Alterações epigenéticas no abortamento
espontâneo
Dissertação de Mestrado em
Biotecnologia para as Ciências da Saúde
Sara Virgínia Fernandes Sampaio de Vasconcelos
Orientação: Professora Doutora Sofia Dória Príncipe dos Santos Cerveira
Co-orientação: Doutora Cristina Joana Moreira Marques
Alterações epigenéticas no abortamento
espontâneo
Dissertação de Mestrado em
Biotecnologia para as Ciências da Saúde
Sara Virgínia Fernandes Sampaio de Vasconcelos
Orientação: Professora Doutora Sofia Dória Príncipe dos Santos Cerveira
Co-orientação: Doutora Cristina Joana Moreira Marques
Composição do Júri:
Declara-se sob compromisso de honra que este trabalho foi expressamente elaborado pelo autor, como dissertação original, para o efeito da obtenção do grau de Mestre em Biotecnologia para as Ciências da Saúde, na Universidade de Trás-os-Montes e Alto Douro. Todas as contribuições não originais foram devidamente identificadas com indicação da fonte.
"Toda a nossa ciência, comparada com a realidade, é primitiva e infantil e, no entanto, é a coisa mais preciosa que temos."
Gostaria de expressar toda a minha profunda, sincera e eterna gratidão a todos aqueles que direta ou indiretamente contribuíram para a realização do presente trabalho. Agradeço a dedicação, amizade, companheirismo, apoio, compreensão e sabedoria pois sem isso seria impossível.
Ao Serviço de Genética da Faculdade de Medicina da Universidade do Porto, na pessoa do Professor Alberto Barros, expresso o meu profundo agradecimento por toda a simpatia e disponibilidade dos meios para a realização deste trabalho.
À Professora Doutora Sofia Dória, minha orientadora, pelo apoio, carinho e dedicação imprescindíveis. Soube sempre como relativizar os contratempos e foi, sem dúvida, uma constante motivação. Obrigada por tudo.
À Doutora Joana Marques, minha co-orientadora, agradeço de forma especial pela partilha de experiência e conhecimento. A realização deste trabalho seria impossível sem a Doutora Joana, que tanto aturou os meus momentos de pânico e me fez crescer com cada um. Obrigada por tudo.
À Direção do Mestrado, Professora Isabel Gaivão, pela disponibilidade e prontidão que sempre demonstrou na resolução de qualquer problema.
À Doutora Carla Ramalho, por toda a disponibilidade do material biológico e informação clínica.
Ao Diogo, Ritinha, Carlita, Dianita, Miguelito, Andreia, por terem sido sempre uns super parceiros e por todos os momentos de puras gargalhadas.
A TODAS as pessoas do Serviço de Genética que me fizeram rir todos os dias, sempre prontas ajudar e a ensinar. Sorte a minha de ter tido a oportunidade de aprender tanto com pessoas tão bonitas que me fizeram apaixonar ainda mais por esta área. Obrigada do fundo do coração!
A todos os meus grandes amigos que, felizmente, seria difícil enumerar, obrigada pela amizade eterna, pela confiança e por estarem sempre presentes incondicionalmente.
Agradeço de uma forma só minha, à minha Mãe, que apesar da distância e da saudade, está sempre presente no meu coração.
Aos Homens da minha vida, ao meu Pai e ao meu Irmão, agradeço tudo. Ao meu pai, a minha balança, que me equilibra melhor do que ninguém e que tem o poder de fazer desaparecer todos os problemas da minha vida. Ao meu irmão, a minha bússola, que me orienta sempre e que tem a opinião mais verdadeira e com mais valor. Obrigada por serem o meu porto seguro todos os dias, a minha força incondicional e os meus maiores exemplos.
À minha especial Tia Nela, por me dar sempre a oportunidade de me realizar a todos os níveis e por cuidar sempre de mim.
Ao Tiago, meu companheiro de todas as jornadas da minha vida. Obrigada pelos oito anos de amor, apoio e confiança incondicionais.
O abortamento espontâneo é considerado a complicação mais comum na gravidez. Embora apresente uma natureza heterogénea, as razões por trás de 40-50% dos abortamentos espontâneos recorrentes são ainda mal compreendidas. A epigenética tem um papel central na regulação do crescimento e desenvolvimento fetal e, por isso, torna-se relevante o estudo do papel da epigenética na etiologia da perda de gravidez humana. Os genes de imprinting estão descritos como genes cruciais no desenvolvimento intrauterino, surgindo como possíveis candidatos para este estudo. De acordo com a origem parental, a expressão destes genes pode ser controlada pela metilação do DNA, através de regiões diferencialmente metiladas. Além disso, recentemente, foi descrita outra modificação epigenética, a 5-hidroximetilcitosina (5-hmC), mas o seu papel desempenhado durante a gravidez ainda é pouco compreendido.
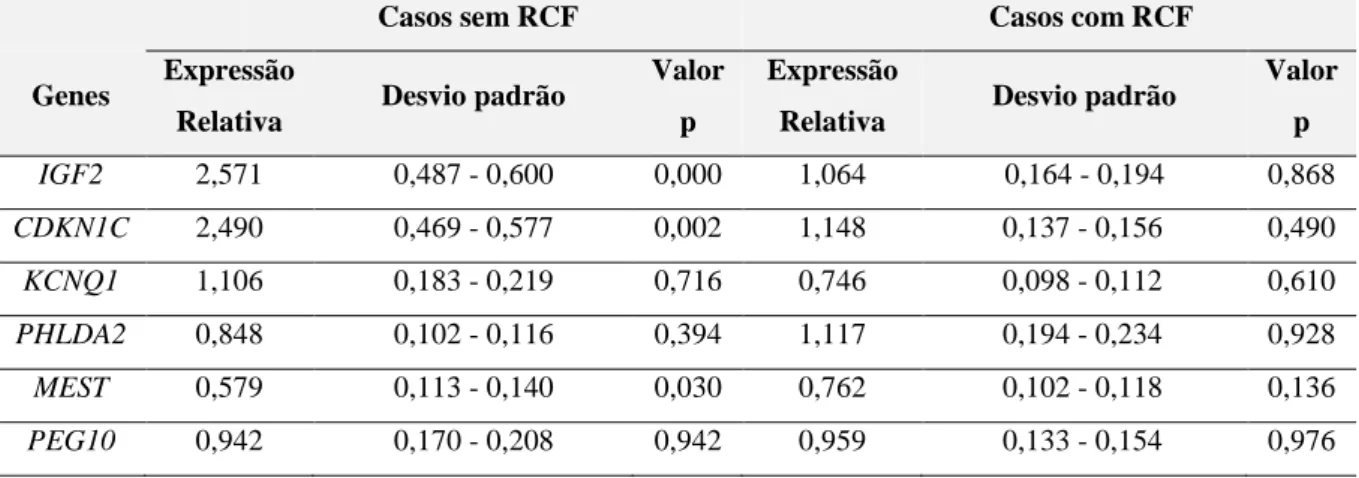
O objetivo principal deste estudo foi encontrar possíveis alterações epigenéticas em placenta e em tecido fetal derivadas de produtos de abortamento espontâneo idiopático (AEI) do segundo trimestre da gravidez, de modo, a contribuir-se para um aumento do conhecimento da etiologia da perda de gravidez humana. Para realizar o objetivo proposto, foram selecionadas um total de 70 amostras de placenta e tecido fetal de gestações perdidas entre as 12 e as 24 semanas de gestação. A análise da expressão de genes envolvidos na metilação (DNMTs) e na hidroximetilação (TETs), de seis genes de imprinting (IGF2, CDKN1C, KCNQ1, PHLDA2, MEST e PEG10) e de um gene não-imprinting (LEP) foi realizada. Além disso, foram estudados padrões de metilação de uma região promotora do gene MEST e de duas regiões controlo de imprinting (H19 DMR e KvDMR1), assim como também os níveis globais de 5-hmC na placenta e no tecido fetal de AEI.
Tanto a expressão dos genes codificantes das enzimas TET como das enzimas DNMTs revelou estar desregulada nas amostras de AEI, o que sugeriu a existência de uma dinâmica rede epigenética envolvida no abortamento espontâneo. Foram também observadas mudanças na expressão de três genes de imprinting, uma subexpressão do MEST e uma sobre-expressão do IGF2 e do CDKN1C. O IGF2 mostrou desempenhar um papel crucial tanto no desenvolvimento placentário como no desenvolvimento fetal, pelo menos durante o segundo trimestre de gravidez. A ausência de uma possível associação significativa entre a expressão génica e o perfil de metilação pode ser justificada pelo número reduzido de amostras ou pela ação de outro mecanismo epigenético. Pela primeira vez, os níveis globais de 5-hmC foram avaliados em ambos os tecidos (placenta e tecido fetal) derivados de AEI, sendo que se
revelaram mais elevados no tecido placentário. Além destes resultados, mudanças na expressão do gene LEP também foram observadas, o que sublinhou a complexidade molecular subjacente ao abortamento espontâneo humano.
Em conclusão, os resultados obtidos durante este trabalho demonstraram uma possível desregulação epigenética no AEI que, juntamente com outros mecanismos moleculares, podem conduzir em última instância à perda da gravidez ou ser apenas uma consequência de um desenvolvimento anormal da gravidez.
Palavras chaves: Abortamento espontâneo idiopático, Epigenética, Metilação do DNA, Hidroximetilação do DNA, Imprinting Genómico
Spontaneous miscarriage is considered the most common complication in pregnancy. Despite its heterogeneous nature, the reasons behind 40-50% of recurrent miscarriages are still poorly understood. Given the fundamental role of epigenetics in the regulation of growth and fetal development, it becomes important to study its influence in human spontaneous pregnancy losses. Imprinted genes are described as crucial for intrauterine development, making them possible candidates for this study. The expression of these genes is regulated by DNA methylation, through differential methylation in the two parental alleles. Moreover, the new epigenetic modification, 5-Hydroxymethylcytosine (5-hmC), was recently described, but its role in pregnancy is not well understood.
