J.
Phj.s.
II France 6(1996)
1759-1780 DECEMBER 1996, PAGE 1759Theory of Long-Range Interactions in Polymer Systems
A-N- Semenov
(~'~'*)
(~)
Department
ofApplied Mathematics, University
of Leeds, Leeds LS2 9JT, U-K- (~)Nesmeyanov
Institute ofOrgano~Element Compounds
of RussianAcademy
ofScience,
28 Vavilova Str., Moscow l17812, Russia
(Received
15July1996~
received in final form 12August
1996,accepted
2September1996)
PACS.36.20.~r Macromolecules and
polymer
molecules PACS.68.45.~v Solid~fluid interfacesAbstract. A mean-field theory of inhomogeneous polymer systems is
developed
in order toinclude the finite molecular
weight
effects. Ageneral analytical expression
for the conformational free energyproviding
abridge
between strong and weaksegregation
limits is derived. Thegeneral
results are further
employed
to obtain thedependence
of interfacial tension inpolymer
blendson the molecular
weights
of the components. Along-range
repulsion(induced by
the presence ofpolymer ends)
between solid walls inpolymer
solutions and interfaces inpolymer
blends is predicted, the effectbeing formally
due to finite molecularweight
corrections. It is shown that thisrepulsion
can lead to a stabilization of colloidal systems with small amount of addedpolymer
even in the absence of any specific interactions between polymer and particles. It is also shown that the end~induced repulsion can lead tofreezing
of domain structures at late stages of demixing in polymer blends.1. Introduction
Interfacial and surface
properties
ofmultiphase polymer systems
areimportant
both for manyapplications (such
asadhesion,
lubrication and stabilization ofcolloids)
and for academic rea~sons. In
particular,
interfaces inbinary polymer
blends have beenintensively
studied bothexperimentally [1-5]
andtheoretically [6-16]
for thepast
several decades. Theproperties
of the interfaces were discussed in terms of theFlory
interaction parameter x, the number of links per chain N and othergeometric
characteristics ofpolymer
chains. A mean~field the- ory of interfaces between immisciblehomopolymers (A
andB)
in thestrong segregation limit, xN
»1,
wasproposed by
Helfand and co-workers[7-9].
Their results for interfacial tension ~f and half-thickness A of an interface betweensymmetric polymers (with geometrically
identicallinks)
are~f =
~~)~x°.~,
zl =ax~°.~ (i)
where a
=
b/@,
b is the statistical segment ofpolymer
chains, v is the volume perlink,
T thetemperature.
Theprofile
of volume concentration ofA-links, #(z)
e#A(z),
is#(z)
=p(z)
e 0.5(1+
tanh(I)j (2)
(* e-mail: [email protected]
where z
= 0
corresponds
to the interfaceplane;
theprofile
for the Bcomponent
is#B(z)
=
1
d(z)
since the system isincompressible.
The interfacial thickness in the
strong segregation
limit(SSL)
is thus much smaller than thetypical
size of apolymer
chain: A < R =aN°.5,
which iswhy
this limit is often called theregime
of narrow interfaces. Note that A can be considered as an effective correlationlength:
for distances z » A the effect of the interface
exponentially decays
and becomesnegligible (see Eq. (2)).
Thus correlations due topolymer
chainconnectivity, corresponding
to the scaleR,
are screened out
by
monomer interactions.(The
samescreening
is known for any concentratedpolymer system
where the bulk correlationlength
is smaller than the chain size Rii?].)
Thesame
screening
is alsodisplayed by
asystem
of twoparallel
interfaces whichnearly
do not affect each other if the distance betweenthem, h,
is muchlarger
than the correlationlength (see
Ref.[18j):
the mean-fieldtheory predicts
that the interaction energy isexponentially
small if h » £h(in particular
the interaction isnegligible
for hr~
R).
The last statements however are
strictly
validonly
in the limit of infinite molecularweight,
N - cc. Below we show that the effect of chain ends
(which
arepresent
for a finiteN)
results notonly
in a renormalization of the interfacial tension(and
other interfacialproperties)
but alsogives
rise to animportant long-range
interaction between any localinhomogeneities
in concentratedpolymer systems.
Inparticular
we find that twoparallel
interfaces do interact on the distances hr~
R.
In the next
section,
wedevelop
ageneral
mean-fieldtheory taking
into account corrections to the free energy due topolymer
chain ends. From a formalpoint
of view thetheory
is an ex-tension of the
ground
state dominanceapproach [17,19, 20]
tosystems
which are characterizedby
a continuous spectrum. Thegeneral
results are tested in theregime
of weakinhomogeneity
and are then
applied
topredict
the molecularweight dependence
of the interfacial tension. In the fifth section the results are furtherapplied
to treat interactions betweenparallel
surfaces and also between small solid orliquid
inclusions in a concentratedpolymer system.
We show how thepredicted long
range forces may lead to a stabilization of colloids(emulsions)
with- outadding
of anyspecial stabilizing agents.
Theproblem
of interaction between interfaces inpolymer
blends is considered as well. In the last section we discuss another effect of the pre- dictedlong-range
interaction: apossible
termination of the domaingrowth (effective freezing
of the domain coalescence
process)
at a certainstage during demixing
in blends ofincompatible polymers.
2. Free
Energy
of a Finite MolecularWeight Polymer System
This section is devoted to a mean field consideration of
thermodynamic properties
ofa two-
component polymer
mixture(a generalization
of the results to the case of any number ofcomponents
isstraightforward).
Note that the mean-fieldapproximation adopted
in this pa- per is valid ifv/b~
< 1(see
Ref.[20j
for moredetail):
the corrections due to fluctuationsof the monomer
density profile (not
consideredhere)
areroughly proportional
to the smallparameter
u/b~.
