Je remercie également Abhirami Ratnakumar et Matthew Webster de l'Université d'Uppsala avec qui nous avons travaillé au sein du consortium européen LUPA (que je remercie également) pour rechercher des signatures pour la sélection artificielle et avec qui nous continuons à travailler pour identifier les différences entre les races pour caractériser et recherche d'haplotypes liés aux caractéristiques phénotypiques de la race. L'espèce canine représente donc un modèle pour l'étude des maladies génétiques et pour l'étude de l'évolution.
Introduction générale
Depuis 1995 et le premier séquençage du génome de la bactérie Haemophilus Influenzae (Fleischmann, et al., 1995), le nombre d'organismes dont le génome a bénéficié d'un séquençage complet (virus, bactéries, eucaryotes multicellulaires, mammifères) a augmenté. manière spectaculaire. Nous disposons ainsi des outils génomiques nécessaires pour (i) étudier l’impact de la sélection naturelle sur la séquence du génome du chien (Lindblad-Toh, et al., 2005) et (ii) étudier l’impact de la sélection artificielle sur les différentes races de chiens.
Le concept d’évolution
- Les forces de l’évolution
- Les polymorphismes génétiques
- La détection de la sélection
- L’évolution des espèces
La sélection positive entraînera une augmentation de la fréquence de ce variant dans la population jusqu'à ce que l'allèle favorable soit fixé. L'objectif est d'identifier les cibles de sélection positive au sein d'une espèce en utilisant l'approche dN/dS.
Le modèle génétique canin
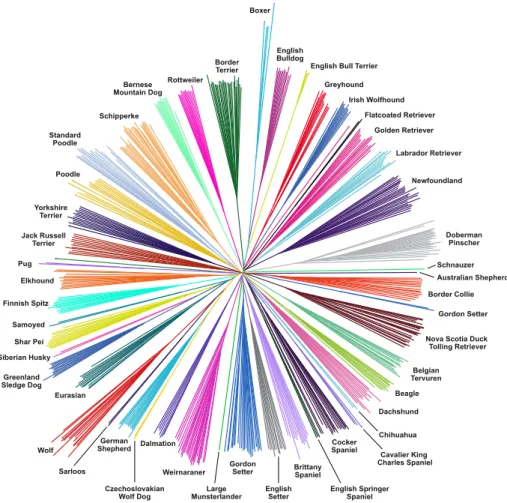
Le chien, modèle de diversité phénotypique
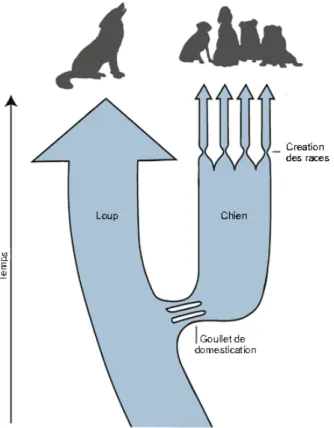
Les données actuelles sur l'origine de la domestication canine sont en parfaite corrélation avec celles déjà évoquées par J. Le but est de tirer parti de la diversité et de la structure de la population canine en général.

Le génome canin
La taille totale du génome du chien, estimée à environ 2,41 Go, est nettement inférieure à la taille des génomes humain et murin (Wade et al., 2006). Il est fort concevable que cette famille de séquences répétitives joue un rôle important dans la plasticité et la dynamique du génome canin (Kirkness et al., 2003).

Le chien, modèle d’étude des maladies génétiques
En conséquence, plus de la moitié des maladies sont spécifiques à une race et sont subdivisées en une ou un petit nombre de races (Switonski et al., 2004). Le groupe d'Awano a identifié le gène responsable de la myélopathie dégénérative, une maladie de la moelle épinière, chez la race Pembroke Welsh Corgi (Awano et al., 2009).