The aim of this study was to find epigenetic alterations in the placental and fetal tissue from idiopathic spontaneous abortion (ISA) products from the second trimester of pregnancy, in order to extend the knowledge of the etiology of human pregnancy losses. In this study, a total of 70 placental and fetal tissue samples from pregnancies loss between 12 and 24 weeks of gestation were colleted. Gene expression analysis of the genes involved in methylation (DNMTs) and hydroxymethylation (TETs), six imprinted genes (IGF2, CDKN1C, KCNQ1, PHLDA2, MEST and PEG10) and one non-imprited gene (LEP) was performed. Methylation patterns of a promoter region of the MEST gene and of two imprinting control regions (H19 DMR and KvDMR1), as well as the global levels of 5-hmC in placental and fetal tissue from ISA were also studied.
The expression of the genes encoding the TET and the DNMT enzymes revealed to be unregulated in the ISA samples, which suggests the existence of a dynamic epigenetic network involved in spontaneous abortion. Changes in the expression of three imprinted genes, downregulation of the MEST and upregulation of IGF2 and CDKN1C, were also observed. IGF2 appears to play a crucial role in both placental and fetal development, at least during the second trimester of pregnancy. The absence of a possible significant association between gene expression and methylation profile can be justified by the reduced number of samples or by the action of another epigenetic mechanism. For the first time, global levels of 5-hmC were evaluated in both tissues (placental and fetal tissue) derived from ISA, which were found to be higher in placental tissue. In addition to these results, changes in LEP gene expression were also observed, which underlines the molecular complexity underlying human spontaneous abortion.
In conclusion, the results obtained during this work demonstrated a possible epigenetic deregulation in ISA that, together with other molecular mechanisms, may ultimately lead to the loss of pregnancy or be only a consequence of an abnormal development of pregnancy.
Keywords: Spontaneous pregnancy losses, Epigenetics, DNA Methylation, DNA Hidroxymethylation, Genomic Imprinting
AGRADECIMENTOS ... V RESUMO ... VII ABSTRACT ... IX ÍNDICE DE FIGURAS... XIII ÍNDICE DE TABELAS... XV LISTA DE ABREVIATURAS ... XVII
1 INTRODUÇÃO ... 2
1.1 ABORTAMENTO ESPONTÂNEO -DEFINIÇÃO, EPIDEMIOLOGIA E ETIOLOGIA DA PERDA DE GRAVIDEZ HUMANA ... 2
1.2 EPIGENÉTICA E O DESENVOLVIMENTO PLACENTÁRIO E FETAL ... 5
1.2.1 Metilação do DNA ... 7
1.2.2 Hidroximetilação do DNA ... 11
1.3 IMPRINTING GENÓMICO DURANTE O DESENVOLVIMENTO INTRAUTERINO ... 13
1.3.1 Genes de imprinting do cromossoma 11 e o desenvolvimento intrauterino ... 17
1.3.1.1 Região Controlo de Imprinting - H19 DMR ... 17
1.3.1.2 Região Controlo de Imprinting - KvDMR1 ... 19
1.3.2 Genes de imprinting do cromossoma 7 e o desenvolvimento intrauterino ... 20
1.3.2.1 Gene de imprinting MEST e gene de imprinting PEG10 ... 20
1.4 GENES NÃO-IMPRINTING NO DESENVOLVIMENTO INTRAUTERINO – O EXEMPLO DA LEPTINA ... 21
2 OBJETIVOS ... 24
3 MATERIAL E MÉTODOS ... 26
3.1 SELEÇÃO DE AMOSTRAS ... 26
3.2 EXTRAÇÃO DE RNA E DNA ... 27
3.3 TRATAMENTO DO RNA COM DNASE E SÍNTESE DE CDNA ... 29
3.4 PCRQUANTITATIVO EM TEMPO REAL PARA ANÁLISE DA EXPRESSÃO GÉNICA... 29
3.5 SEQUENCIAÇÃO DO DNA TRATADO COM BISSULFITO DE SÓDIO PARA ANÁLISE DE PADRÕES DE METILAÇÃO ... 31
3.5.1 Tratamento com Bissulfito de sódio ... 32
3.5.2 PCR para amplificação da região de interesse ... 33
3.5.3 Clonagem e Transformação de bactérias DH5α ... 34
3.5.4 PCR para amplificação dos clones ... 35
3.6 PURIFICAÇÃO DO DNA COM ETANOL ... 37
3.7 MS-MLPA(METHYLATION-SPECIFIC MULTIPLE LIGATION-DEPENDENT PROBE AMPLIFICATION) PARA ANÁLISE DE PADRÕES DE METILAÇÃO ... 37
3.8 ELISA(ENZYME-LINKED IMMUNOSORBENT ASSAY) PARA ANÁLISE DE PADRÕES DE HIDROXIMETILAÇÃO ... 39
3.9 ANÁLISE ESTATÍSTICA DOS RESULTADOS ... 40
4 RESULTADOS ... 44
4.1 EXPRESSÃO GÉNICA ... 44
4.1.1 Placenta ... 44
4.1.2 Tecido Fetal ... 47
4.2 ESTUDO DOS PADRÕES DE METILAÇÃO ATRAVÉS DA TÉCNICA DE SEQUENCIAÇÃO APÓS TRATAMENTO COM BISSULFITO E CLONAGEM.. ... 51
4.2.1 Placenta ... 51
4.2.2 Tecido Fetal ... 55
4.3 ESTUDO DOS PADRÕES DE METILAÇÃO ATRAVÉS DA TÉCNICA DE MS-MLPA ... 56
4.3.1 Placenta ... 56
4.3.2 Tecido Fetal ... 58
4.4 HIDROXIMETILAÇÃO GLOBAL NA PLACENTA E NO TECIDO FETAL ... 60
5 DISCUSSÃO ... 66
6 CONCLUSÃO E PERSPETIVAS FUTURAS ... 76
7 BIBLIOGRAFIA ... 80
FIGURA 1:CONVERSÃO DE UMA CITOSINA PARA 5-METILCITOSINA PELA AÇÃO DA DNA METILTRANSFERASE (DNMT).ADAPTADO DE
(PASTOR ET AL.,2014). ... 7
FIGURA 2:DISTRIBUIÇÃO DA METILAÇÃO EM DINUCLEÓTIDOS CPG NO GENOMA.ADAPTADO DE (GAO &DAS,2014). ... 8
FIGURA 3:VIAS DE DESMETILAÇÃO ATIVA ESQUEMATIZADAS.ADAPTADO DE (MESSERSCHMIDT ET AL.,2014). ... 13
FIGURA 4:REPROGRAMAÇÃO EPIGENÉTICA SUBJACENTE AO IMPRINTING GENÓMICO.ADAPTADO DE (ISHIDA &MOORE,2013). ... 16
FIGURA 5:REGIÃO CONTROLO DE IMPRINTING DOS GENES IGF2 E H19.ADAPTADO DE (PLASSCHAERT &BARTOLOMEI,2014). .... 18
FIGURA 6: REGIÃO CONTROLO DE IMPRINTING DOS GENES PHLDA2, CDKN1C E KCNQ1. ADAPTADO DE (PLASSCHAERT &
BARTOLOMEI,2014). ... 19
FIGURA 7: REPRESENTAÇÃO ESQUEMÁTICA DA SEPARAÇÃO DA AMOSTRA EM TRÊS FASES COM O REAGENTE TRIZOL
(HTTPS://THEORY.LABSTER.COM/DNA_ISOLATION/ ACEDIDO A 15.07.2018). ... 27
FIGURA 8:ESQUEMA REPRESENTATIVO DO TRATAMENTO COM BISSULFITO DE SÓDIO.ADAPTADO DE EPITECT®BISULFITE HANDBOOKE
(QIAGEN). ... 32
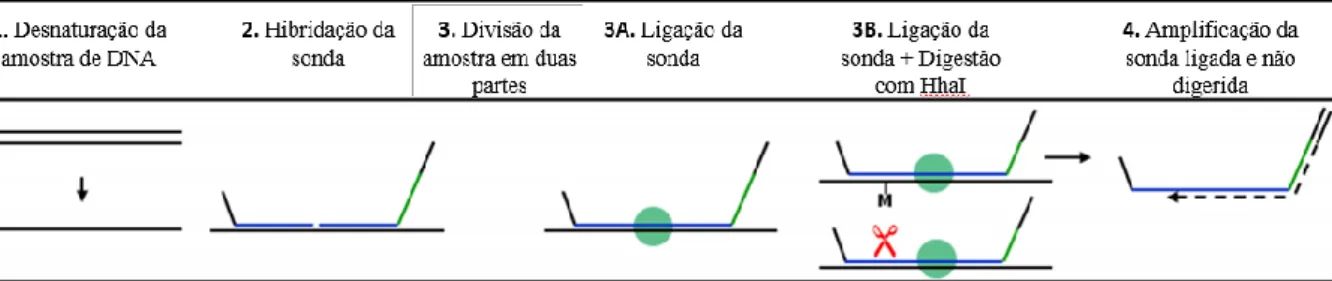
FIGURA 9: ETAPAS DO PROCEDIMENTO DE MS-MLPA. ADAPTADO DE MS-MLPA® GENERAL PROTOCOL ( MRC HOLLAND,
AMSTERDAM,THE NEDERLAND’S). ... 39
FIGURA 10:VISÃO GERAL DAS CAMADAS ESTABELECIDAS DURANTE A METODOLOGIA DA TÉCNICA ELISA PARA A DETEÇÃO DE 5-HMC. ADAPTADO DE QUEST5-HMCTMDNAELISAKIT. ... 40
FIGURA 11:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DOS GENES CODIFICANTES DAS ENZIMAS DE HIDROXIMETILAÇÃO (TET1,TET2, TET3) E METILAÇÃO (DNMT1,DNMT3A,DNMT3B) NA PLACENTA.*REPRESENTA P<0,05(TESTE U DE MANN-WHITNEY). ... 45
FIGURA 12:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DOS GENES DE IMPRINTING NA PLACENTA.*REPRESENTA P<0,05(TESTE U DE
MANN-WHITNEY). ... 46
FIGURA 13:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DO GENE CODIFICANTE DA LEPTINA NA PLACENTA.*REPRESENTA P<0,05(TESTE
U DE MANN-WHITNEY). ... 47
FIGURA 14:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DOS GENES CODIFICANTES DAS ENZIMAS DE HIDROXIMETILAÇÃO (TET1,TET2,
TET3) E METILAÇÃO (DNMT1,DNMT3A,DNMT3B) NO TECIDO FETAL.*REPRESENTA P<0,05(TESTE U DE MANN -WHITNEY). ... 48
FIGURA 15:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DOS GENES DE IMPRINTING NO TECIDO FETAL.*REPRESENTA P<0,05(TESTE U
