D'une part, Protea, consacré à la prédiction de séquences codantes homologues, et d'autre part, carNAc, consacré à la prédiction de structures secondaires conservées. La deuxième partie de ce chapitre est consacrée à l'application de Protea et du carRNA pour l'annotation de séquences génomiques.
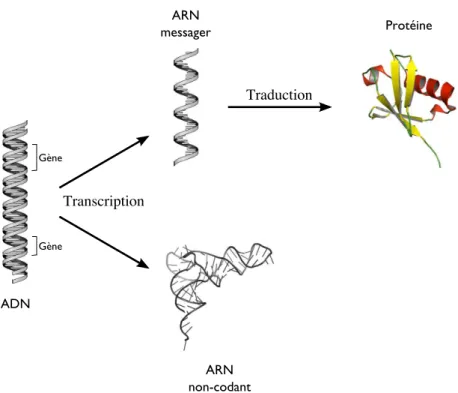
L’ARN au sein de la cellule
- Les organismes vivants
- Le dogme central de la biologie mol´eculaire
- Les acides nucl´eiques
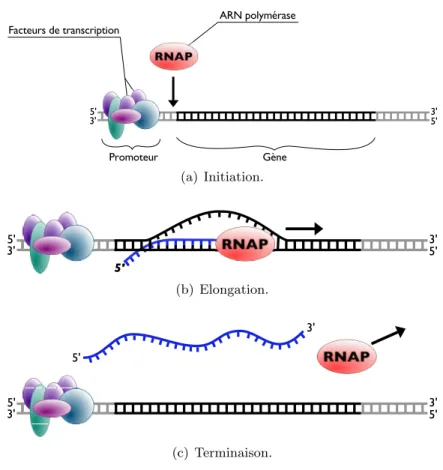
- La transcription d’un g`ene en un ARN
- La maturation de l’ARN
Ce rôle de médiateur de l’information génétique constitue le premier rôle de l’ARN qui est alors appelé ARN messager. La composition des nucléotides n’est pas le seul élément qui diffère entre l’ADN et l’ARN.

Les ARN codants
Les prot´eines
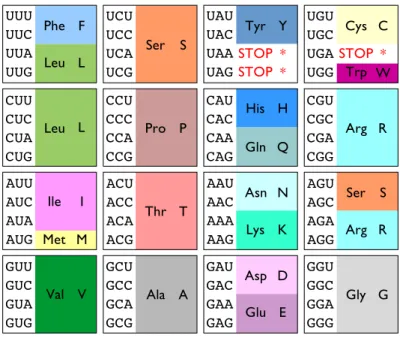
La traduction en prot´eine
Même si la grande majorité des organismes vivants utilisent le code génétique standard, il existe quelques exceptions à cette règle chez certains organismes pour lesquels les acides aminés codés ne sont pas les mêmes. . Par exemple, chez les champignons Candida le codon CUG habituellement traduit par leucine correspond à la sérine, ou encore chez certains procaryotes où le codon STOPUAG code parfois pour un acide aminé supplémentaire, la pyrrolysine.

La r´egulation de la transcription
Les ARN non-codants
La structure de l’ARN
La stabilité d'une molécule d'ARN est mesurée par son énergie libre dérivée des principes de la thermodynamique. La stabilité apportée par une tige dépend de sa longueur et de la nature de ses appariements : les paires canoniques (G≡C,A=U et G=U) sont plus stables que les paires non canoniques (G−A,C−U,.
Les familles d’ARN non-codants
La stabilité apportée par une tige dépend de sa longueur et de la nature de ses appariements : les appariements canoniques (G≡C,A=U etG=. L'observation de structures tridimensionnelles réelles a notamment permis le développement du Scor réalisé[KTHB02] base de données contenant plus de 8 000 modèles répétitifs.

L’´evolution des acides nucl´eiques
- G´en´eralit´es
- Les m´ecanismes de l’´evolution
- L’´evolution des g`enes codants
- L’´evolution des g`enes ` a ARN
Pour les séquences codantes, cette pression de sélection est liée à la fonctionnalité de la protéine produite. Une insertion ou une suppression dans la séquence codante peut augmenter ou diminuer la longueur de la protéine codée.
L’analyse comparative de s´equences nucl´eiques
- L’alignement de s´equences comme support de l’analyse comparative . 24
- Mise en œuvre bio-informatique
- Le cadre ouvert de lecture
- Les autres signaux li´es ` a la structure du g`ene
- Les biais de composition de la s´equence codante
- Les mises en œuvre logicielles
La sous-figure (a) de la figure 1.17 représente l'alignement de deux fragments de séquences codantes homologues. La sous-figure (b) de la figure 1.17 représente l'alignement des séquences d'ARN de transfert homologues.

