Universidade do Minho
Escola de Ciências
Paulo César Fernandes da Silva
novembro de 2017
The yeast DEL assay with dominant
genetic markers
Paulo César Fernandes da Silva
The y
eas
t DEL assa
y wit
h dominant genetic mark
er
s
Universidade do Minho
Escola de Ciências
Paulo César Fernandes da Silva
novembro de 2017
The yeast DEL assay with dominant
genetic markers
Trabalho realizado sob orientação do
Professor Doutor Björn Fredrik Johansson
e do
Professor Doutor Rui Pedro Soares de Oliveira
Dissertação de Mestrado
ii D E C L ARAÇÃO
Nome: Paulo César Fernandes da Silva
Endereço Electrónico: pcfernandesdasilva@gmail.com Telemóvel: 933196427
Número de identificação civil: 14551372
Título da Dissertação: The yeast DEL assay with dominant genetic markers Orientadores:
Professor Doutor Björn Fredrik Johansson Professor Doutor Rui Pedro Soares de Oliveira Ano de Conclusão: 2017
Designação do Mestrado: Mestrado em Genética Molecular
É AUTORIZADA A REPRODUÇÃO PARCIAL DESTA DISSERTAÇÃO, APENAS PARA EFEITOS DE INVESTIGAÇÃO, MEDIANTE DECLARAÇÃO ESCRITA DO INTERESSADO, QUE A TAL SE COMPROMETE;
Universidade do Minho, ___/___/______
iii A G RA DEC IMENTOS
Agradeço calorosamente,
Aos meus orientadores Björn Johansson e Rui Oliveira por toda a disponibilidade, motivação, confiança, Ciência e Razão, que foram os grandes pilares do nosso trabalho.
Aos meus colegas de trabalho, Flávio, Humberto, Miguel, Gabriel, Bia, Alexandra, Cláudia, Rosana, Pedro, Eduardo, Helena, Filipa, Sofia, Leandro e Mário por todo o companheirismo, entreajuda, boas conversas e gargalhadas.
Aos técnicos do Departamento de Biologia, e em particular à Núria e ao sr. Luís por toda a assistência indispensável e sempre oportuna.
À minha família, à Marisa e aos meus amigos por todo o amor, apoio e orientação que tão importantes foram em todos os momentos da minha vida.
v A B S TRAC T
The yeast DEL assay is a genotoxicity test that works by measuring the frequency of reversion
of two disrupted his3 alleles sharing approximately 400 bp of homology by intrachromosomal
recombination. This event leads to recovery of histidine prototrophy and deletion (DEL) of the
intervening LEU2 marker and leucine auxotrophy. This assay has provided results displaying high
correlation for well-known carcinogens with standardized and validated in vitro genotoxicity assays such as the Ames test. In this work, a novel version of the yeast DEL assay containing dominant genetic markers (dDEL) using antibiotic resistance genes KanR and Hph was developed to expand its application to wild-type yeast strains. The dDEL system enables quantitative measurement of genotoxicity using industrially relevant yeast strains lacking auxotrophic markers. For the construction of the dDEL assay, a shuttle plasmid was assembled from seven linear DNA fragments as a genetic support of the dDEL system, part of which was subsequently integrated in the HIS3 locus of the desired strains. The genetic constructions were performed through the yeast homologous recombination machinery. The yeast dDEL system was established in the laboratory strains CEN.PK 112-3A and RS112 (substituting the original DEL cassette), and in the industrial ethanol production strain PE-2. The functionality of the dDEL assay was assessed with hydrogen peroxide in a comparison with the original DEL assay. The DEL and dDEL systems in laboratory strains performed in a similar manner, and, interestingly, dDEL presented the highest relative increase in the recombination frequency. The dDEL assay in the PE-2 strain was used to evaluate the genotoxicity of furfural, which is a main fermentative inhibitor present in lignocellulosic hydrolysates. In this work, furfural was positively detected as genotoxic by the RS112 strain, although it displayed no clear genotoxic damage in the PE-2 strain, perhaps reflecting differences in the DNA repair machinery. This result supports the notion that performing genotoxicity resistance and other tests in the desired relevant biotechnological strains is vital in order to select stronger and more robust industrial yeast strains for a specific purpose.
vii RE S UM O
O ensaio DEL em levedura é um teste de genotoxicidade que funciona através da medição da
frequência de reversão de dois alelos his3 interrompidos, partilhando aproximadamente 400 pb
através de recombinação intracromossomal. Este evento leva à recuperação da prototrofia para a
histidina e a supressão (DEL) do marcador interruptor LEU2 e à auxotrofia para a leucina. Este
ensaio forneceu resultados com alta correlação para carcinogéneos bem conhecidos com ensaios in vitro de genotoxicidade padronizados e validados, tal como o teste de Ames. Neste trabalho, foi desenvolvida uma nova versão do ensaio DEL em levedura, contendo marcadores genéticos
dominantes (dDEL) usando os genes de resistência a antibióticos KanR e Hph, para expandir a
sua aplicação a estirpes de levedura selvagens. O sistema dDEL permite a medição quantitativa de genotoxicidade usando estirpes de leveduras industrialmente relevantes sem marcadores auxotróficos. Para a construção do ensaio dDEL, foi elaborado um plasmídeo vaivém a partir de sete fragmentos lineares como suporte genético para o sistema dDEL, parte do qual foi depois
integrado no locus genómico HIS3 nas estirpes desejadas. As construções genéticas foram
desempenhadas através da maquinaria de recombinação homóloga de levedura. O sistema dDEL foi construído nas estirpes laboratoriais CEN.PK 102-3A e RS112 (para substituir a cassete DEL original), e na estirpe industrial produtora de etanol PE-2. A funcionalidade do ensaio dDEL foi avaliada com peróxido de hidrogénio em comparação com o ensaio DEL original. Os sistemas DEL e dDEL em estirpes laboratoriais tiveram um desempenho semelhante e, curiosamente o dDEL apresentou o mais elevado aumento relativo da frequência de recombinação. O ensaio dDEL na estirpe PE-2 foi usado para avaliar a genotoxicidade de furfural, que é um dos principais inibidores presente em hidrolisados lenhocelulósicos. Neste trabalho, o furfural foi positivamente detectado como genotóxico pela estirpe RS112, contudo não demonstrou danos genotóxicos evidentes na estirpe PE-2, possivelmente reflectindo diferenças na maquinaria de reparação de DNA. Este resultado apoia a noção de que o desempenho de testes de resistência à genotoxicidade, entre outros, nas estirpes industriais desejadas é crucial para se selecionar estirpes industriais de levedura mais robustas para um propósito específico.