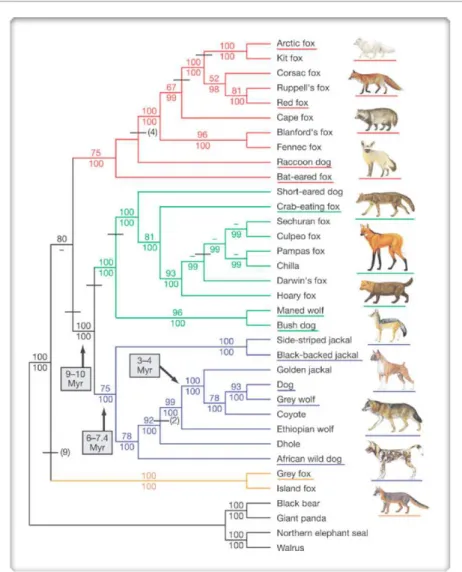
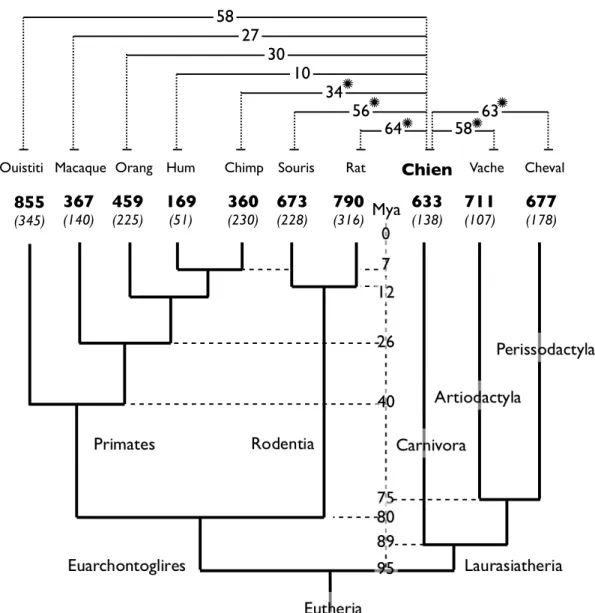
Le contexte phylogénétique
L'identification de cibles de sélection positive au sein d'une espèce par l'approche dN/dS vise à identifier les gènes et les réseaux de gènes impliqués lors de l'évolution de l'espèce considérée. Un seuil de valeur p de 0,05 est utilisé pour identifier les gènes candidats sous sélection positive dans chaque espèce.
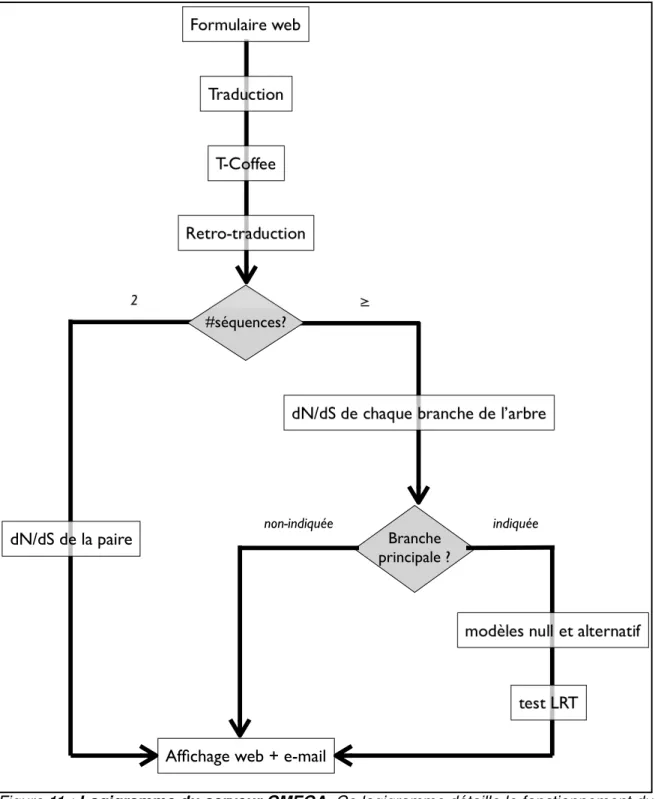
Les gènes canins sous sélection positive dans les autres espèces
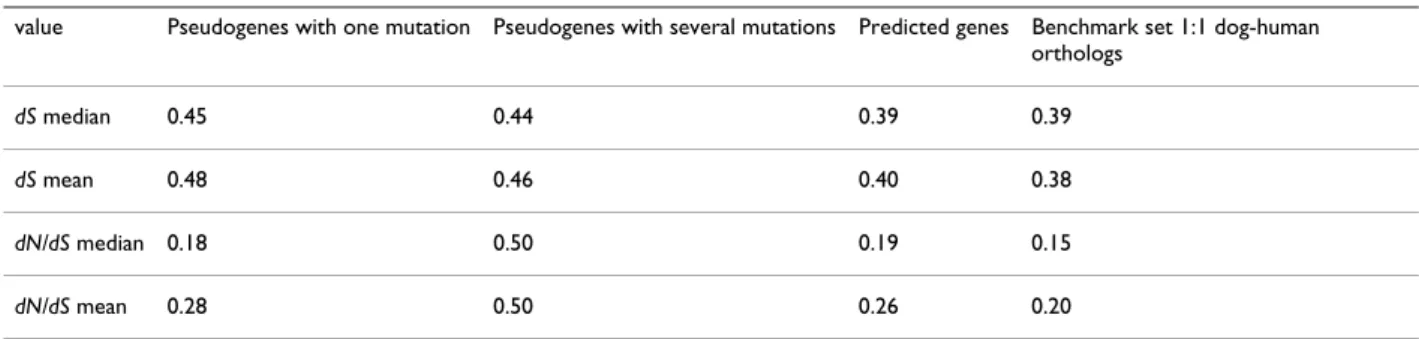
Les chiffres en gras au-dessus du nom de l'espèce représentent le nombre de gènes détectés sous sélection positive par le test LRT parmi les 10 730 gènes testés. Le nombre de gènes sous sélection positive communs aux chiens et aux autres espèces varie de 10 à 64, comme le montre la figure 