Les approches par homologie de s´equence
Similarit´e avec des s´equences peptidiques
Même lorsqu'une protéine similaire est disponible, il est difficile de déterminer la structure complète d'un gène, notamment les limites des extrémités 5' et 3'. De plus, les sites d'épissage dans GeneWise et GenomeScan sont détectés à l'aide d'un HMM, fortement inspiré de Genscans.
Similarit´e avec des s´equences transcrites
Le séquençage « classique » d'un ADNc permet d'obtenir de manière fiable la séquence complète. En effet, les ADNc étant issus d'ARN transcrits, ils contiennent, en plus de la séquence codante, les extrémités 5' et 3' non traduites.

S´equences g´enomiques
Les approches par analyse comparative
Protea
Le mod`ele sur deux s´equences
Étant donné deux séquences coeurs, il faut donc comparer 36 paires de cadres de lecture, et donc 36 paires de séquences d'acides aminés possibles. Enfin, on compte le nombre de paires de séquences pour lesquelles la paire de trames de lecture correctes obtient le meilleur score parmi les 36 paires comparées.

L’extension ` a une famille de s´equences, le graphe des cadres de lecture 42
Grâce à cette définition de la traduction d'un alignement, des décalages de cadre de lecture positifs sont pris en charge entre des séquences regroupées dans la même méta-séquence. Sur l’exemple de la figure 2.3, la bonne paire de cadres de lecture pour les lignes de V est (1,1).
Mise en œuvre logicielle
Dans le cas de petits GCL, la détection d'ensembles de séquences codantes homologues se fait par un seuil sur le z-score du meilleur score global de tâche. 2.5 – En raison de la redondance du code génétique, les mutations dites silencieuses (positions grises) n'ont aucun effet sur les acides aminés codés.
R´esultats exp´erimentaux de Protea
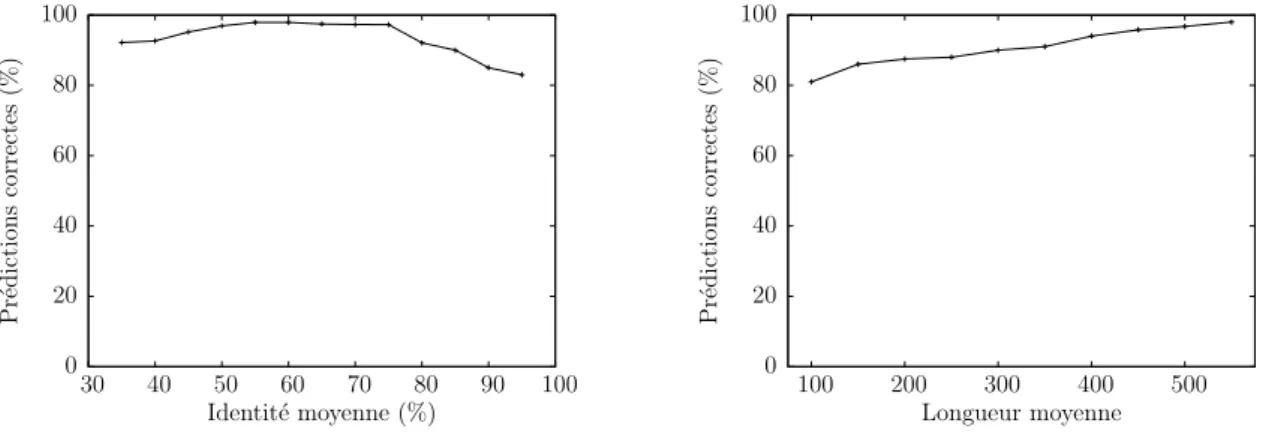
L’´evaluation des performances de Protea
De manière analogue, la spécificité Sp dans un ensemble de données est donnée par. où. Ces résultats sont également présentés graphiquement dans la figure 2.7. a) Répartition des prédictions de « codage » dans l'ensemble de données CODANT. Fig.2.7 – Répartition des prédictions « codantes » de Protea dans les groupes de 11 séquences des jeux de données CODING (a) et NON-CODING (b).