DE MANN-WHITNEY). ... 49
FIGURA 16:GRÁFICO DA EXPRESSÃO GÉNICA RELATIVA DO GENE CODIFICANTE DA LEPTINA NO TECIDO FETAL.*REPRESENTA P<0,05 (TESTE U DE MANN-WHITNEY). ... 50
FIGURA 17:PADRÕES DE METLAÇÃO DO GENE MEST NA PLACENTA..REPRESENTADAS DUAS AMOSTRAS CONTROLO E DUAS AMOSTRAS DE AEI SEM RCF.OS CÍRCULOS REPRESENTAM AS CPGS: QUANDO PREENCHIDOS A PRETO -CPGS METILADAS E QUANDO PREENCHIDOS A BRANCO -CPGS NÃO METILADAS.CADA COLUNA REPRESENTA UMA CPG ESTUDADA E CADA LINHA REPRESENTA UM CLONE ESTUDADO... 52
FIGURA 18:PADRÕES DE METLAÇÃO DA KVDMR1 NA PLACENTA.REPRESENTADAS DUAS AMOSTRAS CONTROLO E DUAS AMOSTRAS DE
AEI SEM RCF.OS CÍRCULOS REPRESENTAM AS CPGS: QUANDO PREENCHIDOS A PRETO -CPGS METILADAS, E QUANDO PREENCHIDOS A BRANCO -CPGS NÃO METILADAS.CADA COLUNA REPRESENTA UMA CPG ESTUDADA E CADA LINHA REPRESENTA UM CLONE ESTUDADO... 53
FIGURA 19:PADRÕES DE METLAÇÃO DA H19DMR NA PLACENTA. REPRESENTADAS DUAS AMOSTRAS CONTROLO E DUAS AMOSTRAS DE AEI SEM RCF.OS CÍRCULOS REPRESENTAM AS CPGS: QUANDO PREENCHIDOS A PRETO -CPGS METILADAS, E QUANDO PREENCHIDOS A BRANCO -CPGS NÃO METILADA.CADA COLUNA REPRESENTA UMA CPG ESTUDADA E CADA LINHA REPRESENTA UM CLONE ESTUDADO... 54
FIGURA 20:PADRÕES DE METLAÇÃO DA H19DMR NO TECIDO FETAL. REPRESENTADAS DUAS AMOSTRAS CONTROLO, DUAS AMOSTRAS DE AEI SEM RCF E UMA AMOSTRA DE AEI COM RCF.OS CÍRCULOS REPRESENTAM AS CPGS: QUANDO PREENCHIDOS A PRETO -CPGS METILADAS, QUANDO PREENCHIDAS A CINZA - NÃO CONSIDERADAS POR ERRO DE LEITURA NA SEQUENCIAÇÃO E QUANDO PREENCHIDOS A BRANCO -CPGS NÃO METILADA.CADA COLUNA REPRESENTA UMA CPG ESTUDADA E CADA LINHA REPRESENTA UM CLONE ESTUDADO... 55
FIGURA 21:CURVA PADRÃO OBTIDA DE 3 CONTROLOS FORNECIDOS PELO FABRICANTE DO KIT QUEST5-HMCTMDNAELISA(ZYMO
RESEARCH,USA) PARA DETERMINAÇÃO DA PERCENTAGEM GLOBAL DE 5-HMC. ... 61
FIGURA 22:PERCENTAGEM GLOBAL DE 5-HMC DOS DIFERENTES GRUPOS BIOLÓGICOS NO TECIDO PLACENTÁRIO.ESTÁ REPRESENTADA A MÉDIA DE %5-HMC DE CADA GRUPO COM AS RESPETIVAS BARRAS DE ERRO. ... 61
FIGURA 23:PERCENTAGEM GLOBAL DE 5-HMC DOS DIFERENTES GRUPOS BIOLÓGICOS NO TECIDO FETAL.ESTÁ REPRESENTADA A MÉDIA DE %5-HMC DE CADA GRUPO COM AS RESPETIVAS BARRAS DE ERRO. ... 63
TABELA 1:POSSÍVEIS ETIOLOGIAS DA PERDA DE GRAVIDEZ ESPORÁDICA E RECORRENTE.ADAPTADO DE WARREN &SILVER,2008. .... 4
TABELA 2:ORGANIZAÇÃO DAS AMOSTRAS UTILIZADAS NO ESTUDO. ... 26
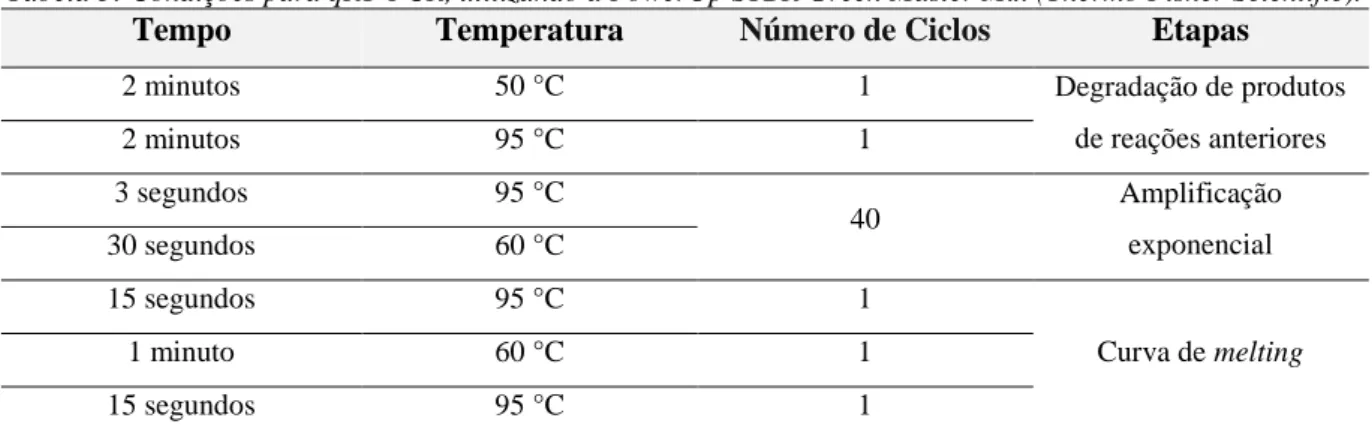
TABELA 3:CONDIÇÕES PARA QRT-PCR, UTILIZANDO A POWERUP SYBRGREEN MASTER MIX (THERMO FISHER SCIENTIFIC). ... 31
TABELA 4:CONDIÇÕES DE TEMPO E TEMPERATURA PARA O TRATAMENTO COM BISSULFITO DE SÓDIO. ... 32
TABELA 5:PRIMERS-BISSULFITO DAS REGIÕES DE INTERESSE PARA AMPLIFICAÇÃO POR PCR. ... 34
TABELA 6:CONDIÇÕES PARA PCR DOS CLONES. ... 35
TABELA 7:CONDIÇÕES PARA PCRMS-MLPA. ... 39
TABELA 8:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NA PLACENTA, RELATIVAMENTE AOS GENES QUE CODIFICAM AS ENZIMAS TET E DNMTS.DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY). ... 45
TABELA 9:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NA PLACENTA, RELATIVAMENTE AOS GENES DE IMPRINTING. DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY)... 46
TABELA 10:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NA PLACENTA, RELATIVAMENTE AO GENE CODIFICANTE DA LEPTINA. DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY)... 47
TABELA 11:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NO TECIDO FETAL, RELATIVAMENTE AOS GENES QUE CODIFICAM AS ENZIMAS TET E DNMTS.DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY). ... 49
TABELA 12:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NO TECIDO FETAL, RELATIVAMENTE AOS GENES DE IMPRINTING. DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY)... 50
TABELA 13:VALORES DE EXPRESSÃO RELATIVA, DESVIO PADRÃO E P NO TECIDO FETAL, RELATIVAMENTE AO GENE CODIFICANTE DA LEPTINA.DIFERENÇAS SIGNIFICATIVAS QUANDO P<0,05(TESTE U DE MANN-WHITNEY). ... 51
TABELA 14:PERCENTAGENS DE METILAÇÃO DE CADA AMOSTRA E DA MÉDIA DE CADA GRUPO BIOLÓGICO PARA O ESTUDO DO GENE MEST NA PLACENTA, OBTIDAS A PARTIR DA TÉCNICA DE SEQUENCIAÇÃO DE SANGER APÓS TRATAMENTO DO DNA COM BISSULFITO E CLONAGEM.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 52
TABELA 15:PERCENTAGENS DE METILAÇÃO DE CADA AMOSTRA E DA MÉDIA DE CADA GRUPO BIOLÓGICO PARA O ESTUDO DA KVDMR1 NA PLACENTA, OBTIDAS A PARTIR DA TÉCNICA DE SEQUENCIAÇÃO DE SANGER APÓS TRATAMENTO DO DNA COM BISSULFITO E CLONAGEM.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 53
TABELA 16:PERCENTAGENS DE METILAÇÃO DE CADA AMOSTRA E DA MÉDIA DE CADA GRUPO BIOLÓGICO PARA O ESTUDO DA H19DMR NA PLACENTA, OBTIDAS A PARTIR DA TÉCNICA DE SEQUENCIAÇÃO DE SANGER APÓS TRATAMENTO DO DNA COM BISSULFITO E CLONAGEM.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 54
TABELA 17:PERCENTAGENS DE METILAÇÃO DE CADA AMOSTRA E DA MÉDIA DE CADA GRUPO BIOLÓGICO PARA O ESTUDO DA H19DMR NO TECIDO FETAL, OBTIDAS A PARTIR DA TÉCNICA DE SEQUENCIAÇÃO DE SANGER APÓS TRATAMENTO DO DNA COM BISSULFITO E CLONAGEM.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 56
TABELA 18:PERCENTAGENS DE METILAÇÃO DA KVDMR1 E DO GENE CDKN1C NA PLACENTA, OBTIDAS A PARTIR DA TÉCNICA DE MS-MLPA.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 57
TABELA 19:PERCENTAGENS DE METILAÇÃO DA H19DMR E DO GENE IGF2 NA PLACENTA, OBTIDAS A PARTIR DA TÉCNICA DE MS-MLPA.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 58
TABELA 20:PERCENTAGENS DE METILAÇÃO KVDMR1 E DO GENE CDKN1C NO TECIDO FETAL, OBTIDAS A PARTIR DA TÉCNICA DE MS-MLPA.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 59
TABELA 21:PERCENTAGENS DE METILAÇÃO DA H19DMR E DO GENE IGF2 NO TECIDO FETAL, OBTIDAS A PARTIR DA TÉCNICA DE MS-MLPA.VALOR DE P ESTATISTICAMENTE SIGNIFICATIVO QUANDO P<0,05(T-TEST). ... 60
TABELA 22:PERCENTAGENS DE 5-HMC DESCRIMINADAS PARA CADA AMOSTRA DE PLACENTA, PERCENTAGENS MÉDIAS DE 5-HMC DE CADA GRUPO BIOLÓGICO E VALOR DE P, SENDO QUE AS DIFERENÇAS SÃO CONSIDERADAS ESTATISTICAMENTE SIGNIFICATIVAS QUANDO P<0,5(TESTE U DE MANN-WHITNEY). ... 62
TABELA 23:PERCENTAGENS DE 5-HMC DESCRIMINADAS PARA CADA AMOSTRA DE TECIDO FETAL, PERCENTAGENS MÉDIAS DE 5-HMC
DE CADA GRUPO BIOLÓGICO E VALOR DE P, SENDO QUE AS DIFERENÇAS SÃO CONSIDERADAS ESTATISTICAMENTE SIGNIFICATIVAS QUANDO.P<0,5(TESTE U DE MANN-WHITNEY). ... 64
TABELA 24:PRIMERS UTILIZADOS PARA O ESTUDO DA EXPRESSÃO DOS GENES ENVOLVIDOS NA HIDROXIMETILAÇÃO E METILAÇÃO, A PARTIR DA TÉCNICA DE QRT-PCR. ... 88
TABELA 25:PRIMERS UTILIZADOS PARA O ESTUDO DA EXPRESSÃO DOS GENES DE IMPRINTING, A PARTIR DA TÉCNICA DE QRT-PCR. 89
TABELA 26:PRIMERS UTILIZADOS PARA O ESTUDO DA EXPRESSÃO DO GENE DA LEPTINA, A PARTIR DA TÉCNICA DE QRT-PCR. ... 89