ix TA B L E OF C ONTENTS Declaração ... ii Agradecimentos ... iii Abstract ... v Resumo... vii Table of contents ... ix List of abbreviations ... xi
List of figures ...xiii
List of tables ... xv
1. Introduction ... 17
1.1. Yeast, our oldest partner in biotechnology ... 17
1.2. DNA damage repair in yeast... 19
1.3. Homologous recombination repair ... 22
1.4. In vitro genotoxicity detection assays... 25
1.5. The yeast DEL assay ... 28
1.6. Validation of the yeast DEL assay ... 31
1.7. The yeast DEL assay with dominant genetic markers ... 33
1.8. Lignocellulosic hydrolysates as source of carbon (and inhibitors)... 36
1.9. The yeast dDEL assay in industrial relevant yeast strains ... 38
1.10. Objectives... 39
2. Material and methods ... 41
2.1. Yeast and bacterial strains ... 41
2.2. Maintenance and growth conditions... 41
2.3. Plasmid and gDNA purification ... 42
x
2.5. Plasmid pPS1 construction ... 44
2.6. Genomic integration of the dDEL cassette ... 45
2.7. Geneticin disc diffusion assay... 47
2.8. Growth curves... 47
2.9. The yeast (d)DEL assays ... 47
3. Results and discussion ... 49
3.1. Plasmid construction ... 49
3.2. Integration of the dDEL cassette in the yeast genome ... 52
3.3. Disc diffusion assays ... 59
3.4. Growth curves of (d)DEL and industrial strains... 60
3.5. The yeast (d)DEL assays using hydrogen peroxide ... 61
3.5.1. Optimization of the recovery time in the dDEL assay ... 62
3.5.2. The DEL assay with RS112 ... 63
3.5.3. The dDEL assay with RS dDEL... 65
3.5.4. The dDEL assay with CEN.PK dDEL ... 67
3.5.5. The yeast dDEL assay with the industrial PE-2 strain ... 69
3.6. The yeast (d)DEL assays using furfural... 70
4. Conclusions... 75
5. Future perspectives... 77
6. References ... 79
7. Appendices ... 91
xi L I S T OF A B BRE VIATIONS Ade Adenine bp Base pairs CA Chromosomal aberration DEL Deletion
dDEL Dominant deletion
(d)DEL Deletion and dominant deletion
DSB Double-strand break
EDTA Ethylenediaminetetraacetic acid
g g force
gDNA Genomic DNA
G418 Geneticin
His Histidine
HPRT hypoxanthine-guanine phosphoribosyl transferase
HR Homologous recombination
Hyg Hygromycin B
H2O2 Hydrogen peroxide
IVMN In Vitro Micronucleus
KAc Potassium acetate
LB Lysogeny broth
Leu Leucine
LiAc Lithium acetate
MLA Mouse lymphoma TK assay
MW Molecular weight
OD Optical density
OECD Organization for Economic Co-operation and Development
PCR Polymerase chain reaction
PEG Polyethylene glycol
ROS Reactive oxygen species
xii
SDS Sodium dodecyl sulfate
SSA Single-strand annealing
ssDNA Single-stranded DNA
TAE Tris-acetate-EDTA
TE Tris-EDTA
TK Thymidine kinase
YNB Yeast nitrogen base
xiii L I S T OF FI G URES
Figure 1. Substrates and products of yeast bioprocesses... 19
Figure 2. DNA damage repair pathways. ... 20
Figure 3. Mechanisms of DSB repair involving homologous sequences ... 23
Figure 4. The standard in vitro genotoxicity test battery ... 26
Figure 5. Construction of the yeast DEL system through the genomic integration of the plasmid pRS6 in the HIS3 locus... 29
Figure 6. The yeast DEL recombination system ... 30
Figure 7. Scaled representation of the plasmid pPS1 ... 34
Figure 8. Mechanistic representation of the genomic rearrangement on the yeast dDEL assay ... 35
Figure 9. Scheme indicating main routes of formation of lignocellulosic inhibitors... 37
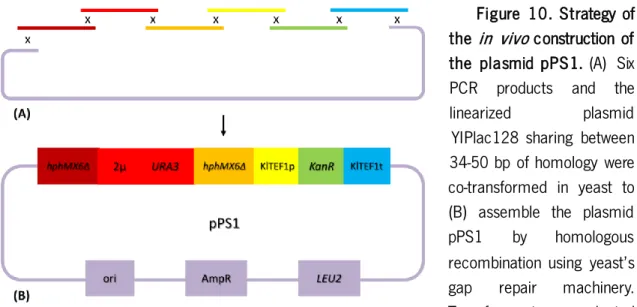
Figure 10. Strategy of the in vivo construction of the plasmid pPS1 ... 45
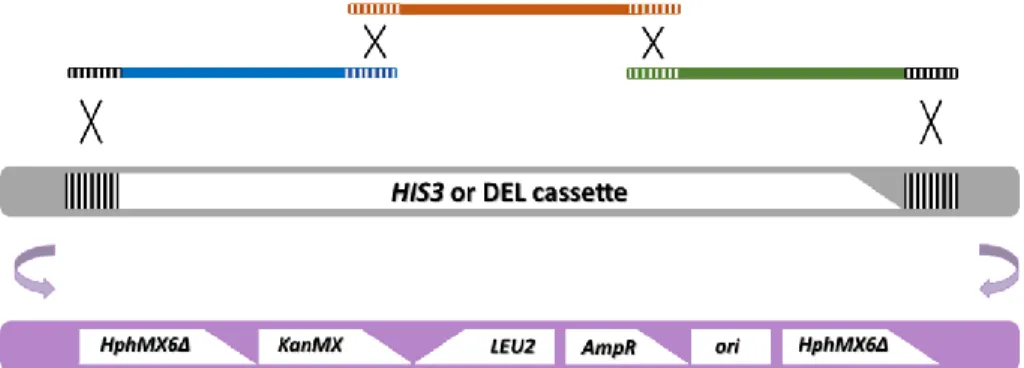
Figure 11. Strategy for the genomic integration of the dDEL cassette... 46
Figure 12. Enzymatic digestion profile of the plasmid pPS1 to assess structure and segment orientation ... 50
Figure 13. PCR amplifications of the plasmid pPS1 to assess structure and segment orientation ... 51
Figure 14. Scaled representation of the dDEL cassette integrated in the HIS3 locus ... 52
Figure 15. Selective media for the selection of CEN.PK dDEL and RS dDEL transformants . 53 Figure 16. Preliminary confirmation of dDEL cassette integration in the genome of the strains CEN.PK 102-3A and RS112 by colony PCR ... 54
Figure 17. Confirmation of the dDEL integration in the HIS3 locus of the strains CEN.PK dDEL and RS dDEL by PCR using purified gDNA... 54
xiv
Figure 18. Assessment of the restoration of the HphMX6 genetic marker coupled with the
deletion of the disrupting sequence by colony PCR ... 55
Figure 19. Confirmation of the dDEL integration in the HIS3 locus of the strain PE dDEL by
PCR using purified gDNA ... 57 Figure 20. Phenotype comparison among all the (d)DEL strains used in this work ... 58 Figure 21. Disc diffusion assays using geneticin (G418) to evaluate the strength of the promoters KlTEF1p and AgTEFp... 60
Figure 22. Growth curves of the S. cerevisiae strains used in the (d)DEL assays and the wild-type strain PE-2 ... 61 Figure 23. Comparison of dDEL frequencies and cell viabilities in the yeast dDEL assays for different recovery times ... 63
Figure 24. Recombination frequencies and cell viabilities in the yeast DEL assay with the strain RS112 treated with 4 mM hydrogen peroxide ... 64
Figure 25. Recombination frequencies and cell viabilities in the yeast dDEL assay with the strain RS dDEL treated with 4 mM hydrogen peroxide ... 66
Figure 26. Recombination frequencies and cell viabilities in the yeast dDEL assay with the strain CEN.PK dDEL treated with 4 mM hydrogen peroxide ... 68
Figure 27. Recombination frequencies and cell viabilities in the yeast dDEL assay with the strain PE dDEL treated with 4 mM hydrogen peroxide ... 69
Figure 28. Recombination frequencies and cell viabilities in the yeast dDEL assay with the strain PE dDEL treated with furfural ... 70
Figure 29. Recombination frequencies and cell viabilities in the yeast DEL assay with the strain RS112 treated with furfural ... 71
xv L I S T OF TA B LES
Tabela 1. Standard PCR conditions per 1kb of product for 30-35 cycles. ... 44 Table 2. DNA segments with respective features used for the assembly of the plasmid pPS1. ... 44 Table 3. Primers used in this work... 91
17 1. I NTROD UCTION
1.1. Ye a st, o ur o ldest partner in biotechnology
Yeasts form a group of unicellular fungi that includes the baker’s yeast Saccharomyces cerevisiae, which is the most exploited unicellular eukaryotic organism in biological sciences and industry, playing important roles in fermentation industries, food and chemical production, environmental technologies, studies on human health, and fundamental biosciences (Walker, 2009). Saccharomyces cerevisiae represents a simple eukaryotic system whose genome can be easily manipulated, displaying several properties that make it suitable for biological research, such
as the rapid growth as dispersed cells, the ease of replica plating and mutant isolation, a well
-defined genetic system as it was the first eukaryotic genome to be sequenced (Goffeau et al., 1996), a remarkable DNA transformation system and a very efficient homologous recombination machinery (Sherman, 2003). In addition, as a eukaryote, yeast shares a complex internal cell structure and mechanisms with higher eukaryotes such as plants and animals, yet with highly compact genome. Therefore, because of the few ethical constraints for its manipulation, and for being a consistent framework onto which analytical methods can be developed, optimized and standardized, yeasts can be considered a representative system of a larger and more complex biological phenomenon or organism (Karathia, Vilaprinyo, Sorribas, & Alves, 2011).
Yeast is regarded as the most ancient biotechnological microorganism, being used for food processing and fermentation of alcoholic beverages for several millennia until the present days.
