Les cellules germinales primordiales nouvellement induites voient toutes leurs marques de méthylation d’ADN complètement effacées puis rétablies de novo. Troisièmement, nous avons constaté des différences structurelles chez l'homme entre les profils de méthylation de novo des cellules EC et des spermatozoïdes. Lors de leur induction, les cellules germinales primordiales subissent une délétion à l'échelle du génome et un rétablissement de novo des marques de méthylation de l'ADN.
We also identified important structural differences in de novo methylation profiles between human sperm and ES cells.
Introduction
Induction and Development of Male Germ Cells in Mammals
- Key Aspects of Early Embryonic Development
- Induction of Primordial Germ Cells from Somatic Tissues at E6.5
- Colonization of Embryonic Gonads by Mitotic and Post Mitotic PGCs between
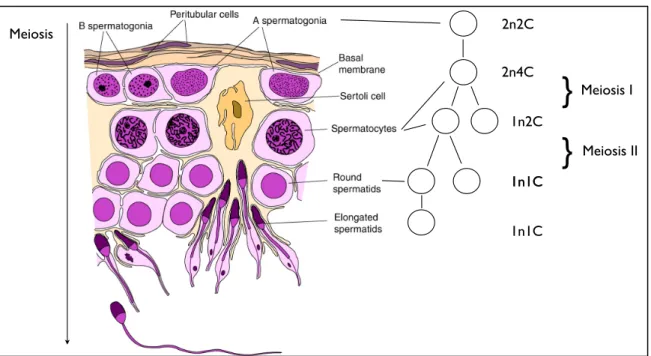
- From Spermatogonial Stem Cells to Mature Sperm
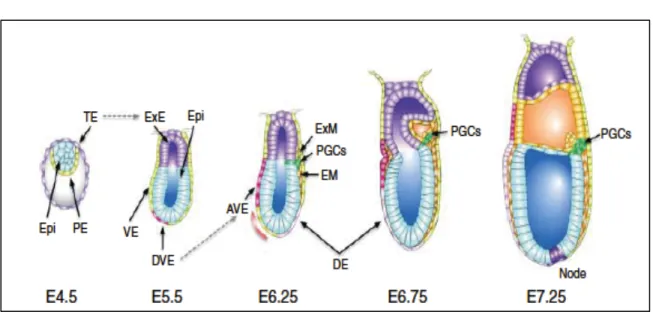
PGCs arise from a competent region located at the junction of the extraembryonic ectoderm (ExE), the visceral endoderm (VE), and the adjacent anterior-proximal epiblast (Figure 1.2, Ginsburg et al., 1990; Lawson and Hage , 1994). In the current model for PGC migration, homotypic cell adhesion and heterotypic repulsion serve to drive cellular migration and directionality (Tanaka et al., 2005). An interferon-inducible transmembrane protein (Ifitm3, also known as Fragilis) is expressed upon induction by BMP signaling during PGC specification ( Saitou et al., 2002 ).
A similar protein, Ifitm1/Fragilis2, is later expressed in the developing PGC and the budding mesodermal cells (Tanaka et al. 2001; Lange et al. 2003).
DNA Methylation Dynamics During Mammalian Development
- Key Aspects of DNA methylation
- DNA Methylation: Functional Relevance and Evolutionary Consequences
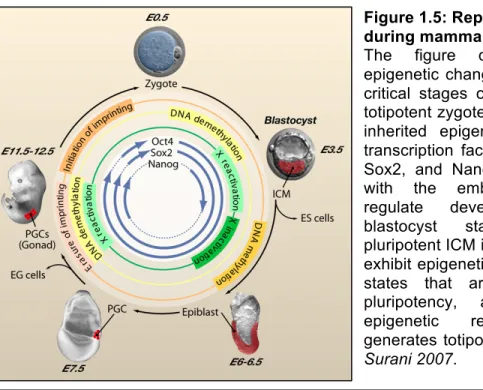
- Germ Cells and ES Cells: Outcome of Epigenetic Reprogramming
Furthermore, animal mutants for TDG (a glycosyase involved in the base excision repair pathway) show demethylation defects during early embryogenesis ( Cortellino et al., 2011 ). In turn, H3K4 methylation inhibits DNMT3L recruitment at promoters and prevents de novo methylation (Ooi et al., 2007). DNMT3B and DNMT1 are found at much lower levels throughout PGC development (La Salle et al., 2004).