TABELA 27:PRIMERS DOS GENES DE REFERÊNCIA UTILIZADOS NO ESTUDO DA EXPRESSÃO GÉNICA A PARTIR DA TÉCNICA DE QRT-PCR. ... 89
AEI – Abortamento espontâneo idiopático AID – Desaminase induzida por ativação BER – Reparação por excisão de base Bp – Base Pair
BWS – Síndrome Beckwith-Wiedemann cDNA – DNA complementar
CHSJ – Centro Hospitalar de São João CpG – Dinucliótido citosina-fosfato-guanina CTCF – CCCTC-binding factor
DMR – Região diferencialmente metilada DNA – Ácido desoxirribonucleico
DNMTs – DNA metiltransferases dNTP – Desoxinucleótido trifosfato DPM – Domínio parcialmente metilado dTTP – Desoxitimidina trifosfato dUTP – Desoxiuridina trifosfato
EDTA – Ácido etilenodiamino tetra-acético ELISA – Enzyme-Linked Immunosorbent Assay
FMUP – Faculdade de Medicina da Universidade do Porto Fw – Forward
ICpG – Ilha CpG
ICR – Região controlo de imprinting
IPTG – Isopropil β-D-1-tiogalactopiranosida lncRNA – RNA não codificante longo mRNA – RNA mensageiro
MS-MLPA – Methylation-Specific Multiple Ligation-dependent Probe Amplification ncRNAs – RNAs não codificantes
PCR- Reação em Cadeia da Polimerase PGC – Célula germinativa primordial PGE – Perda de gravidez espontânea PGR – Perda de gravidez recorrente
qRT-PCR – PCR quantitativo em tempo real RCF – Restrição de crescimento fetal
RNA – Ácido ribonucleico RSS - Síndrome Silver-Russel Rv – Reverse
SAM – S-adenosilmetilcitosina TDG – Timina DNA glicosilase
TETs – Enzimas ten-eleven translocation UNG – Uracilo N-Glicosilase
X-Gal – 5-bromo-4-cloro-3-indoxil-β-D-galactopiranosídeo 5-caC – 5-carboxilcitosina 5-fC – 5-formilcitosina 5-hmC – 5-hidroximetilcitosina 5-hmU – 5-hidroximetiluracilo 5-mC – 5-metilcitosina
Capítulo 1
– Introdução –
1
Introdução
1.1 Abortamento espontâneo -
Definição, epidemiologia e etiolog
ia da
perda de gravidez humana
O abortamento espontâneo é considerado a complicação mais comum na gravidez, tendo um elevado impacto ao nível clínico e social. Esta condição, para além de acarretar circunstâncias altamente emocionais para os casais envolvidos, representa ainda um enorme desafio médico (Qian et al., 2017; Tur-Torres et al., 2017).
Este problema clínico pode definir-se como a perda espontânea de uma gravidez intrauterina sem intervenção externa, até às 24 semanas de gestação (Tur-Torres et al., 2017). Cerca de 10%-15% das gestações clinicamente reconhecidas terminam espontaneamente e está descrito que esta frequência é semelhante entre diferentes populações humanas (Nikitina et al., 2016; Tur-Torres et al., 2017).
A perda espontânea de uma gravidez possui diferentes denominações com base nas diferentes fases do desenvolvimento intrauterino. Quando ocorre um abortamento no período pré-embrionário, que após o primeiro dia do último ciclo menstrual dura cerca de 5 semanas, estamos perante uma perda anembrionária ou restos ovulares. A fase de desenvolvimento que se segue é o período embrionário que se prolonga da sexta à nona semana de gestação. Quando um embrião não expressa atividade cardíaca entre estas semanas de gestação verifica-se uma morte embrionária. Por último, a partir da décima semana de gestação inicia-se o período fetal que se prolonga até ao final da gravidez, sendo que perdas a partir desta semana podem ser denominadas como mortes fetais. Esta distinção e determinação tornam-se importantes, uma vez que, a causa de perda de gravidez pode variar ao longo da idade gestacional (Warren & Silver, 2008).
O abortamento espontâneo pode tratar-se ainda de um evento esporádico ou repetitivo, sendo que aproximadamente 1-3% dos casos correspondem a uma perda de gravidez recorrente (PGR) (Tur-Torres et al., 2017). Não existe consenso a partir de que número se considera uma PGR, contudo, vários investigadores e clínicos apoiam-se na definição da Sociedade Europeia de Reprodução Humana e Embriologia, que considera corresponder a três ou mais perdas consecutivas de gravidez espontânea (Jauniaux et al., 2006; Rai & Regan, 2006). Por outro lado,
que a partir de duas ou mais perdas de gravidez já se considera uma PGR, o que faz aumentar a frequência destas para 2-5% (American Society for Reprodutive Medicine, 2013; Nikitina et al., 2016). A acumulação de evidências sugeriu que o risco de uma mulher sofrer novamente um abortamento espontâneo aumenta sucessivamente após cada perda de gravidez. Quando uma mulher sofre dois abortamentos espontâneos consecutivos, o risco de voltar a ter uma perda de gravidez na próxima gestação aumenta para 30% e após três abortamentos espontâneos consecutivos o risco pode atingir os 40-45% (Ogasawara et al., 2000; Qian et al., 2017; Rai & Regan, 2006; Royal College Obstetricians and Gynaecologists, 2011). Este aumento é parcialmente explicado pelo facto de as mulheres progressivamente tentarem engravidar em idades mais tardias (Rai & Regan, 2006). Ainda que este risco se refira a perdas de gravidez sucessivas, não podemos negligenciar e excluir a hipótese de uma mulher poder vir a ter um abortamento espontâneo após ter conseguido completar uma gravidez com sucesso (Tur-Torres et al., 2017).
Esta complicação clínica reflete uma natureza heterogénea pois pode suceder-se a inúmeros fatores, tais como alterações endócrinas não controladas clinicamente, doenças infecciosas maternas, idade materna avançada, alterações imunológicas, fatores ambientais adversos, malformações anatómicas, distúrbios trombofílicos e, ainda, alterações genéticas (Tabela 1) (Christiansen et al., 2008; Hanna et al., 2013; Nikitina et al., 2016; Tur-Torres et al., 2017).
Tabela 1: Possíveis etiologias da perda de gravidez esporádica e recorrente. Adaptado de Warren & Silver, 2008.
Causas Exemplos
Causas Genéticas
Anomalias cromossómicas fetais
Anomalias cromossómicas equilibradas nos progenitores Doenças monogénicas
Mosaicismo confinado à placenta Anomalias morfológicas/Defeitos estruturais
Causas Anatómicas Maternas
Malformações uterinas Anomalias cervicais Miomas Aderências intrauterinas Causas Trombofílicas Causas Endócrinas Diabetes mellitus Patologia da tiroide Patologias hormonais Síndrome do Ovário Policístico Causas Imunológicas Síndrome Antifosfolipídico
Fatores autoimunes maternos Causas Ambientais Álcool Tabaco Drogas e químicos Infeções Virais Bacterianas
Apesar das diferentes causas, os fatores genéticos são considerados os principais responsáveis pelo abortamento espontâneo esporádico e recorrente. As alterações cromossómicas fetais representam a causa mais comum e estão amplamente descritas, surgindo maioritariamente as alterações cromossómicas numéricas que ocorrem de novo e que sobressaem relativamente às alterações herdadas. (Royal College Obstetricians and Gynaecologists, 2011; Tur-Torres et al., 2017).