2.I. BACKGROUND. Let us consider a
system
of twohomopolymers
A and B in aquasi-
equilibrium
state characterizedby
some known(given)
distributions of A and B monomers,#A(r)
and@B(r).
Thethermodynamic potential (effective
freeenergy) corresponding
to thisstate can be
represented
as a sum of three basic termsaccounting
for the conformationaldistributions of the
polymer
chains and monomer-monomer interactions[20j
~ [lbA,
~Bj
"Finf
[lbAj +F$nf
[lbBj +tint (3)
N°12 THEORY OF LONG-RANGE INTERACTIONS IN POLYMER SYSTEMS 1761
where
F)~~
is due to a decrease of conformationalentropy (which
also includes translationalentropy)
of A-chains in theinhomogeneous
state characterizedby
the distribution PA(r); F$~~
is the
analogous
conformationalfree
energy of B-chains. The last terml§nt
=l~nt (IA, 48)
represents the free energy of interactions between the monomers. It is the term
l~nt
that determines thecompressibility
of thesystem
and also thedegree
ofincompatibility
of A and B links. The standardFlory-Huggins
modelii?]
assumes that the blend isincompressible
4A(r)
+iB(r)
" I
and also that the
density
of the interaction energy isgiven by
aquadratic
form of#A
and @B1tint
"~~~~ / lbA(r)@B(r)d~r (4)
U
where u is the effective volume per one link
[21j.
Although
the effects of finitecompressibility
and alsohigher
order terms in thedensity
of the interaction energy
might
beimportant
in some cases[13j,
theFlory-Huggins
modelprovides (at
least semiquantitatively)
anadequate description
of mostpolymer systems.
In fact many of the resultspresented
in this paper areactually
insensitive to theparticular
form of the interaction free energy. We use theFlory-Huggins equation (4)
in all of theexamples
considered below in order to
keep
thestrongest
link withprevious
theoretical studies.The conformational free energy in the limit of
long
A chains(NA
-cc)
is[17,19,20j:
~2 (v j
)2F)~~ [#A)
"
~
kBT
~d~r (5)
4U
#A
The contribution of B chains is
given by
ananalogous expression. Equation (5)
is valid if the characteristic scale ofinhomogeneity, I,
is muchlarger
than the link size aA(the
continuous chainlimit),
and is much smaller than the Gaussian chainsize, RA
"
N(/~aA:
aA<I<RA
The conditions I » aA, I » aB are assumed
throughout
this paper.Note that for a small
amplitude inhomogeneity, #A
=(IA +6#A, b#A
<(#A), equation (5)
can be derived
using
randomphase approximation (RPA) ii?],
however thisequation
is validmore
generally
forarbitrary amplitude b#A.
Below
(to
the end of Sect.2)
the conformational free energy of theA-component only
is considered. The size a= aA is chosen as a unit
length,
andkBT
as a unit energy. We also omit index A in order to make notationssimpler.
Equation (5) implies
the so-calledground
statedominance;
it is exact in the limit of in-finitely long chains,
N=
NA
- cc(and
also for a continuous model ofpolymer chains).
Ageneralization
of theground
state dominanceapproach
for a finite chainsystem
is considered below. It is assumed that the chains are stilllong enough:
the coil size R is muchlarger
than thetypical inhomogeneity
scale I(say,
the thickness of interfacial or surfacelayer), aN~/~
» l.For
simplicity
we first consider a one-dimensional(d
=
I)
casej ageneralization
of the results toarbitrary
d isstraightforward
and isperformed
later. We calculate two correction terms to the dominant term,equation (5),
which areformally proportional
to1/,V
and1/N~+~/~
Following
the lines firstproposed by
Lifshitz[19j
we note that the conformational free energyof,
say,A-component, Fcanf [4A),
isactually equal
to the free energy of asystem
of A-chains with no excluded-volume interaction(ideal system)
in anon-equilibrium
state characterizedby
a non-uniform concentration
profile, #(z
=
#A(z).
We will assume first that aninhomogeneity
of the monomer distribution is localized in the
region
(z(£ I,
so thatp(z)
-do
in theregion
(z( » I. It is useful to consider the idealsystem
under externalfield, U(z),
which induces thegiven
non-uniform monomerdensity profile,
withU(z)
- 0 for z - cc[22j.
The conformational free energy
(per
unit area in x, yplane along
which thesystem
is homo-geneous)
is thenFca~f [#j
= F[Uj @Udz (6)
where
fl [Uj
= In
2
is thethermodynamic potential
of thesystem
under the external field U As the chains do notinteract,
thepartition
function is2
=
Zf IN!,
whereZi
=
Zi [Uj
is thesingle-chain partition
function andM
=
/ d(z)dz (7)
NV
is the total number of chains. Thus in order to find
Fcanf
we need to calculated U andfl [Uj.
The rest of the section is devoted to this task.
2.2. CORRELATION FUNCTIONS OF IDEAL POLYMER CHAINS. A conformation of a
single
chain is characterized
by
coordinates of all monomers,R(s),
0 < s <N,
where s is themonomer
position along
the chain. Thecorresponding single-chain
Hamiltonian is[23j
H[Rj
=jm ff ((f)~
ds +f/
U(R(s))
ds. The statisticalweight
of a chain withgiven
endpositions,
z and z'
(the
Greenfunction)
is:G(N,
z,z')
=
j
exp
j-H jRi) biz R(0))b(z' R(N))
DjRi
The Green functionobeys
thefollowing equation [20, 24j
with the initial condition
G(0,
z,z')
=
b(z z').