However, alcoholic fermentation process remained a mystery until the 19th century when Louis
Pasteur developed his vast work on microbiology. In 1857, he published his first paper on alcoholic fermentation providing very important practical and fundamental contributions based on meticulous and ingenious experiments (Barnett, 2000; Pasteur, 1857). Later, Pasteur’s experimental results demonstrated that fermentation resulted from microbial physiological functions, which led him to state in an elegant fashion that “the chemical changes of fermentation are associated with a vital activity, beginning and ending with the latter. I believe that alcoholic fermentation never occurs without either the simultaneous organization, development and multiplication of cells or the continued life of cells already formed”. His work also provided very important findings that remained to the present days as basic principles on yeast cultivation, such as the establishment of a cause-effect relationship between the presence of oxygen and the inhibition of the fermentation, which is presently known as the Pasteur effect: “[…] If, however, in
18 a similar experiment, contact with the air is allowed over a large surface area, much more yeast is produced for the same quantity of sugar consumed. The air loses oxygen as a result of its absorption by the yeast. The yeast grows vigorously in these conditions, but its capacity to ferment tends to disappear. For one part of yeast formed, only 4 to 10 parts of sugar are transformed. The yeast nevertheless retains its capacity to cause fermentation. Indeed, it appears greatly increased if it is again cultured with sugar in the absence of free oxygen.”
Although until the mid of the 20th century bacteria and filamentous fungi were the most
commonly used organisms in the development of bioprocesses, novel strategies and advances in genetic engineering enabled the emergence of new applications in yeast biotechnology (Fig. 1). These applications aimed to satisfy the demand for many products, including the production of primary and secondary metabolites, the synthesis of recombinant proteins, and biochemical transformations (Mattanovich, Sauer, & Gasser, 2014). The main reasons for the increasing use
of S. cerevisiae in industrial biotechnology are focused on the outstanding capacity of yeast to
ferment sugars as well as its tolerance to low pH values, high sugar and ethanol concentration, which in turn lower the risk of contamination. In addition, yeast is relatively resistant to inhibitors present in biomass hydrolysates, is able to grow anaerobically (Nevoigt, 2008) with high substrate uptake rates (Mattanovich et al., 2014). The robustness against stressful conditions and the high fermentative performance promotes the production of industrially relevant biochemicals such as glycerol, organic acids, sugar alcohols, steroids and isoprenoids (Nevoigt, 2008). Furthermore, the classification of S. cerevisiae as a GRAS organism (generally regarded as safe) and the combination of the industrially selected traits with the genetic engineering approaches have been providing enhanced product quality and amount in biotechnological processes.
19 Figure 1. Substrates and products of yeast bioprocesses. Main carbon sources employed in yeast bioprocesses are derived from (A) corn starch, (B) cane or beet sugar, (C) lignocellulose (corn stover, straw, wood etc.) and (D) crude glycerol from biodiesel production. Different native and engineered yeast strains convert the substrate to products of (E) primary or (F) secondary metabolism, or (G) recombinant proteins. Whole cell biocatalysis is a special case where (H) a complex substrate is biochemically transformed to (I) a product by the metabolic activity of yeast cells. Chemical structures are illustrative images only. Obtained from Mattanovich, Sauer, & Gasser (2014).
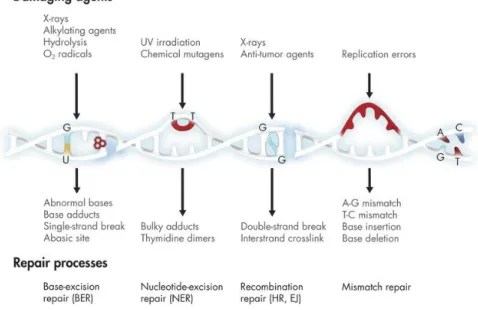
1.2. D NA damage r epair in yeast
Living organisms have developed several DNA repair mechanisms to maintain the integrity of the genomic DNA that can be damaged by genotoxic agents. These agents, or genotoxins, arise spontaneously during cellular metabolism and include reactive oxygen species (ROS) that are a by-product of aerobic metabolism and cause both base damage and strand break, hydrolytic loss of bases (especially purines) from the phosphodiester backbone and the deamination and alkylation of bases; as well as from exogenous sources including ionizing radiation which produces clusters of ROS that create single and double-strand breaks (DSB), ultraviolet (UV [UVB and UVC]) radiation which generates cyclobutane pyrimidine dimers (CPDs) and oxidative base damage, and base-damaging chemicals which alter or destroy base-pairing capacity (Boiteux & Jinks-Robertson, 2013). It is critical that cells respond efficiently to all sorts of DNA insults in order to prevent mutagenic, clastogenic (breakage of a chromosome) or aneugenic (loss of a chromosome) events that can cause genomic instability, diseases and death. As so, several mechanisms for DNA repair
20 have evolved in eukaryotic cells that ensure the accurate transmission of genetic information from one cell to its daughters, through the minimization of the number of heritable mutations.
DNA repair mechanisms are specifically activated according to the type of the damage detected. Single-strand DNA (ssDNA) damage can be repaired by direct reversal of the damage, base-excision repair (BER), nucleotide-excision repair (NER) and mismatch repair (MMR), while double-strand DNA damage is repaired by recombination repair (homologous recombination [HR] in yeast or illegitimate recombination in vertebrates [Fig. 2]). The simplest and most accurate repair mechanism is the direct reversal of damage which, in contrast to other DNA damage repair pathways, is performed in one single-step reaction. This mechanism it able to repair CPDs which are the major products of UVB and UVC radiation activated by DNA photolyase, as well as methylated bases caused by alkylating agents. In yeast, these damages are respectively repaired by a CPD-specific photolyase (PHR1) and a methyltransferase (MGT1). CPD repair occurs through
the binding of the PHR1 to the CPD-containing DNA that is then activated by light resulting in the
breaking of the covalent bond to restore DNA structure. Methylated bases are repaired through the
irreversible transference of the methyl group to a cysteine residue in the active site of the MGT1
enzyme that is subsequently degraded (Boiteux & Jinks-Robertson, 2013).
Figure 2. DNA damage repair pathways. Exposure to ionizing radiation, free radicals, and genotoxic chemicals as well as routine DNA replication, can result in DNA damage and/or mutations, activating several DNA repair pathways as well as cell cycle arrest, apoptosis and development of many diseases. Obtained from the on-line publication of Ehrlich (2017).
21 BER pathway efficiently corrects most non-bulky DNA base damage that are not addressable by direct reversal (Bauer, Corbett, & Doetsch, 2015). This mechanism is mostly concerned with endogenous damage that result from oxidative stress, hydrolysis, alkylation and deamination. BER is initiated by the recognition and hydrolysis of the damaged base by DNA N-glycosylase abasic (AP) site. Then, AP endonucleases bind to AP sites in duplex DNA and nick the phosphodiester backbone immediately 5’ of the lesion, followed by the removal of the remaining deoxyribose phosphate residues by a 3’- or 5’-phosphodiesterase. Finally, a DNA polymerase fills the gap created and a DNA ligase seal the remaining nick (Boiteux & Jinks-Robertson, 2013).
NER pathway is a multistep process that is used to repair helix-distortion lesions that interfere with base pairing and consequently impede DNA replication and transcription. This pathway is significantly important for preventing the deleterious effects of environmental mutagens such as UV radiation and mutagenic chemicals that cause CPDs and bulky base adducts (Boiteux & Jinks-Robertson, 2013). NER can be divided in two sub pathways, transcription coupled NER (TC-NER) and global genome NER (GG-NER), that only differ in the mean of detecting DNA damage. TC-NER is activated when a RNA polymerase is stalled at a CPD, signaling for the CPD repair, while GG-NER relies on a specific complex that is formed by Rad16/Rad7 and the autonomously replicating binding factor I (ABF1) (Waters, Evans, Bennett, Yu, & Reed, 2012). After lesion recognition, Rad14 and replication protein A (RPA) are recruited to form the pre-incision complex that verifies the lesion and unwinds the DNA. The endonucleases Rad1-Rad10 and Rad2 make a dual incision, 5’ and 3’ of the lesion, respectively, leaving a nucleotide gap that is filled by repair synthesis and ligation (Boiteux & Jinks-Robertson, 2013).