Na verdade, estima-se que mais de 99% das gestações associadas a alterações cromossómicas fetais resultam num abortamento espontâneo e que estas alterações são responsáveis por cerca de 50-60% dos casos de perdas de gravidez espontâneas (PGE). No entanto, 40-50% dos casos de PGR estão associados a um cariótipo normal e as suas causas são ainda mal compreendidas e preocupantes (Nikitina et al., 2016; Qian et al., 2017). De acordo com a literatura, a probabilidade de ocorrer uma nova PGE em pacientes com história clínica de abortamentos espontâneos de embriões com cariótipo normal parece ser maior do que em pacientes com história clínica de PGE de embriões com cariótipo não normal, ou seja, a perda de uma gravidez anterior de um embrião com cariótipo normal torna-se um fator preditor de um próximo abortamento espontâneo (Ogasawara et al., 2000). Recentemente, Nikitina et al. (2016)
analisaram cerca de 900 embriões resultantes de abortamentos espontâneos esporádicos e recorrentes e verificaram uma maior frequência de cariótipos normais nos embriões de mulheres com abortamentos recorrentes. Este estudo confirmou que outros fatores não citogenéticos são mais comuns em PGR idiopáticas e que estes se traduzem habitualmente a um mau prognóstico (Nikitina et al., 2016).
Apesar de toda a investigação e interesse da comunidade científica e médica, não é ainda possível prever uma perda de gravidez, devido principalmente à falta de conhecimento sobre os mecanismos moleculares subjacentes ao abortamento espontâneo idiopático (AEI) (Pereza et al., 2017; Qian et al., 2017; Tur-Torres et al., 2017). Na verdade, a presença de um cariótipo normal no embrião/feto não significa que exista um equilíbrio funcional do seu genoma, uma vez que, para além de outras causas genéticas não cromossómicas que possam existir, também erros no estabelecimento de marcas epigenéticas e alterações nos mecanismos epigenéticos podem ocorrer, tornando-se relevante o estudo do papel da epigenética na etiologia da perda de gravidez humana (Hanna et al., 2013; Nikitina et al., 2016; Sazhenova et al., 2012).
1.2 Epigenética e o desenvolvimento placentário e fetal
Todas as células do nosso organismo contêm a mesma sequência de DNA, no entanto, a morfologia e as funções desempenhadas por estas são diferentes entre si. Este facto é possível devido à epigenética, que pode ser definida como um mecanismo capaz de regular a expressão génica sem que ocorram alterações na sequência de DNA. Efetivamente, as marcas epigenéticas são essenciais para a diferenciação celular e o facto de serem transmitidas durante a divisão celular mitótica, permite que a informação epigenética passe para as células-filhas e reforce os perfis de transcrição específicos de cada tipo celular (Adalsteinsson & Ferguson-Smith, 2014; Monk, 2015b; Nelissen et al., 2011; Reik & Kelsey, 2014; Vaiman, 2017).
Durante a reprodução e o desenvolvimento embrionário ocorrem dois momentos principais de reprogramação epigenética no genoma, que serão explicados com mais detalhe em subcapítulos seguintes. Estes eventos compreendem uma remoção dos padrões epigenéticos e um subsequente restabelecimento dos mesmos (Canovas et al., 2017; Sundrani et al., 2016). No decorrer da formação das células germinativas ocorre uma reprogramação epigenética necessária para que estas células se tornem funcionais e capazes de, posteriormente, dar origem a um embrião. Neste sentido, quando ocorrem erros nesta etapa, os embriões resultantes da fecundação destas células podem desenvolver-se anormalmente e serem perdidos durante a
gestação (Adalsteinsson & Ferguson-Smith, 2014). Outro evento preponderante de reprogramação epigenética acontece durante o desenvolvimento embrionário inicial, o qual se demonstra importante para se iniciar a diferenciação embrionária. Assim, na fase do blastocisto surgem duas linhas celulares morfologicamente diferentes e altamente distinguíveis. Nesta fase é possível evidenciar células estaminais embrionárias na cavidade interna do blastocisto, que darão origem aos diferentes tecidos do embrião, e, numa camada mais externa, células estaminais trofoblásticas, que darão origem à placenta (Sundrani et al., 2016; Vaiman, 2017).
A placenta é um órgão materno-fetal temporário que, desde o início até ao final da gravidez, se revela crucial para o desenvolvimento intrauterino. Durante as diferentes semanas de gestação, em resposta às necessidades fetais e exposições ambientais, este órgão sofre várias mudanças moleculares, estruturais, morfológicas e funcionais (Lim et al., 2017). São várias as funções desempenhadas por este órgão, entre as quais, a secreção de hormonas e fatores de crescimento, a proteção contra o sistema imunológico materno e, ainda, o transporte de nutrientes, oxigénio e resíduos entre a mãe e o feto (Lambertini et al., 2013; Lim et al., 2017; Monk, 2015a; Nelissen et al., 2011). É evidente que se a formação e o desenvolvimento da placenta não ocorrerem devidamente poderão prejudicar o feto e até conduzir a um abortamento espontâneo (Logan et al., 2013). Deste modo, a placenta consiste num tecido extraembrionário originado a partir do blastocisto e que, sendo predominantemente regulada pelo genoma fetal, se torna capaz de fornecer informações sobre o próprio estado (epi)genético do feto (Bianco-Miotto et al., 2016; Lambertini et al., 2013). Consequentemente, a placenta representa o tecido apropriado para se analisar o perfil da expressão de determinados genes associados ao crescimento e ao desenvolvimento fetal (Lambertini et al., 2013).
Nos últimos anos tem surgido um crescente interesse em estudar os diferentes mecanismos epigenéticos que podem estar subjacentes a possíveis alterações da atividade génica. Estes provocam modificações potencialmente reversíveis que podem ser classificadas de duas formas, aquelas que criam um estado ativo da cromatina, regulando positivamente a expressão génica, ou aquelas que criam um estado repressivo da cromatina, regulando negativamente a expressão génica (Piedrahita, 2012). Dentro destas modificações epigenéticas destacámos a metilação e a hidroximetilação do DNA, no entanto outros mecanismos epigenéticos podem ser distinguidos como as modificações de histonas, a ação de RNAs não codificantes (ncRNAs) e a remodelação da cromatina (Kang et al., 2015; E. Li, 2002; Logan et al., 2013; Nelissen et al., 2011; Sundrani et al., 2016).
1.2.1 Metilação do DNA
A metilação do DNA tem sido a marca epigenética mais estudada, consistindo numa modificação epigenética de vital importância na regulação da expressão génica ao longo do desenvolvimento de mamíferos. Para além de estar envolvida na formação das diferentes linhas celulares formadas durante a gravidez, a metilação também representa um papel crucial em diversos processos biológicos, como a inativação do cromossoma X e o imprinting genómico (Monk, 2015a; Sundrani et al., 2016; Tanaka et al., 2014; G. Zhang & Pradhan, 2014).
Esta marca epigenética consiste na adição de um grupo metil (CH3) do dador
S-adenosilmetionina (SAM) ao quinto carbono de uma citosina, originando uma 5-metilcitosina (5-mC), também conhecida como a “quinta base” do código genético (Figura 1) (Messerschmidt et al., 2014; Monk, 2015b). Normalmente, este mecanismo ocorre num dinucleótido citosina-fosfato-guanina (CpG), no entanto, mais raramente e sem ainda se perceber a sua função exata, a metilação também pode ocorrer noutros locais designados de CpH, onde H pode corresponder a uma adenina, a uma citosina ou a uma timina (Canovas et al., 2017; Messerschmidt et al., 2014; Monk, 2015b).
Figura 1: Conversão de uma citosina para 5-metilcitosina pela ação da DNA metiltransferase (DNMT). Adaptado de (Pastor et al., 2014).
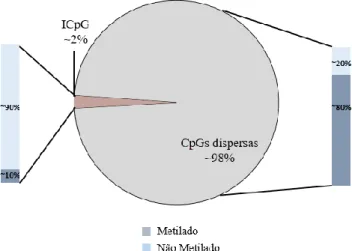
O metiloma mostra que a metilação em CpGs é simétrica, o que significa que cada dinucleotídeo CpG metilado possui uma díade GpC na cadeia complementar com uma citosina também metilada. Além disso, este segue uma distribuição bimodal, uma vez que, a metilação pode ocorrer em regiões de 500 a 2000 pares de bases ricas em CpGs, denominadas ilhas CpG (ICpG), ou em CpGs singulares fora destas regiões. Em 98% do genoma, as CpGs estão presentes, aproximadamente, uma em cada 80 dinucleótidos, enquanto que as ICpG compreendem apenas 1-2% do genoma. Normalmente, as ICpG concentram-se em promotores, onde geralmente permanecem hipometiladas. Pelo contrário, CpGs fora destas ICpG, geralmente, encontram-se 60% a 90% hipermetiladas, dependendo do tipo celular onde se
encontram (Figura 2) (Adalsteinsson & Ferguson-Smith, 2014; Canovas et al., 2017; Koukoura, et al., 2012; Messerschmidt et al., 2014). Neste sentido, das 28 milhões de CpGs que existem no genoma humano, a maioria é metilada (Robinson & Price, 2015).
Figura 2: Distribuição da metilação em dinucleótidos CpG no genoma. Adaptado de (Gao & Das, 2014).
A metilação do DNA nas regiões promotoras dos genes pode influenciar o nível de expressão génica, uma vez que afeta a acessibilidade do DNA a modificadores de cromatina, fatores de transcrição e outros elementos da maquinaria de transcrição (Hanna et al., 2013; Sundrani et al., 2016).