Hammoud and colleagues recently showed in human sperm that about 4% of the genome is still packed in nucleosome-conserving domains ( Hammoud et al., 2009 ).
Germ Cell Associated Small RNA Pathways
- Overview of RNAi
- PIWI Proteins and Germ Cell Specific RNAi
- PiRNA Biogenesis during Male Germ Cell Development
- Link between piRNAs and De Novo Methylation in Male PGCs
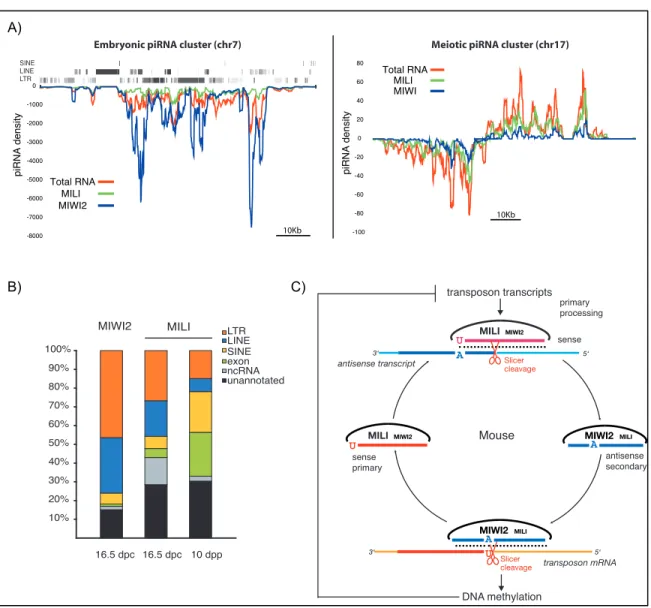
Similarly, MILI is detected in the female germline shortly after PGC migration and is continuously expressed in both arrested and growing oocytes (Aravin et al., 2008). Secondary piRNAs can in turn mediate the cleavage of a complementary transcript, generating the 5' end of a new piRNA that closes the loop on itself (Figure 1.7C) (Brenecke et al., 2007; Aravin et al., 2008). Recently, catalytically inactive mutants of all three PIWI proteins were generated in mice (De Fazio et al., 2011; Reuter et al., 2011).
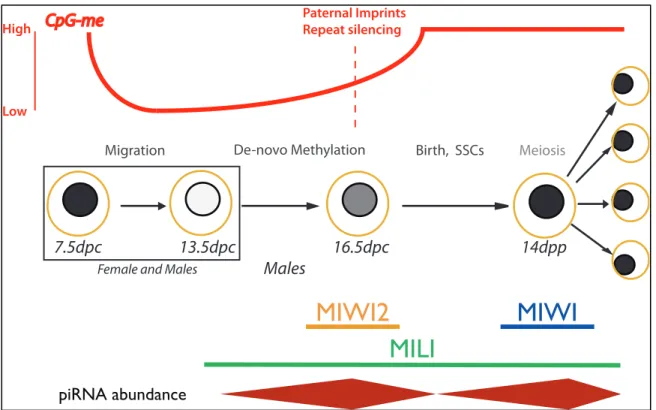
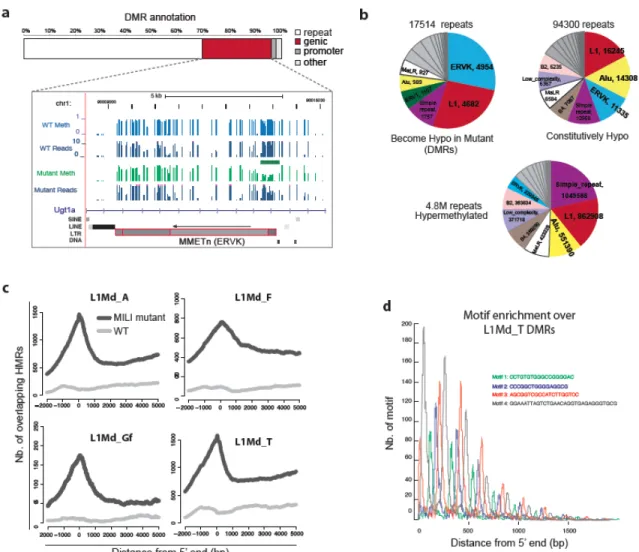
In addition, these mutants show a reduction in transposon de novo methylation in PGCs as early as 16.5 dpc ( Kuramochi-Miyagawa et al., 2008 ).