Thegeneral
solution ofequation (8)
can be written in the formG(N,
z,z')
=
~j ~fim(z)~i$(z')e~~'"
~(9)
m
where
~fim(z)
andEm
are theeigenfunctions
andeigenvalues
of theequation -~lclz)
+Ulz)~mlz)
=Em~l'mlz).
Equation (9) implies
summation over alleigenstates.
Below we are interested in the case of continuous rather than discrete spectrum. Moreover
we will assume that the spectrum does not contain any discrete branch at all. This
assumption
is validated in Section 2.3. In this case two
eigenfunctions ~k(z)
and~l-k(z) correspond
to each E= k~
(note
that the lowerboundary
of the continuousspectrum
is E=
U(oo)
=
0):
~l'+ (U k~) ~k
= 0
(10)
Since the
complex-conjugated
function~((z)
also must be a solution of the sameequation,
we canalways
chooseeigenfunctions
so that~l-~(z)
=
~li(z) iii)
N°12 THEORY OF LONG-RANGE INTERACTIONS IN POLYMER SYSTEMS 1763
Accordingly
we rewriteequation (9) keeping separately
the termcorresponding
to theground state,
E = 0:~~~'~'~'~ j~~~i~~~~
~
/_~ ~~~~~~~~~~'~~
~~~~~~~
The
eigenfunctions obey
thefollowing
normalization condition/ ~l~(z)~li>(z)dz
=
2ii(k k') (13)
Note that
equation (10)
for k= 0
implies
that~o(z)
isnearly
constant in theregion
)z) » I.We also assume that
~o loo)
=
~fio(-oo) (see
Sect.2.3).
It is convenient to normalize~o
with thefollowing
condition:~§o
(oo)
= 1. Below in Section 2.3 we show that the function
~,o(z)
then becomes a continuation of thefamily ~§k(z)
in the limit k- 0. The
point
k = o is excluded fromintegration
in the second term in the r-h-s- ofequation (12).
The reasonwhy
the E= 0
term is treated
separately
inequation (12) (in spite
of the fact that this term isvanishing
in themacroscopic limit,
V-
oo)
is considered below.Integrating
the functionG(N,
z,z')
over the second coordinate we find the statisticalweight
of an N-chain with agiven position
of one endonly:
Wh~~~
v =
/ ~((z)dz
~~~~
is
nearly
the total volume(length)
of thesystem (as il,o(oo)
=1)
and we defineCo
=f~o (I ~o) de, Ck
"
fi'((z)dz
for k#
0. Note that bothCo
andCk
remains finite inthe limit V - oo. Note also that both definitions are
actually
consistent since the scalarproduct
of two differenteigenfunctions
must be zero:J ~§o(z)~§((z)dz
=0,
k#
0. Thereforewe can
generally
write:Ck
"/ ~((Z)11 #0(Z))
dZ(16)
We notice now that the first term for
G(N,z) (corresponding
to E=
0)
is notvanishingly
small relative to the second one: the E
= 0 term
gains
additional factor I'during integration
over z'. That is
why
this term must be treatedseparately.
Thesingle-chain partition
function isZi
"
f G(N, z)dz; using equation (14)
we thusget
Zi
" V(1+ ~°
+/
~~)Ck)~ e~~~~ (17)
1'
~
2K
Let us now calculate the
profile
for the overall monomer volumeconcentration, #(z).
Theprobability
to find the n-th link of apolymer
chain atpoint
z ispn(z)
=
Zi In, z)/Zi>
whereZi In, z)
is the statisticalweight
of a chain with fixed n-th link. Two chain parts(0, n)
and(n, N)
can be consideredindependently (as they
do notinteract)
as subchains of n and N nlinks,
one end of each subchainbeing
fixed atpoint
z. ThereforeZi In, z)
=
Gin, z)G(N
n,z).
Summing
over all monomers of allfit independent
chains we obtain#(z)
=
NV J pn(z)dn.
Taking
into account thatufif/Zi
=
#o IN
in themacroscopic
limitII'
-
oo)
wefinally get
the overall monomer concentration:#(z)
=~° dnG(n, z)G(N
n,z) j18)
N
~
Note that in the limit N
- oo
(in
theground
statedominance)
the second term inequation (14) vanishes,
so thatG(N
-oo,z)
=
~§o(z)
for V - oo and therefore#(z)
=
#oi'((z)
which isa result of the
ground
statetheory [25].
However for a finite N the second termpresents
acorrection:
G(N, z)
=
~§o(z)
+b~§(z).
In order to estimate this correction we note that thecorresponding integral (see Eq. (14)
isexponentially
cut off for k > ko "N~~/~,
and also thatin the
region
k£
ko theamplitude Ck
is of order I(which
is thetypical
range ofintegration
in
Eq. (16) ).
Thereforeb~l(z) ~ ~(
~
for z
~ N~/~ Using equation (18)
we thusget:
~~~~
~°~~(~~
~ ~jl12
where the correction term is small since I < R
=
N~/~
2.3. CALCULATION OF END-CORRECTIONS. In order to calculate
Fconf [#], equation (6),
we first need to find the external field
U(z)
which inducesexactly
thegiven
monomerprofile
when it is
applied
to the idealsystem
ofpolymer
chains.Obviously
this is acomplicated
taskas itself. It is therefore convenient to
perform
the calculation in aslightly
different way. We first find the fieldUo(z)
defined in thefollowing
way: the monomerdensity profile
calculated for U= Uo in the
ground
stateapproximation
mustexactly
coincide with thegiven
distribution#(z).
Then we calculate the free energy of thesystem
under the field Uo and find correctioniii (z)
to the monomer concentrationprofile
on thetop
of theground
stateapproximation.