The basic role of the MMR pathway is to remove helical distortions that arise from misincorporation errors introduced by the DNA polymerase during replication or when non-identical duplexes exchange strands during recombination (Boiteux & Jinks-Robertson, 2013). Originally, MMR genes were identified as “mutators” (Mut in bacteria or MutS protein homolog, MSH, in yeast) because unrepaired replication errors resulted in a 100-1000 fold increase in spontaneous mutation rates (Hanne, Liu, Lee, & Fishel, 2013; Siegel & Bryson, 1967). In this mechanism, mismatched nucleotides are recognized by Msh2-Msh6, and then Msh2-MSH6, Mlh1-Pms1 and EXOl degrade the mismatched strand. The RPA protects the single-stranded DNA gap, while the replication machinery reassembles to synthesize the complementary strand and the remaining nicks are sealed by DNA ligase (Hanne et al., 2013).
22 1.3. H o mologous r ecombination r epair
All excision repair processes – direct reversal, BER, NER and MMR – rely on the presence of an undamaged strand opposite to the lesion which is used as template to restore the original sequence in the damaged strand. However in the case of DSBs, both strands are damaged so that no strand is available for repair synthesis, implying that the sequence information has to be restored by different means (Pfeiffer, Goedecke, and Obe 2000). For this, several different repair processes have evolved which can be divided in two groups: homology-dependent mechanisms (related to HR) which require extensive regions of sequence homology, varying from few dozens to hundreds base pairs (bp); and homology-independent mechanisms (related to illegitimate recombination) which dispense regions with homology, but often involves microhomology patches of ≤10-bp (Pfeiffer et al. 2000).
The mechanisms of HR repair prevail in yeast, while illegitimate recombination processes are more efficient in vertebrates. In addition, the fact that the frequency of DNA repair mediated by HR in yeast is directly related to the appearance of DSBs (T.-C. Wu & Lichten, 1994) led to the conclusion that most, if not all, recombination events are induced by DSBs (Pfeiffer, Goedecke, & Obe, 2000). In mitotic cells, the primary function of HR is to repair DSBs which arise frequently as consequence of normal metabolism or ROS derived from oxygen metabolism. On the other hand, deleterious DSBs can be caused by environmental insults, such as chemicals or radiation, yielding the most detrimental forms of DNA damage since, if unrepaired, DSBs result in broken chromosomes and loss of genetic material, genomic alterations or cell death (Aylon & Kupiec, 2004).
There are at least three mechanisms of HR that can be used to repair a DSB in a mitotic yeast cell (Pâques and Haber 1999): gene conversion, break-induced recombination (BIR), and single-strand annealing (SSA). A summarized mechanistic insight on these processes is described below (Fig. 3). DSB repair initiates with the exonucleolytic resection of the ends of the DSB in the 5’-to-3’ direction to produce 3’ single-stranded tails. In gene conversion and BIR, 3’ tails invade a homologous template creating a D-loop, where 3’ ends prime new DNA synthesis (Slade & Radman, 2011).
23 Figure 3. Mechanisms of DSB repair involving homologous sequences. DSB repair commences with the exonucleolytic resection of the ends of DSBs in the 5’-to-3’ direction to produce 3’ singlestranded tails. Gene conversion further branches into two different mechanisms. In synthesis -dependent strand annealing (SDSA), newly synthesized DNA strands formed by D-loop migration are displaced from the template and anneal to each other to restore a contiguous chromosome in a noncrossover configuration. In the homologous recombination (HR) model, two Holliday junctions are formed as the D-loop created by strand invasion pairs with the other side of the DSB and the 3’ end of the noninvading strand is extended by DNA synthesis. If both Holliday junctions are cleaved in the same way, gene conversion is not associated with crossover whereas differential cleavage results in crossover. BIR occurs when only one end of a DSB is available for recombination. Such one-ended strand invasion results in extensive DNA synthesis. Intrachromosomal single-strand annealing (SSA) enables the repair of a DSB that occurs between two flanking homologous regions (direct repeats). Resection produces single-stranded tails in which complementary strands of the duplicated sequence are exposed and can reanneal, resulting in a deletion of the intervening sequence. Obtained from Slade and Radman (2011), based on Pâques and Haber (1999).
Gene conversion is defined as a nonreciprocal transfer of genetic information from one molecule to its homologue, usually occurring between two alleles of a gene. Gene conversions can
24 be explained by two different families of models. The double-strand break repair (DSBR) model (or HR model), elaborated by Szostak and coworkers (Sun, Treco, & Szostak, 1991), and attempts to account for the strong association of gene conversions with DNA crossovers. In this model, two Holliday junctions are formed as the D-loop created by strand invasion pairs with the other side of the DSB (second-end capture) and the 3’ end of the noninvading strand is extended by DNA synthesis. The two Holliday junctions can branch migrate to enlarge the heteroduplex region. Then, Holliday junctions can be cleaved by a resolvase by cutting either the two noncrossed strands or the two crossed strands. If both Holliday junctions are cleaved in the same way, gene conversion is not associated with crossover, whereas differential cleavage results in crossover (Slade & Radman, 2011).
However, because many mitotic gene conversions were infrequently associated with crossing over and to explain observations that were not predicted by the model of Szostak et al., a second family of gene conversion models emerged, named as synthesis-dependent strand annealing (SDSA; Pâques & Haber, 1999). In SDSA, newly synthesized DNA strands formed by D-loop migration are displaced from the template and return to the broken molecule, allowing the two newly synthesized strands to anneal to each other in a noncrossover configuration. This process may be mediated by topoisomerases or helicases that disrupt the replication structure or it may occur due to the small size of the replication “bubble”. Moreover, in this mechanism DNA synthesis is conservative because all the new synthesized sequences are on the same molecule, instead of semi-conservative as in the Szostak et al. model (Pâques & Haber, 1999).
Most DSB repair events occur by a non-crossover mechanism limiting loss of heterozygosity (LOH) for markers downstream of the site of repair and preventing chromosome rearrangements (Llorente, Smith, & Symington, 2008). Thus, gene conversion usually involves short conversion segments. However, very long DNA conversions have been observed that cannot be explained by gene conversion pathways. BIR is the repair pathway responsible for these events and its central feature is that only one end of a DSB invades the homologue or sister chromatid and initiates leading and lagging strand synthesis (Pfeiffer et al., 2000). This mechanism is used to repair DSBs that arise by the collapse of the replication fork or by erosion of uncapped telomeres as only one free end is available. As BIR results in a long tract of LOH, it is suggested that it is suppressed when DSBs have two ends so that the repair is carried out by a more conservative mechanism (Llorente et al., 2008).
25 If a DSB occurs between two flanking homologous regions, the repair of the broken chromosome is very efficient and results in a deletion containing a single copy of the repeated sequence. SSA is the mechanism that appears to account for these events. SSA depends on the activity of an exonuclease that will hydrolyze the tails formed by the DSB to produce long single-stranded tails until complementary regions are exposed and can reanneal. This mechanism occurs in competition with gene conversion and previous results have revealed that deletions caused by SSA were three or four times more frequent than gene conversions which can also explain the deletions between dispersed repeated sequences in the human genome (Fishman-Lobell, Rudin, & Haber, 1992). In yeast, SSA mechanism has been found to be nearly 100% efficient when homologous regions flanking DSB are at least 400-bp, but efficiency decreases to less than 10% when the repeats are only 63-bp (Sugawara, Ira, & Haber, 2000). Moreover, in a study comparing the repair mechanism choice during homologous recombination, SSA accounted for the 99.5% of
the events for the recombination between two URA3 alleles when the double-strand break occurred
within the 6-kb integrating sequence (Agmon, Pur, Liefshitz, & Kupiec, 2009). This process is still efficient even if the repeats are separated by as much as 15-kb (Pâques & Haber, 1999).
1.4. I n v itro g enotoxicity de tection a ssays
According to the Organization’s for Economic Co-operation and Development (OECD) Genetic Toxicology Guidance Document (OECD, 2015), the purpose of genotoxicity testing is to identify substances that can cause genetic alterations, either mutations or structural damage, in somatic and/or germ cells, since chromosomal mutations are more likely to be deleterious than beneficial. These tests are designed to assist on the comprehension and evaluation of the potential risk of novel substances on human health providing information to support further regulatory decisions.