10 et résumé dans le tableau 1. La lignée menant au chien partage un plus grand nombre de gènes sous sélection positive avec les lignées d'autres espèces. Laurasatheria et autres espèces. rongeurs que prévu avec des chevauchements aléatoires.
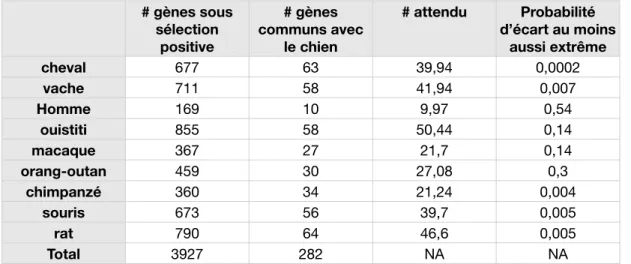
Développement d’un serveur d’analyse des contraintes sélectives des séquences
Lorsque les jeux de données sont constitués d'un nombre de séquences supérieur à deux espèces, le serveur calcule les trois ratios pour chaque espèce et pour chaque branche interne de l'arbre (modèle = 1 NSsites = 0). Pour évaluer la sélection positive au sein d'un gène d'une espèce, le serveur calcule le modèle par site appelé « site secondaire » (modèle = 2, NSsites = 2). Le serveur calcule donc le test à l'aide de l'outil !2 présent dans le package PAML modifié pour être utilisé dans un script.