After that the
corresponding
correction to the field 6U=
WI
needed to compensate forb#i
will be determined.
Finally
the correction to the free energy related to[Vi
is calculated(~).
According
to the definitions above theground
stateeigenfunction, ilo(z), corresponding
to the fieldUo(z),
isstrictly
related to thegiven density profile:
~Yiiz)
=) jig)
so that in
particular
~Yol-cxJ)
=lYo(co)
= 1Using equation (10)
~n.e thusget
q/,,( ~2
~°~~~
"v)())
"
'~°'~~~~WI~°~~~~
~~°~Note that the field Uo defined in
equation (20)
does notproduce
any discrete spectrum(with
E <
0)
sinceilo(z)
is real andpositive everywhere
and therefore mustcorrespond
to theground
state.
Let
I
[U] be the exactequilibrium
monomerdensity profile
inducedby
the field U:#
[U] =if [U] /bU,
wherefl [U]
is defined afterequation (6).
Inparticular #
[Uo]=
#+ iii
where#(z)
is the
given density profile
andiii
is a small correction to theground
state result.Adding
acorrection W to the field we write
(in
the linearapproximation) #
[Uo +ll'j
cf#
+b#i l/l§",
where
l/
isa linear operator,
I/H~(z)
eJ I[(z, z')W(z')dz'.
Therefore the external field weare
looking for,
U= Uo +
WI,
isspecified
in the mainapproximation by
theequation
l/l§1
=
h#1 (21)
(~) The same
general
strategy wasadopted
in reference [16].N°12 THEORY OF LONG-RANGE INTERACTIONSIN POLYMER SYSTEMS 1765
Let us define the free energy functional F
[U, ii
=
fl [U] J U#dz,
where both U and#
areformally
treatedindependently.
The conformational free energy(Eq. (6))
is thusindirectly
defined
by equations Fconf [#j
= F(U, I [U]j, #
[LTj =
#. Using general
relation bF [U,#j
~~ "
# (U) #
and the relations above we find that
~~
~~)/j ~'~~
cf-k (W lf~i
in the main
approximation. Integrating
back the functional derivative weget
Wilz)
F~~nf iii
= F [Uo +lf'i, II
=
Fo
+/
dz
/ dIV(z)
K(WI W)
=
Fo
+Fi (22)
o
where
Fo
+ F[Uo, II
and~ / ~~~~~~~~~~~~
~~~~Below we show that
Fi
istypically proportional
to1/N~/~ (for
one-dimensionalcase).
There- fore we need to calculate this term in the mainapproximation only
sincehigher-order (in 1IN)
corrections are
neglected
here.Using equations (17, 7, 15, 20)
weget
Fo
= F* +~
dz ~
(li 4)
d=~°
~~)Ck)~ e~~~~ (24)
4u
/
NV
/
NV
/2K
where we take into account that
V#o
"
fifNv (see Eq. (19)) [26].
Here F*
=
fifIn )
is theideal-gas
free energy of thecorresponding homogeneous
sys-tem. The second term
(which
isexactly equal
toJ Uo4dz)
is the well-knownoutput
of theground
stateapproximation (compare
withEq. (5)).
The last two terms represent the finite N corrections which areproportional
to1IN
andko IN
mJ
1/N~/~ correspondingly.
Let us turn to calculation of the last correction,
Fi Using equations (14, 18)
we find in the mainapproximation (and
in the limit V -oo):
~~~
~~~ "j~
lYo(z) /~
d~/
dk ~ ~~° ~~ ~~
~~k(~)
~~where the functions
ilk (z)
are defined inequation (lo)
with U=
Uo,
andilo(z)
is defined inequation (19).
We now have to find the functions
4fk(z).
Note that sincetypical
n r~ A~ in the r-h-s- ofequation (25),
weonly
need to consider smallenough
k~
ko =1/N~/~:
theintegral
is cut-off forlarger
wave-numbers.Taking
into accountequation (20)
and the fact thatilo(z (Eq. (19)
tends to
unity
in theregion
)z) »I,
we conclude thatUo(z)
vanishes in thisregion (in
the mostinteresting
cases the decrease ofUo(z)
isexponential).
It is useful to
split
theproblem
and consider first the behavior of the functionsilk(z)
in the outsideregion
)z) » I. Hereequation (lo)
reduces toil[(z)
+k~ilk(z)
" 0
implying
ageneral
solution in the form:ilk(z)
= Ae~~~ +
A'e~~~~,
z <
0; ilk(z)
=
Be~~~
+B'e~~~",
= > 0.Obviously
A and A' must be linear combinations of B and B':Here we take into account that both
ilk(z)
andil((z)
are solutions ofequation (10).
We thus define afamily
of solutions ofequation (10)
whichadditionally debend
on two freeparameters
B and B'.Taking
into account that substitutions k --k,
B -B',
B' -B,
A -A',
A'- A
together
does notchange
theeigenfunction
at all, weget
thefollowing general
relations:a(-k)
=
a*(k), fl(-k)
=
fl*(k) (2i)
Another
general property
of the functions a andfl
follows from energy conservation[27j (within analogy
ofEq. (10)
andSchr6dinger equation):
lal~
= I +lfll~ 128)
Let us
form611y
set B=
1,
B'=
0,
and consider the limit k- 0; the z > 0
asymptotics
is thenilk(z)
= e~~~ - 1.
Obviously equation (10)
with U= Uo
(Eq. (20)) implies
theonly
solution with this
asymptotics, namely
the functionilo(z) (Eq. (19) ).