Genetic alterations differ from other types of toxicity because they may be displayed after long periods of time following the exposure, or the disease endpoint can be caused by DNA damage that occurs in a single cell at low exposures. These alterations can result in a phenotype that is then amplified as the cell divides, creating a dysfunctional group of cells within a tissue or an organ leading to health consequences, including cancer, infertility, development abnormalities or heritable genetic diseases (M. Lynch et al., 2011). A full evaluation of a chemical’s genotoxicity includes tests that can detect three major endpoints: gene mutations, structural chromosomal aberrations and numerical chromosomal abnormalities.
26 To adequately cover all the genetic endpoints, multiple tests – a test battery – must be used as no individual test outcome is fully representative for all endpoints (OECD, 2015). The standard in vitro test battery contains 1) the bacterial reverse mutation assay, 2) the in vitro mammalian
chromosomal aberration (CA) test, 3) the in vitro mammalian cell gene mutation test (MLA and
HPRT) and 4) the in vitro mammalian cell micronucleus (IVMN) test (Corvi & Madia, 2016).
Currently, the assessment of genotoxic hazard to humans usually begins with basic in vitro tests
followed by an in vivo test battery that need to cover the same endpoints as the precedent positive in vitro tests. In vivo testing will not be addressed here; only the four previously mentioned in vitro tests will be briefly described (Fig. 4).
Figure 4. The standard in vitro genotoxicity test battery. The standard Ames test uses
prokaryotic systems to detect gene mutations through the prototrophic reversion of a mutated gene. MLA/HPRT, micronucleus, and chromosomal aberration assays use mammalian cells to respectively detect gene mutations, nucleus aberrations and changes in the structure of chromosomes. Obtained from Corvi & Madia (2016).
The bacterial reverse mutation assay (also known as Salmonella or Ames assay/test) is a
short-term bacterial test carried out on Salmonella typhimurium, specifically designed to detect a wide
range of chemical substances that can produce genetic damage that leads to gene mutations,
which in turn may lead to cancer. There are several strains of S. typhimurium with mutations in
different genes involved in histidine biosynthesis that can be used in this test (Mortelmans & Zeiger,
2000). S. typhimurium cells are treated with a range of concentrations of a test chemical to
determine whether the compound can induce mutations that revert or suppress the original mutations in the histidine biosynthesis genes (Gatehouse, 2012). Therefore, bacteria that revert to
27 histidine independence can grow and form colonies in minimal media agar plates containing a trace of histidine, and mutagenic potential of the test agent is directly proportional to the frequency of phenotypic reversion.
In the in vitro mammalian chromosomal aberration test, mammalian cell lines (such as rodent cell lines and human peripheral lymphocytes) are exposed to the test substance, and then fixed, stained and observed microscopically to measure the frequency of asymmetrical structural chromosome aberrations (deletions and rearrangements). In this assay, exposure to the test substance commences approximately after 48 hours after culture initiation so that cells are at all stages of the cell cycle, as it affects the sensitivity to mutagens. Due to the requirement of cells to undergo through S phase in order to most chromosomal damage to be manifested, after exposure, cells are incubated overnight and then the cell cycle is arrested in metaphase prior to microscopic analyses. Individual cells are observed and all the information on the type of chromosomal aberration is recorded, however numerical aberrations are not detected in this test (Clare, 2012). The mouse lymphoma TK assay (MLA) and the hypoxanthine-guanine phosphoribosyl
transferase (HPRT) gene mutation assay are both in vitro mammalian cell mutation tests that have
the potential to detect mutagenic events at the thymidine kinase (tk) and HPRT genes loci, respectively. In the MLA assay, a cell line derived from a leukemia tumor in mouse with a heterozygous phenotype for the thymidine kinase gene (tk +/−) is used. The thymidine kinase (TK) is not an essential protein as it catalyses the phosphorylation of thymidine deoxyriboside (dThd) to
form deoxythymidylate (dTMP) in the salvage pathway for pyrimidine biosynthesis while de novo
synthesis is still functional (and it is TK independent). This base-sugar complex is incorporated into DNA forming a phosphodiester bridge with the adjacent sugar (Lloyd & Kidd, 2012). The MLA experimental design starts with the treatment of cells with the test substance followed by a recovery
period providing time for tk gene expression. Then, the culture is transferred to a medium
containing trifluorothymidine (TFT) and the number of colonies is counted to assess the frequency of gene damage and/or mutation. TFT is a thymidine analogue that can be incorporated into DNA
by the salvage pathway but its -CF3 group blocks base pairing and inhibits DNA replication. Thus,
if the TK enzyme is functional, cell incorporates the TFT into DNA, leading to death, and only cells with recessive homozygous phenotype (tk-/-), caused by an inactivating mutation, survive to TFT due to the cessation of the salvage pathway.
28 The HPRT gene mutation assay follows very similar methodology to the TK assay as both tests aim to detect DNA damage through forward mutations that confer resistance to a toxic chemical. The HPRT is a transferase that catalyzes the conversion of hypoxanthine to inosine monophosphate and guanine to guanosine monophosphate in the purine biosynthesis salvage pathway. After the toxicological treatments, cell culture is transferred to a medium containing 6-thioguanine (6-TG),
which similarly to TFT blocks the purine salvage pathway and only the HPRT mutants can grow
(Johnson, 2012). As the HPRT is encoded in the X chromosome, the mutants derived from males
are naturally heterozygotic and therefore easy to select for loss of function. The test can be performed in a variety of established cells lines, from which the Chinese hamster cell lines are the most widely used (OECD, 2015).
The in vitro micronucleus assay aims to identify chemicals that induce aneugenic or clastogenic activity in mammalian cells. A micronucleus is formed when a fragment of a chromosome or an intact chromosome is unable to migrate to the mitotic poles in the anaphase phase of the cell division, resulting in its separation from the main nucleus. Thus, this assay is sensitive to structural and numerical aberrations. Many cell lines are appropriate to use in this assay, including mouse lymphoma cells and primary human lymphocytes (Doherty, 2012). In this assay, cells are exposed to the test compound and then fixed and stained before being observed under the microscope for micronuclei scoring. Two major types of the micronucleus assay can be performed: the mononucleate assay and the binucleate assay. The first version of the assay analyzes cells that have completed a cell division and therefore contain only one nucleus and, in the case of a positive event, an additional micronucleous. In the second version, cytochalasin B (CB) is added to the cell culture to block cytokinesis through the inhibition of the actin filaments formation. Thus, cells will present two nuclei and, in positive events, two additional micronuclei.
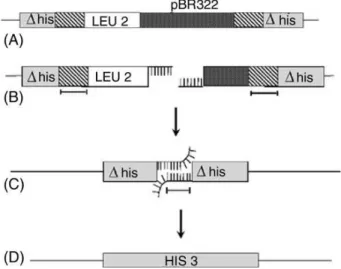
1.5. The ye ast D EL a ssa y
Due to the association among genome rearrangements, deletion and carcinogenesis, a system selecting for intrachromossomal recombination was constructed in S. cerevisiae. Such system was created through the genomic integration of the plasmid pRS6 with an internal fragment of his3 and LEU2 markers in the HIS3 locus leading to the disruption of the chromosomal HIS3 gene (R. H. Schiestl, Igarashi, & Hastings, 1988). This resulted in two copies of the his3, each having a terminal deletion (Fig. 5). This system works by measuring the recombination frequency between the two
29 his3 alleles sharing approximately 400-bp of homology with each other (Fig. 6). The recombination assay starts with a genetic insult that directly or indirectly generates a double-strand break that is further extended by exonucleases deleting the intervening sequence until homologous single stranded regions are exposed. Then, strand annealing occurs and nonhomologous single stranded overhangs are cleaved by the RAD1/RAD10 endonuclease and the ends are ligated to restore a
functional HIS3 gene. As this system results in the deletion (DEL) of the integrating plasmid it is
denominated as the yeast DEL assay (Robert H. Schiestl, 1989).
Figure 5. Construction of the yeast DEL system through the genomic integration of the plasmid pRS6 in the HIS3 locus. This resulted in two his3 deletion alleles sharing about 400-bp of homology separated by an intervening segment containing the plasmid pBR322 and the LEU2 gene. Obtained and adapted from R. H. Schiestl, Igarashi, & Hastings (1988).