Données expérimentales
Un ensemble de requêtes peut être utilisé pour extraire ces informations de la base de données OMEGA. En plus de confirmer des associations déjà connues, cette étude identifie de nouvelles associations pour les phénotypes « tail curl » et « boldness ». Cette analyse a permis de définir un lien entre courbure de la queue et un locus sur le chromosome 1 entre 96,26 et 96,96 Mb.
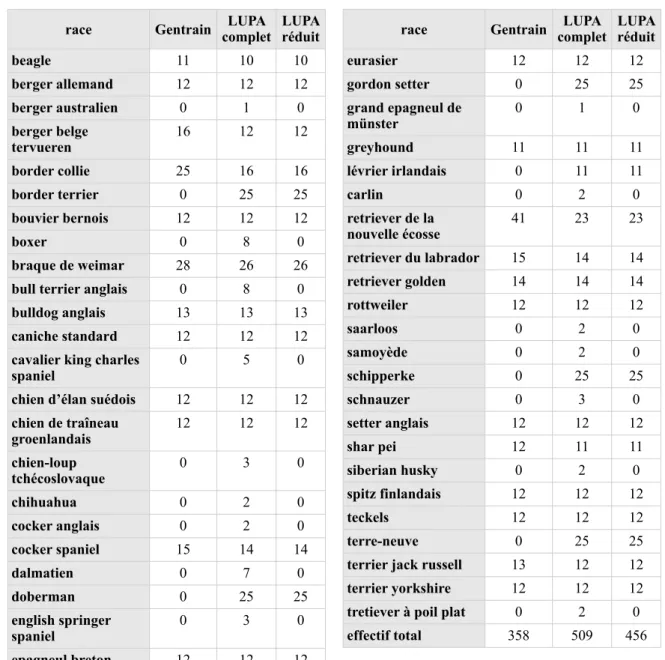
Recherche de régions de différenciation génétique : la méthode Fst-di
Vila` C, Savolainen P, Maldonado JE, Amorim IR, Rice JE, et al. 1997) Multiple and ancient origins of the domestic dog. The assembled sequence of the domestic dog (Canis familis) genome and a catalog of more than two million single nucleotide polymorphisms (SNPs) were completed and published in 2005 (Lindblad-Toh et al. 2005). Identification of small RNAs, termed microRNAs, led to a new appreciation of the role of RNA in the regulation of gene expression (Lee et al.1993; Bartel2004,2009).
Description des régions identi ées
Recherche de perte d’hétérozygotie
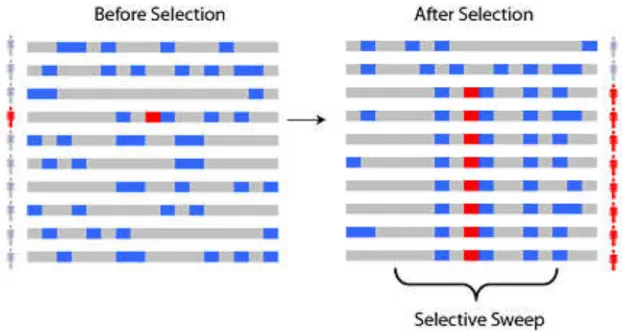
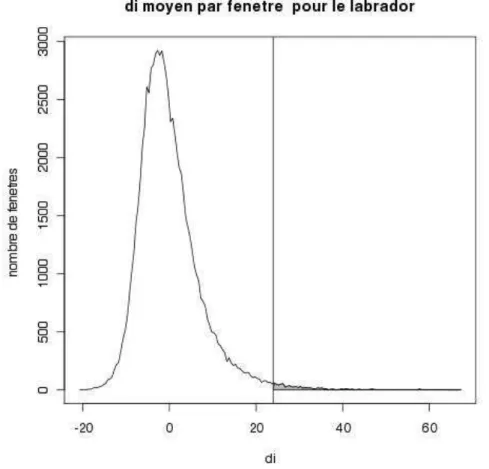
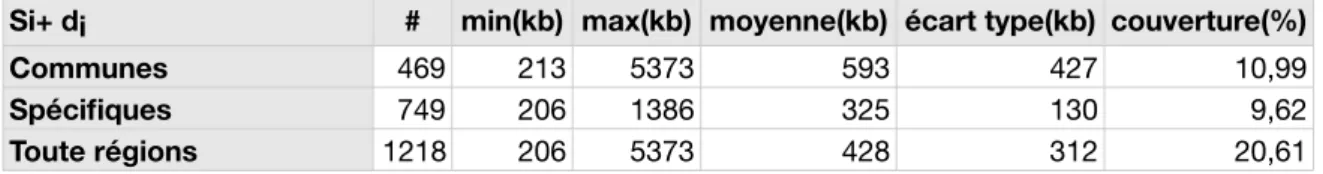
Par exemple, la comparaison entre les races beagle et labrador revient à calculer l'hétérozygotie relative de la race beagle sur la fenêtre considérée, divisée par la somme des hétérozygoties relatives des deux races. A l’inverse, comparer les races Labrador et Beagle sur une base revient à calculer l’hétérozygotie relative de la race Labrador sur la base divisée par la somme des hétérozygoties relatives des deux races. L'indice Si permet d'identifier 44 à 76 régions d'hétérozygotie réduite par race, soit 1804 régions au total qui, après projection, forment un ensemble final de 990 régions de perte de Si.
Intégration des approches di et Si
