas pelos componentes descritos na tabela 2.6 foram incubadas durante 5 min à temperatura ambiente e depois transferidas para gelo.
Tabela 2.6: Componentes das reacções de ligação para o vector pENTR/D-TOPO.
Reagentes Volume por 3 µL de
reacção Concentração final
Produto de PCR 0,25 - 2 µL razão molar 1:1
Solução Salina 0,5 µL NaCl 0,2 M e MgCl
2 0,01M
H2O estéril até 3 µL Vf -
Vector pENTR/D-TOPO 0,5 µL 1,25 ng/µL
De seguida procedeu-se à transformação de uma alíquota de E. coli One Shot TOP10 (Invitrogen) por cada construção (2.3.3). Os clones positivos foram seleccionados primeiro através de PCR de colónias (2.4.3), e depois confirmados por análise de restrição (2.4.8), utilizando para isso duas enzimas de restrição que fazem a excisão do fragmento de inserção. Todos os vectores obtidos foram sequenciados (2.4.9) utilizando o oligonucleótidos de iniciação universal M13 Forward e o oligonucleótido de iniciação esquerdo utilizado para amplificação de cada uma das sequências por PCR (tabela 2.2), de modo a confirmar a presença do fragmento de inserção no local e na orientação correctos.
2.5.2 Linearização dos vectores de entrada
Após sequenciação dos vectores obtidos em 2.5.1 procedeu-se à introdução das sequências inseridas no vector pENTR/D-TOPO no vector de destino. Antes deste passo foi necessário linearizar estes vectores com enzimas de restrição adequadas, uma vez que o vector de destino possui o mesmo gene de resistência que o vector de entrada, a canamicina, evitando-se assim o aparecimento de falsos positivos nesta segunda clonagem. Foram usadas diferentes enzimas para os diferentes vectores de modo a evitar o corte do fragmento de interesse e o corte dos locais de recombinação attL (2.4.8). Todas as digestões foram efectuadas durante 12 - 16 h, de modo a garantir que todos os vectores presentes na solução eram linearizados. Os resultados das restrições foram analisadas por electroforese em gel de agarose (2.3.2), e a banda correspondente ao vector linearizado foi excisada e purificada conforme descrito acima (2.3.6).
2.5.3 Obtenção das construcções pAGP::NLS:3xGFP
Para a localização celular das AGPs seleccionadas foi utilizado o vector de destino pNLS3xGFPnost-pGII de Shinobu Takada (Universidade de Tübingen, Alemanha) no qual foi introduzida uma cassete Gateway (Invitrogen) utilizando os locais de corte da enzima de restrição Sal I, (Milly Ron, Universidade da Califórnia, Berkeley). As sequências promotoras foram transferidas para o vector de destino através de uma reacção para cada uma das inserções (Gateway, Invitrogen) utilizando o Gateway LR Clonase II Enzyme Mix (Invitrogen), seguindo as instruções do