The most widely used yeast DEL strain is the diploid strain RS112 which also carries
heteroallelic mutations in both ade2 genes resulting in adenine auxotrophy. Therefore, in addition
to the detection of intrachromosomal events, this strain can also be used to score the frequency of the interchromosomal gene conversion through the retrieval of the adenine prototrophy (Brennan & Schiestl, 2004). The two types of recombination were found to respond differently to various types of mutagenic and nonmutagenic agents (Robert H. Schiestl, 1989) and they are repaired by different pathways involving different genetic control and protein activation (Pâques & Haber, 1999). The haploid yeast strain RSY6 containing the same DEL construct is also available but
30 The yeast DEL assay is a simple and user-friendly genotoxicity test. Briefly, the DEL assay workflow, described in detail by Brennan & Schiestl (2004), can be divided in three steps. First, cells containing the DEL construct cultivated overnight in a synthetic complete (SC) medium lacking leucine (SC-Leu) are counted and then added to the test agent in SC-Leu medium where they are incubated for 16-18 h. Secondly, cells are recovered, counted and spread in SC, SC medium lacking histidine (SC-His) and when desired in SC medium lacking adenine (SC-Ade), to assess viability, DEL recombination frequency, and interchromosomal recombination, respectively. Finally, the number of colonies is counted after 3 days. Percent survival is calculated by dividing the viable colony count (from SC plates) by the number of cells plated following exposure (counted using a hemocytometer). DEL recombination and interchromosomal recombination frequencies are calculated by dividing the number of His+ and Ade+ cells, respectively, by the number of viable cells. Positive responses are considered for at least a two-fold increase in DEL recombination (compared to the control) and a statistically significant increase in DEL recombination frequency.
Figure 6. The yeast DEL recombination system. This assay measures the recombination frequency between two tandem his3Δ alleles (his3Δ3’, his3Δ5’), containing approximately 400-bp of homology with each other indicated by the oblique bars. (A) The DEL assay construct in the RS112 tester strain. (B) DNA insults generate a double strand break (C) leading to bidirectional degradation by exonucleases until homologous single stranded regions are exposed; (D) homologous recombination between the two his3Δ alleles results in the deletion of intervening sequences to give
a functional HIS3 gene and a reversion to histidine prototrophy. In the yeast DEL assay protocol
cells are exposed to the test agents in liquid culture and subsequently washed, counted, diluted, plated, and, after 3 days, scored for survival and HIS3 reversion on selective media plates. Obtained from Kirpnick et al. (2005).
31 1.6. Va lidation o f t he yeast D EL a ssay
The yeast DEL assay was designed to work as a screening tool for detection of clastogenicity to assess the carcinogenic potential of diverse compounds, providing easily understandable, yet reliable, data. This intrachromosomal test is induced by a wide range of compounds that include actinomycin D, camptothecin, 5-fluorodeoxyuridine, cyclophosphamide (Kirpnick et al., 2005) and nitrogen mustard (H. Schiestl, Gietz, Mehta, & Hastings, 1989) used as anti-cancer agents;
acridine, methyl acrylate, resorcinol (Kirpnick et al., 2005), acrylonitrile, o-toluidine, benzene,
safrole (Carls & Schiestl, 1994) and methyl eugenol (Brennan et al., 1996), used in many industrial applications and consisting in environmental contaminants; among other relevant
chemicals frequently used in routine scientific experiments such as hydrogen peroxide (H2O2
[Brennan, Swoboda, & Schiestl, 1994]), formaldehyde, methyl methanesulfonate, ethidium bromide (H. Schiestl, Gietz, Mehta, & Hastings, 1989), and chloroform (Brennan & Schiestl, 1998). The yeast DEL assay is also sensitive to other sources of DNA insults including ionizing and nonionizing radiation (Robert H. Schiestl, Gietz, Mehta, & Hastings, 1989), DNA single strand breaks (Kirpnick et al., 2005) and heat shock (Davidson, Whyte, Bissinger, & Schiestl, 1996).
As the yeast DEL assay is induced by a broad range of DNA insults, one might expect that its results correlate with data from validated genotoxic assays, that have provided solid and comprehensive information on which regulatory decisions have been made. The Ames test is used worldwide as an initial screening tool to detect the mutagenic potential of chemicals because of its highly predictive value for rodent carcinogenicity (Tejs, 2008). Although, many compounds that cause cancer in rodents are poorly detected by this assay. In a list containing the results obtained by the yeast DEL assay and the Ames test for 50 carcinogenic and 10 noncarcinogenic compounds, the yeast DEL assay accurately identified 92% of the chemicals, while the Ames test showed an overall accuracy of 62% (Ku, Aubrecht, Mauthe, Schiestl, & Fornace Jr, 2007). Moreover, the yeast DEL assay displayed 34% more sensitivity and 10% more specificity than the Ames test, that are respectively defined as the proportion of positive and negative outcomes that are correctly identified. Nevertheless, it is important to note that all the results in this assessment were obtained using the same yeast DEL strain, whereas the Ames assays were performed in a variety of strains and a positive result indicate that one of the Salmonella strains was sensitive to a chemical.
The IVMN assay is a chromosome aberration test that has gained widespread international
32 such as the possibility for detection of both clastogens and aneugens, chromosome breakage, chromosome loss and non-disjunction. A working group established by the European Centre for the Validation of Alternative Methods compared the available data from both IVMN and CA assays to carry out a retrospective validation of the IVMN assay (Corvi et al., 2008). The working group found 83% of global concordance between these two genotoxic assays, which led to the regulatory acceptance of the IVMN assay by the OECD. In another independent study (Kirpnick et al., 2005), the IVMN and the yeast DEL assays were compared for their ability to respond to 10 compounds (seven well-established clastogens, one aneugen and two ambiguous and controversial). The concordance between these assays in this study was close to 70% in the presence of S9 fraction (metabolic activation system obtained from an organ homogenate, usually the liver) and close to 80% in the absence of S9 fraction (the influence of S9 fraction was not examined). Owing to the high degree of concordance with the IVMN assay, the authors considered the yeast DEL assay an excellent candidate for the use as a genotoxicity assay for screening of new compounds.
The yeast DEL assay aims to overcome some of the limitations of the other genotoxicity assays, yet displaying high values of sensitivity and specificity to a broad range of genotoxins. Contrarily to the Ames test which is carried out in a prokaryotic system, the DEL assay is performed in the baker’s yeast. This eukaryotic system solves two of the main disadvantages of using a prokaryote in a genotoxicity test: low amount of heritable information and simple genomic structure, and very simple DNA repairing machinery. Moreover, the Salmonella test, the MLA and the HPRT assays were designed to detect only genetic mutations (reverse or forward), which are only one portion of the damages that DNA molecules can suffer. On the other hand, the design of the yeast DEL assay allows for the detection of chemicals that induce direct or indirect DNA strand breaks which are strongly associated with genomic rearrangements that can lead to cancer in mammals.
The yeast DEL assay also demonstrates many advantages when compared to the IVMN assay. As the DEL assay detects a genomic rearrangement, namely a DNA deletion initiated by a DSB, it can provide insight about a compound genotoxic activity, while the biological relevance of micronuclei is not clear yet. In the DEL assay, DNA deletion events are only measured in cells that are viable and capable of forming colonies. The measure of colony forming efficiency allows for a simple and convenient determination of cytotoxicity of a compound, while the IVMN assay requires extensive microscopic examination and quite expertise of the observer. Furthermore, the IVMN assay is more laborious, time consuming and complex than the yeast DEL assay given the use of mammalian cultures and microscopic analyses.
33 The yeast DEL assay is a rapid and robust method specifically designed for measuring the carcinogenic potential of several chemical and physical agents. However, some non-canonical approaches have been developed, which reinforces the versatility of this tool. Some examples will be briefly described. It was observed that the yeast strain RS112 grown with a vector containing the human gene BRCA1 displayed significant high increase in intra- and interchromosomal recombination (Spugnesi et al., 2013); and, in another study, this DEL strain was also used to identify new human proteins involved in homologous recombination, through a screening of a cDNA libraries of human genes (Collavoli, Comelli, Rainaldi, & Galli, 2008). Moreover, a high-throughput liquid version of the DEL assay was developed using the reduction of the MTS tetrazolium compound into a colored formazan product by viable recombinant yeast cells growing in selective medium with the test agent; cell proliferation was then measured by recording absorbance at 490 nm and higher absorbance values corresponded to higher rates of recombination (Hontzeas, Hafer, & Schiestl, 2007). Furthermore, a strategy to measure DEL frequency using quantitative PCR (TaqMan®) was developed; this technique used a fluorescent probe that annealed in one of the flanking sites of the DEL cassette and a set of primers that annealed in both flanking sites, and only yielded a PCR product if a recombination event occurred due to the high distance between the primers when separated by the whole cassette (Li, Cise, & Watson, 2003).