Thereforelimk-o ilk (z)
and
ilo(z)
must coincide also in theregion
z <0,
)z) »I,
whereilo(z)
-1,
so thata(o)
+fl*(o)
=
(2g)
Now
using equations (27-29)
we obtaina(0)
=1, fl(0)
= 0. Both
o(k)
andfl(k)
must beregular functions;
theirexpected
behavior for small k is:a(k)
= 1+iO(k), fl(k)
=
O(ik).
As theonly
characteristic scale in theeigenvalue problem (Eq. (10)
isI,
we can rewrite the aboverelations as
o(k)
= 1+iO(lk), fl(k)
=
iO(lk) (30)
In the relevant
region
k~
ko the corrections are of orderlko
"
I/N~/~
<1,
and thuscan be
neglected
in the mainapproximation.
The actual values of the
parameters
B and B' must be chosen in order tocomply
with the normalization conditions(Eq. (13))
whichimply
thefollowing
relations:AA'+ BB'
=
0;
)A)~ +)B')~
= 1(31)
Equations (31. 26) completely
define theA, A', B,
B' up to a commonphase
which can be fixedby assuming
that B is apositive
real.(We
also assume that )B~) <)B).) Using
alsoequations (30)
we thusget
A = 1+iO(lk),
B= 1+
O(l~k~),
A'=
iO(lk),
B'=
iO(lk).
Omitting
corrections of orderI/N~/~
we thus obtain
ilk(z)
cf e~~~ for )z) » I. Therefore in the limit k - 0 the functionilk (z)
coincides withilo(z)
as claimed above(since
both functionsreveal the same
asymptotic
behavior andobey
the sameEq. (10) ).
It is easy to check now that inside theinhomogeneity region,
)z)mJ
I, ilk (z)
coincides withilo(z) again
within an error of order lk. Therefore in the mainapproximation
weget
the relationilk(z)
cfilo(z)e~~~ (32)
which is valid
everywhere
up to a correctionmJ lk which is small
provided
that k£ ko.
With
equation (32)
the correctioniii (Eq. (25))
can be rewritten asSubstituting equation (32)
intoequation (16)
weget
Ck
"/ ilo(z)
II ilo(z)) e~~~~dz (34)
N°12 THEORY OF LONG~RANGE INTERACTIONSIN POLYMER SYSTEMS 1767
We are now in a
position
to find theoperator l/. According
to its definition(see Eq. (21)
andabove)
thecorresponding
kernel isKjz, z')
i
jji~)~
where
#
=I[Uj
is the monomer distributioncorresponding
to the fieldLT(z).
The fluctuation theorem[28j
then ensures thath'(z, =')
is alsoequal
to the correlation function of monomerdensity
for the idealsystem
under the fieldU(z): K(z, z')
=
(#(z)#(z')) (#(z)) (#(z')),
wheremean the
thermodynamic
average.Taking
into account that the chains do not interact we thusrepresent
the kernel asfif~2 K(z, z')
=
/ dndn'G(n, z)G(n', z')G(N
nn',
z,
z') (35)
21
where three factors in the
integrand
constitute the statisticalweight
of apolymer
chain with n~th link fixed atpoint
z, and(N n')-th
link atpoint
z'. In the mainapproximation
wekeep only
the first term in the r-h-s- ofequation (14): G(n, z)
milo(z). Substituting equation (32)
into
equation (12)
andneglecting
in the lastequation
the first term which vanishes in thethermodynamic
limit, weget: G(n,
z,z')
milo(z)ilo(z') J fle~~l~~~')e~~~'~
Thus we rewriteequation (35)
as~~~'~'~ ~°~~~~~~~~~~~'~ / ~
~~~~~
~'~~~ ~~~~~
~~~~where
fD
is theDebye
functionfD(it)
=
[(it
+ e~~-1) 137)
Now
using equations (21, 23,
33,36)
after some transformations weget
~i
=) / j)
ic~12 (£ (1- -N~2jj
~ ifD(Nk2 (38)
Finally using equations (22, 24, 38)
we obtain[29j
where
e~"(u
+1) f(~)
~~~~"
~ l + e-~
CL
"/ Q(Z)e~~~~dz 141)
~(z)
=w@ <(Z)/<o (42)
As in
equation (24)
the first term here is theideal-gas
free energy of ahomogeneous system,
the last two terms
represent
corrections to theground
statesquare-gradient
free energy dueto finite
length
N ofpolymer
chains. Note that the corrections areformally proportional
to1/N
and to1/N~/~ correspondingly.
The dominant end-correction of order1/N only
waspreviously
calculated in reference[30j using
aslightly
differentapproach
firstsuggested
inreferences
[31,32j.
Thepresent
result(Eq. (39)
is inagreement
with that obtained before[30j.
An
analysis
shows that the result(Eq. (39))
is valid within an error which isproportional
to
1/N~.
The small parameterimplied
isactually Co/R,
where R=
N~/~a
is the coil size,if the
inhomogeneity
of monomer distribution is localized in theregion
of size R. The con- ditionCo /R
< 1 inparticular implies
that either anamplitude
of the function~(z)
is smalleverywhere (max )~(z))
<1,
I-e-)b#(z))
<1,
whereb#(z)
ed(z) #o),
or theinhomogene- ity
ispronounced
but is localized within aregion
which is much more narrow than R. Moregenerally
we can consider also a sequence ofpronounced
localinhomogeneities (of
widthI) separated by
distances muchlonger
than I. The above results are valid ifiii
<lo Using equations (33, 41)
we find thatiii flu
r~
4(z),
wherefi(z)
isgiven by
theintegral
in the r-h-s- ofequation (33): fi(z)
=(~(z).