O acumular de evidências levou à suposição geral de que a perda e a aquisição de metilação do DNA no promotor de um gene, resulta, respetivamente, na ativação e no silenciamento do mesmo (Adalsteinsson & Ferguson-Smith, 2014). De facto, a metilação do DNA tem sido associada à repressão transcricional, no entanto, uma análise detalhada do conteúdo de CpGs e do tamanho da sequência de diferentes promotores, permitiu classificar três categorias de promotores: (1) promotores de alta densidade em CpGs, raramente são metilados, no entanto, quando, pontualmente, estes são metilados, ocorre o silenciamento do gene, estando de acordo com a visão tradicional de que a metilação do DNA reprime a transcrição; (2) promotores que apresentam uma densidade de CpGs intermédia funcionam de igual forma aos anteriores; (3) promotores de baixa densidade de CpGs, geralmente hipermetilados, permanecem transcricionalmente ativos (Messerschmidt et al., 2014; Tanaka et al., 2014). Apesar de toda esta dinâmica epigenética em regiões promotoras, a metilação do DNA é ainda mais prevalente dentro do corpo do gene. Contrariamente ao mecanismo mais comum, a metilação ao longo de um gene está positivamente correlacionada com os níveis de expressão génica. Este paradoxo permanece por explicar e o papel da metilação em determinadas regiões génicas é ainda pouco
compreendido (Gao & Das, 2014; Jjingo et al., 2012). Um estudo recente mostrou esta associação complexa entre a metilação do DNA e a expressão génica em mais de 2000 genes, dos quais 11 representavam genes de imprinting. Este estudo usou amostras de placenta e mostrou precisamente que em diferentes idades gestacionais se verificou uma correlação negativa entre o nível de expressão génica e o nível de metilação do promotor, bem como uma correlação positiva entre o nível de expressão génica e o nível de metilação do corpo do gene (Lim et al., 2017).
As várias regiões genómicas são metiladas diferencialmente e dinamicamente, dependendo do tipo de célula ou tecido e da fase de desenvolvimento, trabalhando conjuntamente para influenciar e/ou responder à maquinaria da transcrição génica (Robinson & Price, 2015; Tan & Shi, 2012). Na maioria dos tecidos, a metilação do DNA torna-se relativamente estável dentro de um determinado tipo celular, podendo deste modo fornecer informações sobre os processos normais de desenvolvimento. No entanto, a metilação do DNA da placenta parece ser inerentemente mais variável devido às suas funções mediadoras entre o feto e a mãe e à sua necessidade de responder a uma variedade de sinais durante toda a gestação. Fatores ambientais adversos durante o desenvolvimento embrionário podem prejudicar o estabelecimento destas marcas epigenéticas e, por isso, estudar a metilação do DNA da placenta pode fornecer informações sobre possíveis alterações intrauterinas no desenvolvimento de uma gravidez (Hanna et al., 2013; Robinson & Price, 2015). Além disso, também a metilação do DNA fetal se deve considerar relevante, uma vez que o crescimento fetal é afetado pela função adequada de muitos genes de imprinting e não-imprinting que estão sujeitos ao controlo epigenético através da metilação dos seus promotores (Koukoura et al., 2012).
Após a formação do zigoto, a reprogramação epigenética que ocorre é caracterizada por uma desmetilação praticamente global, à qual se segue uma fase de nova metilação. Esta metilação de novo que ocorre na fase de blastocisto é restrita à massa celular interna, sendo que a trofectoderme fica desprovida de metilação. Assim, estabelece-se uma desigualdade epigenética nos níveis de metilação do DNA entre as duas linhas celulares, embrionária e extraembrionária, que é mantida durante toda a gestação e que origina o genoma embrionário globalmente metilado e o genoma dos trofoblastos globalmente hipometilado (Koukoura et al., 2012; Logan et al., 2013; Nelissen et al., 2011).
Esta hipometilação verificada na placenta não é uniforme no genoma, ocorrendo principalmente em domínios parcialmente metilados (DPM) que cobrem aproximadamente 40% do genoma placentário e que representam grandes regiões (>100 kb) não metiladas do DNA,
intercaladas com regiões de maior metilação (Bianco-Miotto et al., 2016; Robinson & Price, 2015). A descoberta de que a placenta humana tem DPM e que estes são mantidos durante a gravidez levanta questões sobre o potencial papel destas regiões no desenvolvimento humano normal (Schroeder et al., 2013; Schroeder & LaSalle, 2013). Embora a função dos DPM ainda não seja clara, tem sido sugerido que estas regiões retratam um mecanismo programado para regular a expressão génica específica deste órgão. Assim, perante esta hipótese, uma interrupção desta organização epigenética, poderá levar a uma disfunção placentária significativa e, em última instância, ao abortamento espontâneo (Robinson & Price, 2015; Sundrani et al., 2016).
As enzimas que catalisam a metilação do DNA estão bem caracterizadas e são designadas por DNA metiltransferases (DNMTs). Dentro desta família distinguimos essencialmente três tipos que são enzimaticamente ativos, a DNMT1, DNMT3A, DNMT3B, e ainda uma subunidade reguladora a DNMT3L (Barišić, Pereza, Hodžić, Ostojić, & Peterlin, 2017; Tan & Shi, 2012).
Os padrões de metilação do DNA são herdados fielmente durante o ciclo celular (Zhang & Pradhan, 2014).A replicação semi-conservativa do DNA produz duas cadeias com locais CpGs hemimetilados, isto é, apenas uma das cadeias filhas apresenta estes locais metilados, para os quais a DNMT1 tem alta afinidade. Assim, esta enzima desempenha uma função de manutenção da metilação durante a replicação do DNA (Messerschmidt et al., 2014; Tanaka et al., 2014). A DNMT3A e DNMT3B mostram ter ação na metilação de novo do DNA, enquanto que, a DNMT3L, embora não possua o característico domínio catalítico conservado nos restantes membros da família, funciona como um cofator crucial nas atividades desempenhadas pelas DNMT3A e DNMT3B (Adalsteinsson & Ferguson-Smith, 2014; Tanaka et al., 2014). Por último, sabe-se ainda que existe uma DNMT2 que difere dos outros elementos da família, uma vez que não metila a molécula de DNA mas sim a molécula de RNA (Messerschmidt et al., 2014; Portela & Esteller, 2010).
Todas as DNMTs são indispensáveis na reprodução humana normal e são especialmente ativas durante a gametogénese, decidualização e desenvolvimento embrionário inicial (Barišić et al., 2017). No entanto, presumivelmente, a regulação da metilação em determinados genes e da hipometilação global do genoma da placenta não depende apenas das DNMTs, mas envolve a ação combinada das enzimas ten-eleven translocation (TETs), da modificação de histonas e de ncRNAs (Logan et al., 2013).
1.2.2 Hidroximetilação do DNA
A metilação do DNA em citosinas tem sido considerada a única modificação covalente do DNA de mamíferos. No entanto, descobertas recentes, de enzimas que catalisam a oxidação de uma citosina metilada, começaram a reformular a visão da paisagem epigenética (Tanaka et al., 2014). A existência de uma "sexta base", a 5-hidroximetilcitosina (5-hmC), veio revelar papéis importantes em diversos processos biológicos, intervindo no desenvolvimento embrionário e contribuindo para a regulação da transcrição génica (Messerschmidt et al., 2014; Pastor et al., 2014). A 5-hmC parece estar principalmente associada à eucromatina que contém genes ativamente transcritos (Gao & Das, 2014).
Como já referido, a identidade celular é fortemente influenciada pelos padrões de metilação que são estabelecidos, por isso, a extensão e a localização da metilação no DNA variam especificamente com base no tecido. De igual forma, é sugerido que os níveis de hidroximetilação também exibam padrões específicos de tecido (Green et al., 2016). Li & Liu (2011) encontraram níveis de 5-hmC muito divergentes em diferentes tecidos, sendo que verificaram níveis globais muito baixos na placenta e níveis mais altos em vários tecidos como o cérebro, rim, cólon, reto e fígado (Li & Liu, 2011). Embora a distribuição global da 5-hmC seja muito diferente e dependente do tipo celular, é consistente na literatura que os níveis mais altos de 5-hmC são encontrados no cérebro (Gao & Das, 2014; Pastor et al., 2014).
Em 2009, foi descoberta a atividade enzimática da família das dioxigenases TET responsáveis por oxidar uma 5-mC e, consequentemente, produzir uma 5-hmC. Estas enzimas, a partir de oxidações sucessivas podem ainda originar uma formilcitosina (fC) e uma 5-carboxilcitosina (5-caC) (Hill et al., 2014; Kang et al., 2015). A 5-hmC é significativamente mais prevalente que a 5-fC e do que a 5-caC no DNA, no entanto, ainda não está claro quais são os fatores que intervêm e que determinam a paragem em 5-hmC ao longo da via de oxidação. Uma provável explicação passa por uma possível modulação da acessibilidade das TET à 5-hmC, através de modificações pós-traducionais ou da interação com determinadas proteínas (Kohli & Zhang, 2013).
Dentro desta família foram identificados três membros, TET1, TET2 e TET3, que abrigam um domínio catalítico central característico das oxigenases dependentes de α-oxoglutarato e de Fe(II) (Kang et al., 2015; Kohli & Zhang, 2013; Pastor et al., 2014). Este domínio catalítico constitui apenas uma fração destas enzimas, o que sugere a possibilidade de que domínios não catalíticos possam ter funções reguladoras. Em todas as isoformas das TET, um domínio rico
em cisteína, que precede o domínio catalítico, parece ser necessário para a atividade destas. A TET1 e a TET3, contrariamente à TET2, possuem ainda um domínio CXXC de ligação ao DNA que reconhece os locais CpG. Alternativamente, a TET2 associa-se à IDAX, uma proteína independente que contém o domínio CXXC e que a ajuda a ser recrutada para os locais alvo (Gao & Das, 2014; Kohli & Zhang, 2013).
Esta descoberta inovadora de que as TET podem oxidar a 5-mC forneceu um possível caminho para explicar a desmetilação do DNA e revelou a 5-hmC como um verdadeiro ponto-chave neste processo (Hill et al., 2014; Kohli & Zhang, 2013).Como ainda não foi encontrada nenhuma desmetilase que leve à desmetilação direta do DNA, considera-se que este processo ocorre indiretamente por vias passivas ou ativas (Gao & Das, 2014).