1.7. The ye ast D EL a ssa y with dominant g enetic m arkers
In this work, a novel reversion assay based on the yeast DEL assay was constructed using two dominant genetic markers. This new system was constructed analogously to the original test in
means of structure, localization, and mode of action. The new system is denominated the yeast
DEL assay with dominant genetic markers (yeast dDEL assay) and aims to be a genetic platform that enables the development of broader and more versatile applications of the DEL system by overcoming the major limitation of the DEL assay, which is the use of recessive markers (HIS3 and LEU2). The DEL assay imply the use of the same DEL tester strains which, on one hand, constitutes an advantage for the standardization of the method that is still a maturing technology for genotoxicity testing (M. Lynch et al., 2011), but on the other hand, constrains the biotechnological potential of such genetic system. By using dominant markers, the yeast dDEL assay can be used in wild-type yeast strains with no need for additional genetic modifications, which enables direct
34 genotoxicity testing in industrial or wild-type yeast strains, with the goal of screening the better performing ones.
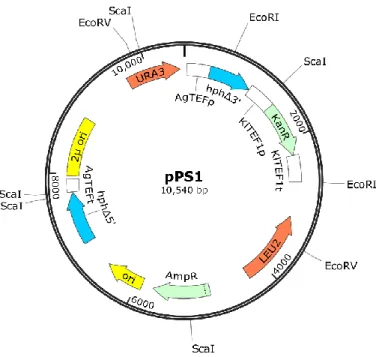
The dDEL cassette was constructed through the assembly of different genetic segments from multiple sources into a shuttle plasmid (Fig. 7). This plasmid, pPS1, contained all the features required for the dDEL assay as well as for the maintenance of the plasmid in yeast and bacteria.
These features are: KanR marker that confers resistance to geneticin (G418) in yeast and to
kanamycin in bacteria, under control of TEF1 promoter (KlTEF1p) and terminator (KlTEF1t) from
the yeast Kluyveromyces lactis; AmpR marker that confers resistance to ampicillin in bacteria;
LEU2 and URA3 genes that encode essential enzymes for de novo synthesis of the amino acid L-leucine and the pyrimidine uracil, respectively; 2-micron (2µ-ori) and pUC19 bacterial ori as origins of replication of the plasmid in S. cerevisiae and E. coli, respectively; and two alleles of the 5’ and 3’ deleted AgTEFp-Hph-AgTEFt (HphMX6) marker that confers resistance to hygromycin B in bacteria and yeast. TEF promoter and terminator were obtained from the filamentous fungus Ashbya gossypii.
Figure 7. Scaled representation of the plasmid pPS1. This plasmid was constructed through
the in vivo assembly of seven fragments by the homologous recombination mechanisms in
Saccharomyces cerevisiae. All the relevant features in the plasmid are identified and colored according to their type: promoters and terminators (white), auxotrophic markers (orange), dominant markers (green), disrupted dominant marker (blue) and origins of replication (yellow).
35 AgTEFt of the other deleted allele. Cutting sites of the restriction enzymes used to evaluate plasmid structure are displayed. This figure was designed using the software SnapGene®.
After construction of the plasmid, the dDEL cassette was integrated in the HIS3 genomic locus
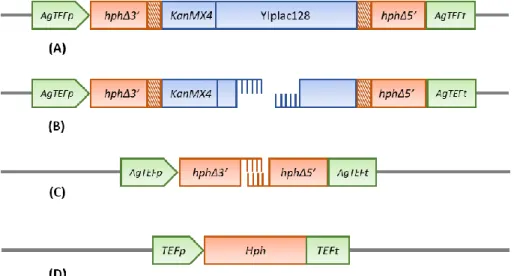
of two laboratory and one industrial strain of S. cerevisiae, by homologous recombination. The
dDEL cassette is constituted by the genetic sequence from the TEF promoter-hphΔ3’ to the hphΔ5’-TEF terminator to make a 7894-bp segment. This assay was designed in order to respond to genotoxic damage that directly or indirectly cause a DSB that is repaired through the SSA repair mechanism of homologous recombination, resulting in the deletion of the integrating sequence and reversion of the Hph gene functionality (Fig. 8). In this work, a novel version of the DEL assay was created, therefore a proof of concept of the dDEL system was carried out prior to further genotoxicity testing. For that, both DEL and dDEL – i.e. (d)DEL, assays were tested by an adapted methodology to assess the functionality of the new assay, using a well-known genotoxic compound.
Figure 8. Mechanistic representation of the genomic rearrangement on the yeast dDEL assay. (A) The dDEL construct is integrated into the HIS3 genomic locus from the partial deleted plasmid
pPS1, resulting in two copies of the hphMX6Δ marker, one with a terminal deletion at the 3’ end
and the other with a terminal deletion at the 5’ end. The striped region indicates 400-bp of homology between the two deleted alleles. (B) DNA strand breakage activates the homologous recombination repair system which leads to bidirectional degradation until single stranded regions
are exposed. (C) Annealing of homologous regions. (D) Reversion of the HphMX6 marker and
36 1.8. L ig nocellulosic hydrolysates a s so urce o f carbon (and inhibitors )
Rapid growth in global energy demand associated with a possible future shortage of fossil fuels have motivated the search for alternative renewable sources that have a lower environmental impact (Ho, Ngo, & Guo, 2014). Nowadays, the most common produced and commercialized biofuel is ethanol which is produced by yeast alcoholic fermentation from food crops with high content of starch or sugar, such as corn and sugarcane (Liao, Mi, Pontrelli, & Luo, 2016), respectively used in the USA and Brazil (Lopes et al., 2016). The sole use of starch- and sugar-rich biomass is not cost efficient and leads to competition between bioethanol and food production for farmland, which generates the idea that large-scale production of ethanol for fuel will require lignocellulosic utilization (Nevoigt, 2008). In fact, lignocellulose is the most widespread and abundant source of carbon in nature and it is the only renewable source that can provide sufficient amount of feedstock to satisfy the world’s energy and chemicals needs (van Zyl, Chimphango, den Haan, Görgens, & Chirwa, 2011). Therefore, certain energy crops or agricultural remains, including corn stover, straw, wood, forestry and paper mill discards may represent attractive low-cost substrates for fuel production (Nevoigt, 2008).
Lignocellulosic biomass is typically non edible and recalcitrant plant material primarily composed by the polysaccharides cellulose and hemicellulose and the phenolic polymer lignin that provides structural strength to the plant (Sluiter, Ruiz, Scarlata, Sluiter, & Templeton, 2010). Thus, the first step of bioethanol production using lignocellulosic material involves the conversion of the polysaccharides into fermentable sugars, such as glucose and xylose (Jayakody, Hayashi, & Kitagaki, 2015). Currently, lignocellulose degradation generally starts with a pretreatment process followed by enzymatic hydrolysis using enzyme cocktails or microorganisms. The pretreatment can be physical, chemical, biological or a combination of these, and aims to remove recalcitrance (i.e. lignin), to increase accessibility of cellulose and hemicellulose to hydrolysis (Ho et al., 2014), to breakdown macroscopic rigidity of biomass and to decrease physical barriers to mass transport (Himmel et al., 2007). Pretreatment is an essential step, and the choice of the technology used for a particular biomass depends on its composition and the byproducts produced as a result of the methodology (Kumar, Barrett, Delwiche, & Stroeve, 2009). However, current pretreatment processes, which usually rely on high temperatures, acid hydrolysis and/or pressure, generate several byproducts with various inhibitory effects on yeast fermentation (Nevoigt, 2008). The main substances that may act as inhibitors of microorganisms include phenolic compounds and other
37 aromatics, furan aldehydes, aliphatic acids, and inorganic ions (Fig. 9; [Jönsson, Alriksson, & Nilvebrant, 2013]).