The operator(
here in the Fourierrepresentation
isS(k)
ill e~/~~~ );
thisoperator
thereforeperforms smoothing
over a scale r~aN°.5
== R. Thus
the most
general
condition ofvalidity
ofequation (39)
is that smoothed~(z)
is small:fi(z)
< 1.Equation (39)
is the main basic result of this paper. It isinteresting
to check the result for the caseif
weakinhomogeneity, )b#(z))
<1,
where the free energy is knownexactly
[17] as anexpansion
inii
=
# #o (up
to 2ndorder):
~°~~ ~~~
~ ~~~~/
~~~~~~~
~~/v#o ~ fD
ji~a2k2
~~~~~ ~~~~
where
b#k
is the Fourierimage
of the function6#(z), Fr~f
=
fi J #o
Infidz
is the reference free energy of thehomogeneous system
with#(z)
e#o,
and pr~f=
fiIn )
is thecorre-
sponding
chemicalpotential. Expanding
r-h-s- ofequation (39)
inanalogous
series up to thequadratic
order weget
the result whichexactly
coincides withequation (43)
as it should be.So far we assumed that
#(z)
tends to#o
in both limits z - oo and z - -oo. It ispossible
to
generalize
the basic result(Eq. (39)
in order to include also the case#(z)
-#o
as z - oo,#(z)
- 0 as z- -oo
provided
that#(z)
vanishesrapidly enough
on the scale I < R. Tosimplify
theargumentation
let us assume that#(z)
e 0 in the left-sideregion
z < zw[33].
Let us shift the coordinate
origin setting
zw = 0 and introduce areflecting
wall to the left of thesystem
at thepoint
zw. It is obvious that the wall would notchange
the free energy of thesystem
since thepolymer
does not penetrate down to zw anyway. Thereflecting
wallboundary condition, fl
= 0 at z =
0,
should be valid notonly
for the totaldensity,
but also for all othermonomer distribution
functions,
inparticular
for theeigenfunctions
ofequation (10).
Let usnow
complement
the monomer distribution#(z)
and the fieldU(z) by
their mirrorimages
in theregion
z < 0 and then remove the wall. Due to the mirrorsymmetry
the new(extended)
system must be characterized
by
both even and oddeigenfunctions.
It is easy to check however that oddeigenfunctions
does not contribute at all to the free energy, whereas even onesjust
coincide with the
eigenfunctions
for theoriginal system
in theregion
z > o. Therefore the one-chainpartition
functionZi
is the same in both cases, and the total free energy of the extendedsystem
isexactly
twice the free energy of theoriginal system.
Next we note that the mirror extended
system
doesobey
the necessary condition:#(z)
-#o
for z - +oo.Obviously
the mirror extensionimplies
thefollowing
transformation of theamplitudes: Ck
-Ck
+C-k Finally
we calculate the free energy of the mirror extendedsystem using equation (39)
with the newamplitudes Cki dividing
the resultby
2 we thusget
N°12 THEORY OF LONG-RANGE INTERACTIONSIN POLYMER SYSTEMS 1769
the conformational free energy of a semi-infinite
system:
~2
(d#/dz)~
= ~~
/ #(~~
~~e~i'~~
~~~
~
~
(44)
~~~~ ~~
lo j°°
~~f(Na~k~ )Ck
+~~~~
~C0
+@
_~
2K
In order to
get
the conformational free energy of aparticular polymer component (say, l~(~~)
we need to
change N, #,
a and#o
toNA, IA,
aA and#oA. Equations (3, 4)
and(39)
or(44)
thus define the free energy of a one-dimensional two-component
polymer system.
3. Molecular
Weight Dependence
of Interfacial Tension inBinary Polymer
BlendsThe results obtained in the
previous
section areapplied
to someparticular systems
below.As a first
example
let us consider a blend of twoincompatible polymers NA, NB
in thestrong segregation limit, XNA
»1, XNB
» 1. In this case thesystem separates
into twomacrophases:
almost pure A and almost pure
B; #A(z)
e#(z)
-I,
z - oo;#B IQ)
= I#(z)
-1,
z - -oo.For
simplicity
we assume that A and B links aregeometrically
similar: aA" aB " a.
The free energy of the
system
is definedby equations (3, 4, 44).
Both~A(z)
efi@
#A(z) (note
that#o
"
1)
and~B(z)
vanish outside the interfacialregion
of widthr~ A. There- fore the
amplitudes Cl
andCf nearly
does notdepend
on k in the relevant(for Eqs. (39, 44) regime,
kr~ ko "
1/R,
sincekoA
< 1.Omitting
the terms which aredirectly proportional
tothe number of chains
(like
F* inEq. (24))
we thus write:F = Fg~ +
F~nd (45)
where Fg~ is the
ground-state
free energyand
F~nd represents
the finite N corrections:where
Cl
=
J (fi #) dz, Cl
=
J (@@
+#)
dz.Minimization of the dominant term Fg~ leads to the well-known characteristic monomer
density profile d(z)
=
ji(z),
wherej(z)
is definedby equation (2).
The correction term,F~nd,
induces aperturbation
of order1IN (here
Nr~
NA
r~
NB).
However since#(z) corresponds
to the minimum of
Fg~,
thecorresponding change
of Fg~ is of order1/N2.
Thus in order toget
the interfacial tension within an error ofOil /N~)
we couldjust
substituteequation (2)
inequations j45-47);
the result is:~ = lo
i
2 in 2 + +
~
~~~ +
~
~~~
+ O
~
(48)
XNA iNB (RNA) (~NB) (XN)
where
~ ~
K
=
~~~~~
/ f(t~)dt
m 0.5921
K -co
and ~o
"
)x~/~
is the zero-order result(see Eq. ii)).