Une application au g´enome humain
Les résultats de Qrna et Protea sur les paires de séquences des ensembles de données CODING et NON-CODING sont présentés dans le tableau 2.2. 2.1 – Résultats de Protea sur les jeux de données CODING, NON-CODING et RANDOM de 3, 5 et 11 séquences. À partir de cet ensemble de données, seuls 0,8 % des ensembles de séquences devraient être « codants ».

Conclusions
Il s’agit d’un signal important qui s’avère très utile pour la prédiction des gènes d’ARN. Dans la section 3.2, nous expliquons ensuite comment ces méthodes s'appliquent à la prédiction des gènes d'ARN. Prédire la structure secondaire d’un ARN est un problème bioinformatique relativement ancien.
La pr´ediction par approche thermodynamique
Chaque cellule E(i, j) de la matrice E correspond à l'énergie libre de la structure minimale d'énergie libre de la sous-séquence s[i.j], avec i≤j, des mots. Par construction, l’énergie libre de la structure minimale d’énergie libre se retrouve dans la cellule E(1, n). M1(i, j) correspond à l'énergie libre de la structure minimale d'énergie libre de la sous-séquence[i.j] sachant que cette sous-séquence fait partie d'une branche multiple comprenant exactement une composante.

La pr´ediction par analyse comparative
Il existe plusieurs implémentations de fonctions de partition pour la prédiction de structure secondaire : RNAfold, Sfold [DL99, DCL04] et une implémentation pour les machines massivement parallèles [FHS00]. Fig.3.5 – Alignement de sept séquences d'ARN de transfert, montrant une structure commune. Un bonus est appliqué en fonction de la corrélation entre les colonnes correspondantes.

BRAliBase I, le benchmark de r´ef´erence
Puis, à l’aide d’une heuristique gloutonne, il construit successivement des ensembles de palindromes tous compatibles entre eux, c’est-à-dire sans croisement ni chevauchement. Une correspondance prédite entre deux bases etl est contradictoire si, et seulement si, il existe une correspondance entre deux bases etl dans la structure de référence telle que k < i < l < j, c'est-à-dire cet ajout de l'appariement basesietj dans la structure de référence produit un pseudo-nœud. Enfin, un appariement prédit entre deux bases i et j est compatible si et seulement s'il n'est pas incohérent ou contradictoire.
La pr´ediction de g`enes ` a ARN
- Les biais de composition en s´equence
- La stabilit´e thermodynamique
- L’homologie de s´equence et de structure
- L’approche comparative, l’existence d’une structure conserv´ee
Dans la section 3.2.1, nous nous intéressons donc à l'analyse de différents biais dans la composition des séquences d'ARN non codants liés à la formation d'une structure. Cette analyse nous amène naturellement vers l’analyse de la stabilité des structures d’ARN non codants présentée dans la section 3.2.2. 3.7 – Répartition de la négation de l'énergie libre z-score des structures de 243 ARN non codants par rapport aux structures optimales de séquences aléatoires de même composition mononucléotidique.

Evolution et enrichissement du logiciel caRNAc
L’existant
3.10 – Avancement de la prédiction d'une structure secondaire conservée entre deux séquences dans caRNAc. Filtrage des barres En fonction des points d'ancrage déterminés à l'étape précédente, les paires de barres pliantes sont répertoriées. En pratique cependant, la complexité spatiale de l'algorithme se réduit à l'hyperdiagonale de la matrice grâce à un examen des tiges incopiables et des points d'ancrage.

Introduction des m´eta-s´equences
L'énergie associée à un méta-flux est définie comme la moyenne des énergies des bâtonnets individuels qu'il contient. L'énergie associée au pliage d'une méta-bâtonnet, ou au co-pliage de deux méta-bâtonnets, est égale à la somme des énergies des bâtonnets individuels repliés en même temps. Par conséquent, nous normalisons l’énergie associée à un méta-flux en prenant la moyenne des énergies des barres individuelles plutôt que leur somme.