En projetant ensemble les coordonnées des régions des différentes races, nous obtenons un total de 1218 régions (voir Tableau 11 et Tableau 12). L'intégration des analyses Di et Si permet la création d'un catalogue exhaustif de régions de différenciation et de perte de diversité génétique qui constitue le catalogue des régions candidates ciblées par la sélection artificielle. L'analyse du contenu génétique de ces locus permet la recherche de voies fonctionnelles clés a priori ciblées par la sélection artificielle.
Recherche d’enrichissements fonctionnels
Nous avons téléchargé les états OMIM de tous les gènes humains en orthologie 1:1 avec le chien et de même taille moyenne que les orthologues des gènes des régions Si et di (230 Ko). L'utilisation de gènes en orthologie 1:1 est à la base d'un paradoxe dans la recherche d'une sélection positive. Parallèlement, l’ajout d’espèces permet d’affiner le contexte phylogénétique et d’améliorer la détection des gènes sous sélection positive dans la lignée conduisant aux chiens.
Limitations de l’analyse de sélection positive par le calcul du dN/dS
Nous avons ensuite obtenu un grand nombre de gènes détectés sous sélection positive pour certaines espèces (1330 chez le chien, 996 chez le rat après correction des p-values par la méthode Bonferroni). Roest Crollius, grâce à l'intégration des alignements de nucléotides et de protéines des exons des orthologues, offre une plus grande fiabilité dans la détection de la sélection positive. Le procédé de détection de sites sous sélection positive dans des gènes implique un type de correction à tests multiples.
Les gènes canins sous sélection positive
Le chien partage des gènes sous sélection positive avec les autres laurasiatheria représentées par le cheval et la vache, qui sont l'espèce la plus proche du chien dans notre ensemble de données, et qui partagent le fait que le chien a été domestiqué. Les chiens partagent également des gènes sous sélection positive avec les rongeurs ; Les rats et les souris, comme les chiens, sont présents dans l’environnement défini par les humains du monde entier. En revanche, le chien ne présente pas plus de gènes en sélection positive comme les primates (sauf le chimpanzé), qui sont phylogénétiquement plus éloignés que les autres Laurasiatheria.