The furan aldehyde furfural, which is commonly found in lignocellulosic hydrolysates, is formed by dehydration of pentose sugars such as xylose and arabinose. This compound inhibits growth of yeast and decreases ethanol yield and productivity (Jönsson et al., 2013) as it inactivates glycolytic and other enzymes such as the alcohol dehydrogenase (ADH), pyruvate dehydrogenase (PDH) and aldehyde dehydrogenase (ALDH), it induces membrane damage, chromatin changes, and DNA damage (R. M. Almeida, Modig, & Petersson, 2007; Caspeta, Castillo, & Nielsen, 2015). Although little is known about the genotoxic mechanisms on cells, furfural and other furan derivatives have been identified as carcinogens as they showed high frequency of chromatid breaks and exchanges in Chinese hamster ovary cells in the absence of a liver microsomal preparation (Stich, Rosin, Wu, & Powrie, 1981). This suggests that furfural may directly interact with DNA without metabolic activation. In addition, furfural was shown to have mutagenic effect on purified plasmid DNA, which
was correlated to the decrease of transformation efficiency in E. coli (Khan, Shamsi, & Hadi, 1995).
Moreover, in a recent study, it was found that furfural causes ROS accumulation in yeast and damages mitochondrial and vacuole membranes and the actin cytoskeleton, as well as it causes nuclear chromatin disorganization and chromatin diffusion (Allen et al., 2010).
Figure 9. Scheme indicating main routes of formation of lignocellulosic inhibitors. Furan aldehydes (HMF and furfural) and aliphatic acids (levulinic acid, formic acid and acetic acid) are carbohydrate degradation products, while lignin is the main source of phenolic compounds. Obtained and adapted from Jönsson, Alriksson, & Nilvebrant, (2013).
38 1.9. The ye ast dDEL a ssay in industrial r elevant yeast strains
Furfural appears to interact with DNA and cause direct and indirect damage, however the role of furfural in genotoxicity is still unclear. As this compound is one of the main inhibitors present in lignocellulosic hydrolysates and given the economic and ecological importance of lignocellulosic biomass on bioethanol production, the deleterious effects of furfural on yeast must be entirely comprehended. Hence, in this work, the clastogenic potential of furfural was evaluated by the yeast DEL assay in the RS112 strain, and by the yeast dDEL assay in the wild-type industrial strain PE-2. This industrial strain was isolated from indigenous yeast populations used in Brazilian bioethanol distilleries, and was found to be one of the best-performing yeast strains in industrial fermentations among the 350 strains analyzed (Basso, De Amorim, De Oliveira, & Lopes, 2008). Due to its high fermentation efficiency with prolonged persistence in the bioreactor, PE-2 is currently used in approximately 30% of Brazilian distilleries, producing around 10% of the world’s bioethanol supply (Argueso et al., 2009; Soares-Costa et al., 2014).
By applying the yeast dDEL system to industrial strains, one is creating the possibility to measure the genotoxicity of a given compound, or a mixture of substances, directly in the desired working strain. This methodology avoids erroneous extrapolation of results obtained in laboratory strains, which were carefully bred and selected for sexual reproduction, optimal growth and easy handling, whereas industrial strains have a much higher degree of genetic complexity and often show aneuploidy and/or polyploidy, poor sporulation efficiency, unstable mating type, and increased robustness against stress conditions (Steensels et al., 2014). In addition, full-genome sequencing of commercial strains and large-scale trait variation experiments have been pointed out the danger of extrapolating gene-trait connections obtained in reference laboratory strains to the species as a whole (Warringer et al., 2011), as new genetic elements were recently discovered
in industrial strains that were not identified in S. cerevisiae. This supports the notion that genetic
variation that underlies phenotypic diversity goes well beyond the single nucleotide polymorphisms (SNPs) or small InDels (Borneman et al., 2011), which resembles the discovery that many bacterial species have a pan-genome (species-wide) larger than that observed in any single strain owing to the evolution of core and strain-specific genes (Kim, Koh, Young Lim, Chung, & Rho, 2017; Lefébure & Stanhope, 2007).
39 The yeast dDEL assay was designed in a simple fashion so it can be applied to any yeast strain. Therefore, this assay can be used for the screening of genotoxic damage of different substances in a particular industrial strain, or, on the other hand, for the screening of resistant (or susceptible) yeast strains to a certain genotoxin or conditions. These evaluations can be performed using a single test agent or a complex broth that may simulate the conditions of the industrial fermentation, or even exposing the cells to other kinds of stress such as mechanical, thermal or radiation. Nevertheless, this technology provides either qualitatively and quantitatively outcomes allowing for objective and reliable analyses and comparisons.
1.10. Objectives
The main objective of this work was the development of a genetic tool – the yeast dDEL assay – that combined the sensitivity to clastogens with the applicability to wild-type S. cerevisiae strains, and that expressly responded to the well-known physiological clastogen hydrogen peroxide, as well as recognized the genotoxic potential of fermentation inhibitors present in lignocellulosic hydrolysates, such as furfural. For this, a series of intermediate tasks were accomplished. Firstly, the dDEL construct was assembled in a shuttle plasmid and, secondly, it was integrated in the genome of laboratory and industrial yeast strains; then, a proof of concept was carried out through the comparative analysis of the both (d)DEL assays using a well-characterized physiological oxidative agent; and finally, the genotoxicity of furfural was evaluated by the yeast DEL and the dDEL assays respectively in the strains RS112 and PE-2.
41 2. M A TE RIAL A ND M E THODS
2.1. Ye a st a nd bacterial strains
Throughout this work, the yeast S. cerevisiae laboratory strains CEN.PK 102-3A (Mata
ura3-52 HIS3 leu2-3,112 TRP1 MAL2-8c SUC2), RS112 (MATa/α ura3-52/ura3-52
leu2-3,112/leu2-Δ98 trp5-27/TRP5 arg4-3/ARG4 ade2-40/ade2-101 ilv1-92/ILV1 HIS3::pRS6/his3-Δ200
LYS2/lys2-801), and the industrial wild-type strain PE-2 was used for the (d)DEL experiments. After genomic integration of the dDEL cassette, these strains were denominated CEN.PK dDEL (HIS3Δ::dDEL), RS dDEL (HIS3Δ::dDEL/his3-Δ200) and PE-2 dDEL (HIS3Δ::dDEL/HIS3). The
yeast strains CEN.PK2-1C (MATa ura3-52 his3-Δ1 leu2-3,112 trp1-289 MAL2-8c SUC2
TGL3Δ::KanMX4) and CEN.PK-1C (PEP4Δ::KanMX4) were used in the disc diffusion assays. The
yeast strain CEN.PK 113-11C (Mata ura3-52 his3-Δ1 LEU2 TRP1 MAL2-8c SUC2) was used as a
control strain.
The bacterial strain Escherichia coli XL1-Blue (endA1 gyrA96(nalR) thi-1 recA1 relA1 lac glnV44
F'[ ::Tn10 proAB+ lacIq Δ(lacZ)M15] hsdR17(rK- mK+)) was used to transform, amplify, store and
extract the plasmids used in this work.
2.2. M a intenance a nd growth conditions
Yeast cultures were grown for general purposes and maintained for short-term storage in rich medium (YPAD; 2% glucose, 2% bacto-peptone [Bacto™], 1% yeast extract [Panreact], 80 mg/L adenine hemisulphate and 2% agar) in a petri dish and in a 1.5 mL tube (“stab” culture). For long term storage, a fresh overnight culture of cells was added to 30% glycerol in equal volume and stored in cryogenic vials at -80 ºC. Throughout this work, all liquid cultures were incubated at 30 ºC with agitation of 200 revolutions/minute (rpm), while cultures in solid medium were grown either at 30 ºC or at room temperature, and stored at 4 ºC for short-term storage.
For the selection of transformants with the plasmid pPS1 or with the dDEL cassette integrated in the genome, rich medium supplemented with geneticin (YPAD+G418, 200 µg/mL; Sigma-Aldrich) was inoculated with the transformant cells. In addition, the synthetic minimal media YNB+HA (2% glucose, 0.67% bacto-yeast nitrogen base without amino acids [Difco™], 80 mg/L histidine and 80 mg/L adenine) and YNB+HUA (2% glucose, 0.67% bacto-yeast nitrogen base