R´esultats exp´erimentaux
Validation sur BRAliBase I
Pour les ensembles de données qui sont en moyenne hautement conservés, les résultats ne changent guère. 3.10 – Résultats BRAliBase I sur les ARN de transfert et sur la RNAse P. a) Résultats sur les petites sous-unités ribosomales Méthode conservatrice. 3.11 - Résultats BRAliBase I pour les petites et grandes sous-unités d'ARN ribosomal.
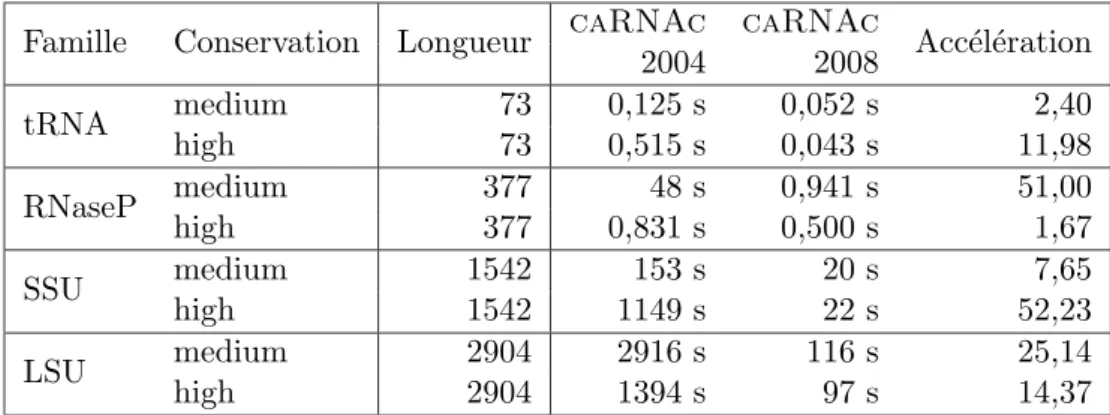
Vers la pr´ediction de g`enes ` a ARN
Pour les familles d'ARN homologues non codants, le z-score moyen des structures prédit par caR-NAc est en moyenne inférieur à celui calculé sur des ensembles de séquences aléatoires. La RNAse a été entraînée à reconnaître les ARN non codants sur les alignements produits par ClustalW. Bien que plus de 90 % des positions dans cet alignement soient correctes, seul caRNAc prédit des séquences d’ARN homologues non codantes.

L’alignement multiple de s´equences nucl´eiques
L’alignement multiple de s´equences codantes homologues
Parmi les méthodes de prédiction de séquences codantes présentées dans la section 2.2, certaines fournissent en sortie un alignement qui prend en compte les séquences d’acides aminés codées. Concernant les méthodes de prédiction par analyse comparative (section 2.3), la situation est différente car presque toutes ces méthodes fonctionnent sur des séquences déjà alignées et ne fournissent donc pas d'alignement de sortie. A notre connaissance, il n'existe finalement qu'un seul logiciel, Dialign2-2 [Mor99] qui réalise l'alignement d'un ensemble de séquences nucléiques en fonction des séquences d'acides aminés, es potentiels qu'elles peuvent coder.
L’alignement multiple de s´equences partageant une structure commune 114
Protea (section 2.4) peut être utilisé pour améliorer l’alignement de séquences codantes homologues, en particulier dans les séquences nucléaires divergentes. Enfin, l'alignement multiple des séquences d'acides aminés est transcrit de manière inverse pour obtenir un alignement multiple des séquences nucléiques de départ. La figure 4.4 montre un exemple d'alignement pour une famille de séquences homologues non codantes partageant une structure commune.

Les r´esultats exp´erimentaux de Magnolia
L’annotation par g´enomique comparative
Le pipeline d’annotation
Tous les alignements obtenus sont ensuite rapportés à la séquence à annoter afin de détecter les régions conservées, c'est-à-dire les régions de la séquence à annoter pour lesquelles il existe plusieurs séquences similaires dans la banque. Comparer la séquence à annoter avec les séquences présentes dans la banque est la première étape du pipeline. Chaque ensemble de séquences doit donc comprendre un fragment de la séquence à annoter et un nombre suffisant de séquences similaires.

R´esultats exp´erimentaux du pipeline
Statistical prediction of single-stranded regions of RNA secondary structure and application to predict effective antisense target sites and beyond. Incorporation of chemical modification constraints into a dynamic programming algorithm for RNA secondary structure prediction. An iterated loop-matching approach for predicting RNA secondary structures with pseudo-nots.