Développement d’un serveur d’analyse des contraintes sélectives des séquences
Le chimpanzé présente une cooccurrence de gènes sous sélection positive avec Laurasiatheria et des rongeurs dans l'ensemble de données ; Cela peut révéler une évolution particulière du chimpanzé ou être un artefact d'analyse, comme Mallick et ses collègues l'ont montré pour 59 gènes qui étaient des faux positifs dans la recherche de gènes sous sélection positive chez le chimpanzé par rapport au génome humain (Mallick, et al. , 2009 ). De futures analyses fonctionnelles permettront de mettre en évidence les voies métaboliques cooptées entre le chien et les espèces ou groupes d'espèces de chacun. OMEGA bénéficiera de deux améliorations principales : l'utilisation des capacités de calcul parallèle des machines de la plateforme bioinformatique GenOuest, qui augmentera la rapidité des calculs de tests en répartissant les différents jeux de données utilisateurs ou calculs de différents modèles sur différents processeurs et en ajoutant une option pour choisir entre la procédure d'alignement actuelle et utiliser un outil d'alignement qui prend en compte les changements de cadre (qui sont alors considérés comme des erreurs sans signification biologique), tel que MACSE (Ranwez, et al., 2011).
La puce CanineHD
2) 1:0 gene ('missing gene' in the dog) is positioned on the reference species of the synteny map. -Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, et al. 2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Most canine genetic diseases are breed specific due to founder effects at the creation of the breed (Parker et al. 2004).
Étude des régions identi ées
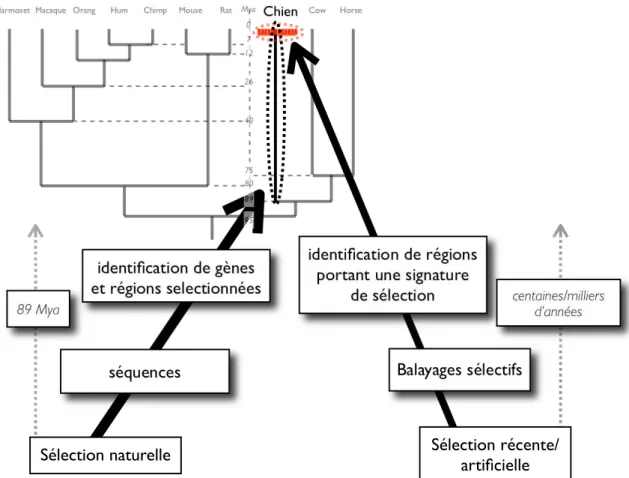
Intégration des signatures des sélections naturelle et arti cielle
Nous avons recherché de manière indépendante les signatures que la sélection naturelle a imprimées sur le génome du chien au cours des 89 millions d'années d'évolution de la branche. Les résultats préliminaires de l'apparition simultanée de la sélection naturelle et artificielle soulèvent la question de l'importance des lieux impliqués dans les événements de la sélection naturelle et artificielle. Dans cet article je décris l'importance de la sélection artificielle dans l'évolution du chien.
Publication en préparation
The domestic dog consists of more than 350 genetically distinct breeds that offer considerable variation in morphology, physiology and susceptibility to disease. Although the dog genome was predicted using a combination of ab initio methods, homology studies, motif analysis and similarity-based programs, it still requires a deep annotation of non-coding genes, alternative splicing, pseudogenes, regulatory regions and gain- and loss events. Such analyzes may benefit from new sequencing technologies (RNA-Seq) to better exploit the advantages of the canine genetic system to detect disease genes.
Publications non liées à la thèse
The amino acid sequences are aligned between the primary structure of the human and canine sequences. This has contributed to the success of the dog as a model for the genetic dissection of phenotypic traits. We repeated these simulations for each of the sample sizes represented in the original data (ranging from 10 to 52 haplotypes), rendering a total of 767 simulated data sets.
Furthermore, much of the variation in traits associated with canine morphology appears to be determined by a small number of large-effect genetic variants (Boyko et al. 2010). The identification of targets of artificial selection in dog breeds therefore allows the detection of the genetic variants that may be involved in phenotypic variations (Akey et al. 2010; Vaysse et al. 2011).