A desmetilação passiva ocorre quando se verifica uma falha na maquinaria da manutenção da metilação durante a replicação do DNA, que pode surgir da ausência de expressão da DNMT1. Assim, este mecanismo resulta numa diluição da metilação do DNA ao longo da replicação do DNA, não podendo ser responsável por uma perda rápida de metilação em células que se dividem lentamente ou que simplesmente não se dividem (Messerschmidt et al., 2014).
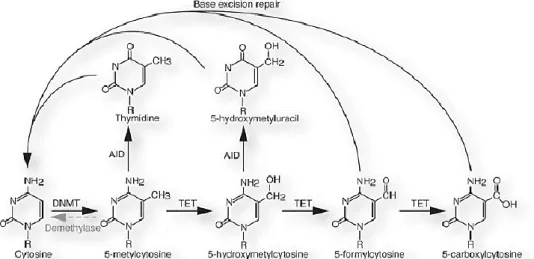
A desmetilação ativa corresponde à remoção enzimática de 5-mC ou das suas formas oxidadas, 5-hmC, 5-fC e 5-caC, à qual se segue a ação de um mecanismo de reparação por excisão de base (BER) que origina novamente uma citosina não modificada (Canovas et al., 2017; Dawlaty et al., 2014; Kohli & Zhang, 2013) (Figura 3). A 5-mC e a 5-hmC podem ser desaminadas pelas desaminases induzidas por ativação (AID), originando, respetivamente, uma
timidina ou uma 5-hidroximetiluracilo (5-hmU) que, consequentemente, criam
incompatibilidades na cadeia do DNA que são reconhecidas pela timina DNA glicosilase (TDG) e que fazem agir o mecanismo de BER (Gao & Das, 2014; Messerschmidt et al., 2014; Wu et al., 2018). Adicionalmente, também pode ocorrer a remoção de 5-fC e de 5-caC, através da ação direta da TDG conjugada com o mecanismo BER. Este processo implica a participação prévia das TETs que ao oxidar estas bases libertam determinados produtos que servem como substrato para a TDG atuar (Canovas et al., 2017). Particularmente, esta enzima demonstrou interagir com inúmeros fatores de transcrição, enzimas modificadoras da cromatina e DNMTs, aumentando a possibilidade de existir um papel funcional para a TDG na modulação da transcrição génica, seja pela sua atividade de glicosilase ou pela função de coativador transcricional (Kohli & Zhang, 2013). Além destas principais vias de desmetilação ativa, foi relatado que em determinadas situações, sob condições redox, as DNMT3A e DNMT3B, além de serem
metilases, podem também desmetilar o DNA, no entanto, a relevância biológica desta reação reversa permanece desconhecida (Kohli & Zhang, 2013, Logan et al., 2013).
Figura 3: Vias de desmetilação ativa esquematizadas. Adaptado de (Messerschmidt et al., 2014).
Embora as pistas tenham emergido rapidamente, ainda permanecem questões sobre a dinâmica da via de desmetilação das citosinas e as funções das moléculas intermediárias envolvidas (Gross et al., 2016). É certo que o significado funcional da 5-hmC ainda está a ser determinado, mas cada vez se torna mais evidente que existe um potencial para a 5-hmC funcionar, não apenas como um intermediário molecular no processo de desmetilação do DNA, mas também como uma modificação epigenética estável e autónoma (Green et al., 2016; Sjöholm et al., 2017; Tanaka et al., 2014).
Tal como as DNMTs, também as TET estão envolvidas na diferenciação e na regulação dos trofoblastos (Logan et al., 2013; Rakoczy et al., 2017). Esta última família é regulada negativamente na placenta, o que provoca, globalmente, quantidades reduzidas de 5-hmC no DNA deste tecido (Green et al., 2016; Logan et al., 2013). Dada a importância das TET na dinâmica do epigenoma placentário, atualmente, para além da metilação, considera-se que também a desmetilação do DNA tem efeitos no desenvolvimento da placenta e do feto (Wu et al., 2018).
1.3 Imprinting genómico durante o desenvolvimento intrauterino
Ao contrário da maioria dos genes autossómicos, que são expressos bialelicamente, existe um conjunto de genes que apresenta uma expressão monoalélica, dependendo da origem
parental (Ishida & Moore, 2013; Tomizawa & Sasaki, 2012). O imprinting genómico é o fenómeno epigenético responsável por esta expressão diferencial entre os alelos materno e paterno, que surge de modificações epigenéticas que ocorrem em regiões específicas do genoma (Hussain, 2017).
A maioria dos mecanismos envolvidos no imprinting, bem como vários genes de imprinting, são conservados entre o rato e o Homem (Kalish et al., 2014; Nelissen et al., 2011). Até ao momento, em humanos, estão caracterizados mais de 100 genes de imprinting (www.geneimprint.com) e a maioria destes está envolvida no desenvolvimento intrauterino, na diferenciação dos tecidos placentários e na regulação do metabolismo de algumas hormonas e de alguns fatores de crescimento (Sazhenova et al., 2017, 2012). O perfil da expressão dos genes de imprinting pode variar ao longo da gestação e são específicos para os diferentes tecidos (Dória et al., 2010; Nelissen et al., 2011).
O imprinting é observado predominantemente em mamíferos eutérios. Esta íntima associação entre a presença deste fenómeno e a presença da placenta levou à formulação de várias hipóteses para explicar o surgimento do imprinting. A teoria do conflito parental é a hipótese mais aceite e prevê que o genoma paterno promove o crescimento fetal e placentário, extraindo o máximo de recursos maternos, enquanto que o genoma materno atua restringindo o crescimento da descendência, limitando os seus recursos para assegurar a sobrevivência e a distribuição equilibrada de nutrientes entre a descendência (Hussain, 2017; Ishida & Moore, 2013; Moore et al., 2015; Piedrahita, 2012). Estas duas forças parentais opostas equilibram-se ao longo de uma gestação, permitindo que o crescimento fetal ocorra normalmente (Piedrahita, 2012).
Os genes de imprinting têm tendência para ocorrerem predominantemente em clusters (com cerca de 1 Mb de comprimento) que podem envolver tanto genes codificantes como genes não codificantes. Estes genes são regulados através de sequências de DNA que se denominam regiões controlo de imprinting (ICRs) (Hussain, 2017; Kalish et al., 2014; Mackay, 2017; Nelissen et al., 2011; Plasschaert & Bartolomei, 2014). Assim, as ICRs regulam a expressão de um ou mais genes proximais, através de mecanismos complexos que envolvem ncRNAs, modificações de histonas e locais de ligação a fatores de transcrição (Ishida & Moore, 2013; Tomizawa & Sasaki, 2012). Todas as ICRs, atualmente conhecidas, incluem regiões diferencialmente metiladas (DMRs) ricas em CpGs, que controlam a expressão dos genes de imprinting de forma a que apenas um alelo parental seja expresso (Hussain, 2017; Mackay, 2017). As DMRs podem ser primárias, se adquirirem a metilação durante a gametogénese, ou
secundárias, se surgirem durante o desenvolvimento a partir da presença das DMRs primárias que passam a regular as DMRs secundárias (Court et al., 2014; Hussain, 2017; Ishida & Moore, 2013).
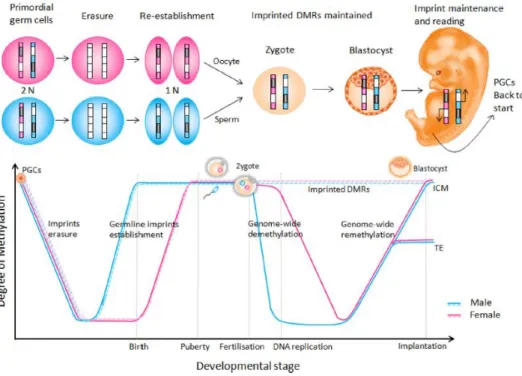
A desmetilação total do genoma ocorre durante a gametogénese quando as células germinativas primordiais (PGCs) do embrião em desenvolvimento entram na crista gonadal (Figura 4). Nesta fase, as DNMTs de novo são silenciadas, o que facilita a desmetilação do DNA. Surpreendentemente, a DNMT1 é abundante nesta etapa, mas o seu cofator principal, Uhrf 1, é transcricionalmente reprimido, o que impede a ação desta. Uma vez que na crista gonadal a expansão destas células ocorre rapidamente, é assegurada a desmetilação passiva global do DNA (Monk, 2015b; Plasschaert & Bartolomei, 2014). Além da regulação que existe no mecanismo das DNMTs, foi sugerido que também a TET1 e a TET2 poderiam desempenhar um papel crítico na remoção das marcas de imprinting nas PGCs (Hackett et al., 2013; Yamaguchi et al., 2014). Posteriormente a esta etapa de desmetilação global, ainda durante a gametogénese, as marcas de imprinting são estabelecidas especificamente com base no sexo do feto, através da ação das DNMTs de novo que deixam de estar silenciadas. Na linha germinativa masculina, a metilação de novo nas DMRs começa a ser estabelecida durante o desenvolvimento fetal tardio. Pelo contrário, a aquisição das marcas de metilação na linha germinativa feminina inicia-se no período pós-natal e ocorre durante toda a vida reprodutiva (Ishida & Moore, 2013; Monk, 2015b) (Figura 4). O modo como os genes de imprinting são especificamente reconhecidos e direcionados para a aquisição de metilação específica conforme o sexo ainda é pouco compreendido. Uma hipótese justificativa baseia-se no perfil de modificações de histonas existentes nos loci de imprinting que surgem como um sinal instrutivo para a maquinaria específica da metilação do DNA (Ishida & Moore, 2013).