Inparticular
for thesymmetric
case,NA
"
NB
"
N,
the molecularweight dependence
of the free energy isgiven by
~(N)
m j~'@
+'~~~~ l(49)
~
(XN)
This
equation
isasymptotically
exact in the limitxN
» 1(within
an error of order1/(xN)~).
However it
apparently provides
agood approximation
even in theregion
wherexN
is notlarge.
In
fact,
thesymmetric system
isseparated
ifxN
> 2[17];
at the criticalpoint, xN
=
2,
thetension must
vanish,
~= 0.
Substituting xN
= 2 in
equation (49)
weget
~ =0.03~o.
It thusseems
likely
that the absolute error of theexpression (Eq. (49))
is notlarger
than3i~
of ~oeverywhere.
The first correction to the tension
(of
order1/~N)
was calculatedpreviously
in several papers[11-13, 30j.
The numericalprefactor
obtained in reference [30] agrees with that inequation (49),
while otherapproaches
lead to differentprefactors
due to some additional ap-proximations
involved(see
Ref. [30] for morediscussion).
As the last comment here we note that the
molecular-weight dependence
of the interfacialtension comes
entirely
from theN-dependence
of the conformational free energy. This con- clusion is indisagreement
with a statement of reference[16]
it is claimed there that if theO(1IN)
correction to ~ is nonzero, then it must be due tomolecular-weight dependence
of the interaction energy(the
fact that the~(N) dependence
was considered in Ref.[16]
at constant pressure rather than constant totaldensity
does not make any difference for anincompressible polymer system).
It seems therefore that thecorresponding
statement of reference[16]
can not begeneral
if valid at all[34].
4. Semidilute
Polymer
Solution near a WallHere we consider another
example
ahomopolymer
solution on theright
to a hard wall at z = 0. The solventquality
is assumed to bemarginal
so that a mean-fieldtheory
can beapplied.
The bulk volume concentration is#(oo)
=
#o.
The interaction free energy in the second virialapproximation (which
is valid if#o
is smallenough)
can be written asl~nt
"~ /#~(z)dz (50)
2u
Here
fl
is a numerical factordepending
on the solventquality.
Local
polymer-wall
interactions can be adsorbed in an effectiveboundary
conditionlj ())
= a
lsi)
z=o
The wall is
repulsive
if o > 0 and isadsorbing (attractive)
if a < 0. The Edwards bulk correlationlength (
=a/@$
is assumed to be much smaller than the coilsize,
R=
aN~/~
Minimization of the free energy
Fiji
=
l~nt
+F~onf
definedby equations (44,
50,51)
leads to thefollowing equilibrium
total monomer distribution:4(z)
=#s(z)
+41(z)
where
~ z
(52)
Is (Z)
"
#o
ta°hfi
~~°°~~
N°12 THEORY OF LONG-RANGE INTERACTIONS IN POLYMER SYSTEMS 1771
is an
output
of theground-state approximation (note
that const heredepends
ona).
The relevantscreening length
isshort, (
<R; #~(z)
cf#o
in theregion
z »(.
The second term isa
relatively
small butlong
range correction which is molecularweight dependent:
~~~~~
~° flj~~3/2
~j~
~~~~~~~~~
~it)
= 2/ ()f(k~)
e~~~
and the reduced
amplitude Co
"
f(/$ Is flu )dz. Obviously Co
~
f.
Thetypical decay
scale for
ii
is thus of order of the coil sizeR,
and itstypical amplitude
is 11 ~lo / (flN#o
)~~~The
long-range
effect of a hard wall had beenpreviously
considered in references[15,16j using
differentpartly
heuristicapproaches. Fortunately
both theprevious
results andequation (53) completely
agrees with each other.It is worth
noting
that for the case of localizedinhomogeneity
the end-corrections to theground
state conformational free energy(last
two terms inEq. (39) depend
on asingle integral
parameter, the
amplitude Co
"
f (fi~ 4/40)
dz. This is due to thefollowing physical
reason which was
originally
formulated(independently)
in references[15j
and[16j
and was thenused as a
starting point
for the theoreticalapproaches developed
in these papers.Within the
ground-state approximation
the monomerdensity
isdirectly
related to theground-state eigenfunction: #(z)
=
#oi'((z).
Let us mark one end per each chain. The distribution of the markedpoints
isc~(z)
=fifG(N,z)/Zi Using
nowequations (14, 17)
and
retaining only
the first(ground-state)
terms in the r-h-s- of theseequations
we obtainc~(z)
=
)~§o(z).
If we now take into account that the volume per chain isNV,
the totalpolymer
volume calculated from end-distribution would beJ
polfio(z)dz,
I.e. different from thevolume
coming
out from the total monomerconcentration, J #(z)dz.
Thedifference,
/[#oi'o(z) #oi'((z)jdz
e#oco (54)
is
just proportional
toCo,
which is thus a measure of a mismatch between two distributions.For the case of say
repulsive
wall(#(0)
=
0)
the difference is related to the fact that theprobability
to find an endpoint
near the wall(which
isproportional
to~§o(z))
ishigher
than that for a middle link(proportional
to~§((z)).
The mismatch(Eq. (54)
indicates an intrinsicerror inherent to the
ground
stateapproximation.
Obviously
both end and total monomer distributions must beslightly
corrected on scaleslarger
than( (note
that a small correction in theregion
zmJ
(
could notpossibly compensate
themismatch)
in order to make them consistent with each other. Since there is no othertypical
scalelarger
than( apart
from the coil size R it isanticipated
that the corrections to the distributions are universal functions ofz/R
with theamplitude proportional
to the initialmismatch,
I.e.proportional
toCo-
5.
