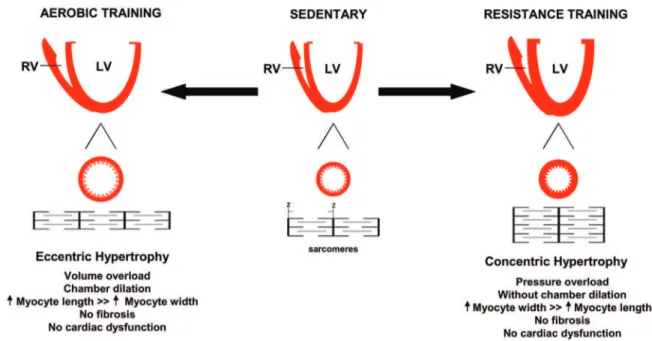
Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants
Texto
Imagem

Documentos relacionados
In the trained animals, the inhibition of NO synthesis attenuated hypertension, induced cardiac hypertrophy and significantly increased myocardial fibrosis, indicating that NO plays
Values expressed as mean ± standard deviation of concentric peak torque (Panel A), Eccentric peak torque (Panel B), Changes (%) of concentric (Panel C) and eccentric (Panel D)
and in the heart, are induced and function during muscle differentiation (7). However, several open questions remain regarding miRNAs and their role in cardiac hypertrophy. 1) What
The concurrent group developed strength exercises (strength training) followed by aerobic exercise, 27 minutes used for resistance training and 30 minutes for aerobic training
Concentric and eccentric contractions of the dorsi- flexors and the plantar flexors are a very important muscle action for long-distance runners and triathletes and must be
Raso et al. showed that the strength produced by isometric muscle actions, concentric, eccentric, isotonic and isokinetic decreases after stopping a training program,
Lista de espécies de plantas cujos frutos são utilizados como sítios de criação de drosofilídeos, na Região Neotropical.. *Dados
Regarding type of training, a study in animals that performed both types of training – aerobic and resis- tance – showed improved cognitive function (memory and spatial learning)