Após a fertilização das células germinativas, o citoplasma do zigoto passa a conter dois pronúcleos que são epigeneticamente distintos. O pronúcleo paterno passa rapidamente por uma desmetilação ativa do genoma, possivelmente através de um mecanismo envolvendo a 5-hmC como intermediária, enquanto que o genoma materno se torna desmetilado por um mecanismo passivo que depende das subsequentes replicações do DNA (Ishida & Moore, 2013; Kalish et al., 2014). No entanto, nesta etapa, a metilação dos genes de imprinting é resistente à desmetilação geral que ocorre no genoma, sendo transmitida às células filhas e assim mantida durante o desenvolvimento (Ishida & Moore, 2013) (Figura 4). Um dos aspetos menos compreendidos no imprinting genómico é como as ICRs mantêm a sua metilação diferencial durante o período de reprogramação pós-fertilização. É provável que uma combinação de certos
fatores de ação cis e trans medeiem a proteção destas regiões contra a desmetilação (Kalish et al., 2014). É sugerido que nesta fase os locais metilados das DMRs sejam protegidos pela ligação de diferentes fatores, como por exemplo, a DPPA3 que protege a metilação no pronúcleo materno da desmetilação proveniente da ação da TET3 ou o ZFP57 que quando se liga a estes locais metilados, recruta a DNMT1 que assegura a manutenção da metilação (Monk, 2015b). Curiosamente, os tecidos extraembrionários e embrionários podem usar mecanismos diferentes para manter as suas marcas de imprinting (Kalish et al., 2014).
Figura 4: Reprogramação epigenética subjacente ao imprinting genómico. Adaptado de (Ishida & Moore, 2013).
Em humanos, a importância fisiológica do imprinting pode ser demonstrada através das doenças que resultam de alterações neste fenómeno epigenético. Estas doenças são raras, mas são frequentemente graves, indicando que expressões apropriadas deste pequeno conjunto de genes são indispensáveis para o desenvolvimento humano normal (Ishida & Moore, 2013). Os fenótipos clínicos destas complicações são diversos, mas envolvem principalmente o desenvolvimento neurológico e o crescimento fetal (Hussain, 2017; Ishida & Moore, 2013). Neste sentido, o papel dos genes de imprinting no crescimento fetal humano pode ser investigado através do estudo das anomalias de crescimento mais comuns, como o frequente fenótipo de restrição do crescimento fetal (RCF) (Ishida & Moore, 2013).
Assim, os genes de imprinting desempenham um papel importante tanto no desenvolvimento e função placentária como no crescimento e desenvolvimento
embrionário/fetal (Hussain, 2017; Lambertini et al., 2013; Piedrahita, 2012). A desregulação do imprinting afeta gravemente o fenótipo embrionário e alterações mais graves nos padrões de imprinting podem até conduzir à infertilidade ou ao abortamento espontâneo (Hussain, 2017; Messerschmidt et al., 2014; Sazhenova et al., 2017; Tomizawa & Sasaki, 2012).
1.3.1 Genes de imprinting do cromossoma 11 e o desenvolvimento
intrauterino
1.3.1.1 Região Controlo de Imprinting - H19 DMR
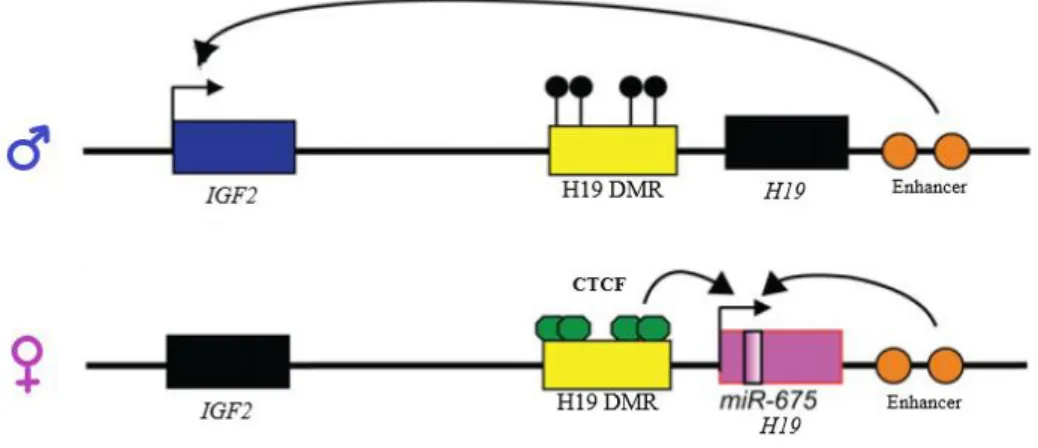
O centro de imprinting H19 DMR, também conhecido como ICR 1, encontra-se localizada na região cromossómica 11p15.5 e regula dois genes importantes no desenvolvimento feto-placentário, o gene maternalmente expresso H19 e o gene paternalmente expresso IGF2 (Insulin-like Growth Factor 2) (Derakhshan-Horeh et al., 2017; Gonzalez-Rodriguez et al., 2016; Kalish et al., 2014; Koukoura et al., 2012) Sabe-se que padrões específicos de metilação do DNA em toda a DMR são necessários para que ocorra uma regulação transcricional adequada de ambos os genes (Gonzalez-Rodriguez et al., 2016). Esta região, encontrada entre os dois genes, é dependente da ligação de um fator de transcrição designado de CCCTC-binding factor (CTCF) (Derakhshan-Horeh et al., 2017; Gonzalez-Rodriguez et al., 2016; Kalish et al., 2014). Em humanos a H19 DMR contém 7 locais de ligação a este fator e, por meio da interação com o mesmo, impede a ativação dos promotores do IGF2 (Kalish et al., 2014). Especificamente, no cromossoma paterno, a metilação da H19 DMR impede a ligação do CTCF e, consequentemente, os enhancers partilhados por ambos os genes, localizados na extremidade 3´do H19, interagem com o IGF2 que passa a ser expresso, enquanto que o H19 é silenciado. No entanto, no cromossoma materno, esta DMR não está metilada e permite que ocorra a ligação do CTCF, que bloqueia o contacto dos enhancers com o IGF2 e favorece a interação destes com o H19, conduzindo assim à expressão do H19 e à repressão do IGF2 (Adalsteinsson & Ferguson-Smith, 2014; Derakhshan-Horeh et al., 2017; Kalish et al., 2014; Koukoura et al., 2012; Plasschaert & Bartolomei, 2014)(Figura 5). Apesar da regulação estabelecida pela H19 DMR, a expressão do IGF2 é adicionalmente modulada pela metilação de outras DMRs (Koukoura et al., 2011; Piyasena et al., 2015).
Figura 5: Região controlo de imprinting dos genes IGF2 e H19. Adaptado de (Plasschaert & Bartolomei, 2014).
O IGF2 é um gene codificante de um fator de crescimento, altamente expresso no tecido fetal e placentário. Este gene de imprinting está envolvido na determinação do tamanho da placenta e na capacidade de esta transferir nutrientes da mãe para o feto, modulando assim o crescimento da placenta e do feto (Kalish et al., 2014; Koukoura et al., 2012). O H19 é também altamente expresso na placenta e em diferentes tecidos fetais (O. Koukoura et al., 2011; Lustig et al., 1994). Este dá origem a um ncRNA longo de 2,3 kb e a um microRNA (miR-675), que se acredita possuírem funções supressoras de crescimento (Li et al., 1998; Plasschaert & Bartolomei, 2014).
Como já foi referido tem sido descrito que genes paternalmente expressos, como o IGF2, tendem a potenciar o crescimento fetal, enquanto que genes expressos maternalmente, como o H19, restringem o crescimento fetal. De facto, a hipometilação desta DMR é observada em pacientes com síndrome de Silver-Russel (RSS), caracterizada por uma restrição de crescimento pré e pós-natal. Este estado hipometilado pode levar à diminuição da expressão do IGF2 e, consequentemente, a este fenótipo. Pelo contrário, uma hipermetilação desta região, associada ao aumento da expressão do IGF2, é observada na síndrome de Beckwith-Wiedemann (BWS), caracterizada por um sobrecrescimento pré e pós-natal (Azzi et al., 2014; Calvello et al., 2013; Kalish et al., 2014; Koukoura et al., 2012).
Uma vez que a H19 DMR controla a expressão destes dois genes conhecidos como moduladores de crescimento, um estado alterado da metilação desta região pode revelar uma potencial ligação entre modificações epigenéticas e um crescimento fetal e placentário anormal (Koukoura et al., 2012).
1.3.1.2 Região Controlo de Imprinting - KvDMR1
O centro de imprinting KvDMR1, também conhecido como ICR 2, situa-se na região cromossómica 11p15.5 e está localizada no décimo intrão do gene KCNQ1 (Potassium Voltage-Gated Channel Subfamily Q Member 1). Este gene de imprinting contém um promotor de um ncRNA longo (lncRNA) - (KCNQ1OT1) - que é transcrito no sentido oposto à sua transcrição e que controla a atividade dos genes de imprinting adjacentes (Chiesa et al., 2012; Sazhenova & Lebedev, 2008).
No alelo paterno, normalmente, esta DMR não é metilada, o que permite a ativação da transcrição do ncRNA que, por sua vez, silencia a expressão dos genes que se encontram em cis no domínio controlador. Contrariamente, a metilação nesta região no alelo materno conduz ao silenciamento do ncRNA o que, consequentemente, permite a expressão dos genes contidos nesta DMR (Kalish et al., 2014; Plasschaert & Bartolomei, 2014) (Figura 6). Dentro deste conjunto de genes de imprinting podemos distinguir por exemplo o CDKN1C (Cyclin Dependent Kinase Inhibitor 1C), o PHLDA2 (Pleckstrin Homology Like Domain Family A Member 2) e, como já referido, o KCNQ1 (Cordeiro et al., 2014).
Figura 6: Região controlo de imprinting dos genes PHLDA2, CDKN1C e KCNQ1. Adaptado de (Plasschaert & Bartolomei, 2014).
Também esta DMR está associada aos casos de BWS. O CDKN1C é um dos genes codificantes deste cluster que impulsiona o fenótipo associado a esta condição clínica. Este produz uma proteína que atua como um inibidor do ciclo celular na transição da fase G1/S, através da inibição dos complexos de ciclinas-cinases que, consequentemente, conduzem a uma regulação negativa do crescimento, da replicação do DNA e da proliferação celular. Assim, para