Long-Range
Interaction in ConcentratedPolymer Systems
At the end of the
previous
section we show that end-effects lead to along
rangeperturbation
ofpolymer density
near a hard wall. Below we consider interaction between surfacesand/or
interfaces caused
by
the same source.5.I. INTERACTION BETWEEN Two PARALLEL SURFACES. Let us consider two
parallel plates
immersed in a semidilute solutionassuming repulsive polymer-wall
interaction(a
- ooin
Eq. (51)).
For a
single plate
the monomerdensity profile
in theground
stateapproximation
is definedby
the well-known lawIi?] given by equation j52)
with const = 0:<(z)
-
<o tallll~ Ill (55)
where z is the distance to the
surface,
and(
=a/@%.
Far
enough
from the surface(z
»()
theperturbation
isexponentially
small:#(z)
m#o 1
4e~~/~j
In the case of two
plates
at z = 0 and z= h
perturbations
inducedby
eachplate
are additive in theregion
z »(,
h z »(:
4(z)
mdo 1 4e~~/~ 4e~lh-z)/fj
The force of interaction between the
plates (per
unitarea), predicted by
theground
statetheory, fgs,
isdirectly
related to the concentration in themiddle,
at z =h/2 [35j: fgs
=) (#(h/2) #o)~.
The minussign
means that the force is
always
attractive. Thus weget
fgs
=-32~#(e~~/~ (56)
u
Let us now calculate the end-corrections to the interaction force.
Unfortunately equation (39)
cannot be
directly applied
to the case ofpolymer
solution confined between twoplates
sincethe
plates completely separate
the confinedsystem
from the outside solution. However it ispossible
togeneralize equation (39)
in order to include the confined caseusing
the idea whichwas
already employed
to treat a semi-infinitesystem (to
deriveEq. (44) ).
Weformally
consider the actualplanes
asbeing reflecting. Considering
allpossible images
of theoriginal
confinedsystem
in these mirrors we thus come to aninfinitely periodic (in
zdirection) system,
withperiodicity
h. It is then easy to show that the free energy of the resultant infinitesystem
perperiod
coincides with the free energy of theoriginal
system. The free energy of the infinitesystem
can be calculatedusing equation (39).
The final result for the free energy of theoriginal system
is:~°~~ ~~ Iv /
~~~~~~
e~u
~~ ~~ / ~~~~~~
~~~~ ~°
~4i~jiu ~j ~~~~~~~~
~~~ ~~~~~~
~~~~where the wave number k
adopts
discretevalues,
km =)n,
n = 0,+1, +2,..
It is the last term that determine thelong-range
interaction(for
h »().
Theamplitudes Ck
can be calculatedwithin the
ground-state approximation. Taking
into account thesymmetry
of thesystem
(j(z)
=#(h z)),
weget Ck
"2C)~~
if k = km with n even, andCk
= 0 if k= km with n
odd,
where C)~~ is theone-plate amplitude
definedby equations (41, 42)
with#(z)
definedby equation (55).
We canneglect k-dependence
of C)~~ since the relevant wave-numbers areN°12 THEORY OF LONG-RANGE INTERACTIONS IN POLYMER SYSTEMS 1773
small,
kr~
1/h
<1If,
where(
is thetypical
localization scalecorresponding
to the function~(z)
for oneplate.
Thus we find C)~~ mCj~~
=2((1
In2).
It is easy to show that the second and third terms in the r-h-s- of
equation (57) nearly
do notdepend
onh,
so thath-dependence
of the free energy is determinedby
the last termalthough
this term is subdominant
according
to its absolute value(the
first termgives
rise to the trivialideal-gas
pressure which isexactly compensated by
similar pressureacting
on theplates
fromoutside). Omitting
constant terms inequation (57)
weget
thelong-range
energy of interaction(per
unitarea)
where
n0.61617, and +
~
T ~ T
is the
reduced
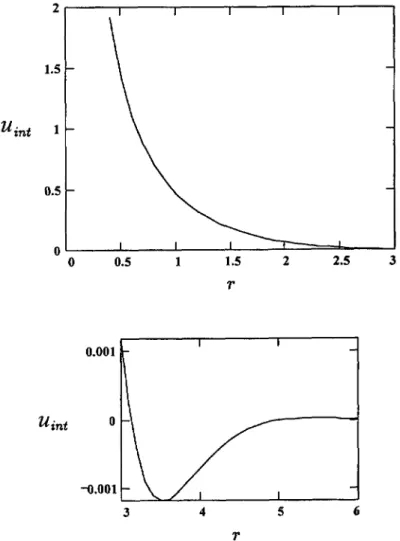
unction lotted in Figure 1.In the
region h
» Rthe
the haracteristic decay
length oforder
[36j.In the
most
(
~~~~~~
~/~v ~~~~~~
~~~°~~~i~fl j
~~~~
Thus end-effects
qualitatively change
the situation here as weget repulsion
instead of weak attractionfollowing
from theground
statetheory.
The nature of thisrepulsive
interaction isqualitatively explained
in the next section: it is related to a formation of virtualend-grafted layers
next to the surfaces. Thelong-range
interaction force isfir
=
-0L§nt/0h.
The total force thus is~°~
~~~ ~~~ ~~~~~
~~~~ ~~~ ~~ ~~~
i~fl /2
The
repulsion
dominates if h >(
In$.
It isinteresting
to note that the energy ofrepulsive
interactions does notdepend
onconcentration,
but doesdepend
on solventquality parameter fl:
the poorer the solventquality
thestronger
theinteraction, U;nt
c~1/fl.
Therefore weshould
expect
thestrongest repulsion
in theta conditions. The free energy of excluded volume interactions in a semidilute theta solution is~,
l~nt
Cf