Epigenetic Inheritance and Evolution some Open Questions
Results
Study of a Transgenic piRNA Cluster in Meiotic Mouse Germ Cells
- Résumé en Français
- Specific contribution to the publication
- Publication reference
They are often strongly enriched in transposon sequences, consistent with a function of the piRNA pathway in transposon control (Vagin et al. We find that critical determinants reside in both the local and long-range sequence environments of of the piRNA cluster complementary to lacZ was recently demonstrated using the RNase protection assay in the ovaries of line P-1152 (Todeschini et al. 2010).
Above is a plot of piRNA read frequencies along the plus and minus strands (indicated) of the element. It produces piRNAs from only one genomic strand and is expressed exclusively in the somatic follicle cells of the ovary. We selected a P[acman] BAC clone that extends from a position z30 kb upstream of the first annotated piRNA z86 kb towards the X chromosome centromere (Venken et al. 2009).
Like flamenco, each region of the ch17 cluster produces piRNAs from only one genomic strand. Importantly, tj generates piRNAs from a discrete segment of its 39-UTR region ( Saito et al. 2009 ). Compared to the patterns derived from independent insertions of the same transgenes in mice (R2 = 0.99) (Fig. 3C), patterns of GFP piRNAs from tjandflamenco appeared quite different (R2.
Furthermore, our data demonstrate that the position of the cluster in the genome is not important, and therefore transgenic piRNA clusters can be created in heterologous genomic locations. Previous bioinformatic analyzes described the generation of individual piRNAs from long precursor molecules as a pseudorandom process with a weak influence of the local sequence environment of individual piRNA species (Betel et al. 2007).
Establishment of De novo Methylation Profiles in Mouse PGCs: interplay between
- Résumé en Français
- Specific contribution to the work
- Manuscript and figures
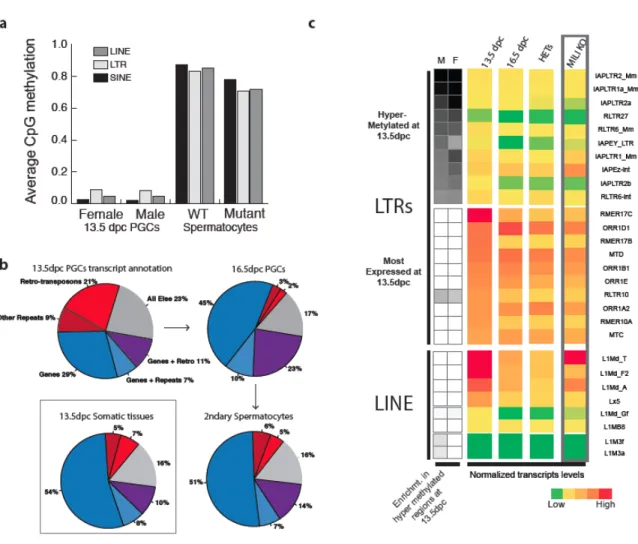
During mammalian germ cell development, DNA methylation is thought to be completely erased and reestablished de novo throughout the genome (Monk M, 1987; Reik et al., 2001; Surani et al., 2007). However, we have previously identified thousands of naturally hypomethylated transposons in primate sperm methylomes, suggesting that some copies escape piRNA-driven de novo methylation (Molaro et al., 2011). Consistent with previous reports, PGCs and spermatocytes represent the two extremes of the methylation spectrum (Popp et al., 2010).
Cytosine methylation is one of the main ways by which retro-transposon transcription is repressed in mammalian genomes (Walsh et al., 1998). We previously reported thousands of transposon copies that naturally avoid de novo methylation in chimpanzee and human sperm methylomes (Molaro et al., 2011). The mili knockout strain was obtained from Haifan Lin (Yale University) and is described in Kuramochi-Miyagawa et al., 2004.
For PGC isolation, Oct4-EGFP mice, described in Lengner et al., 2007, were purchased from Jackson Laboratory (Bar Harbor, Maine). Cell sorting: Secondary and primary spermatocytes were FACS-sorted (Aria II, BD Bioscience) from WT and mili-mutant animals based on DNA content using Hoechst staining, as described in Bastos et al., 2005. Preparation, sequencing and mapping shotgun bisulfite libraries: shotgun bisulfite sequencing was performed as described in (Molaro et al., 2011).
Small RNA cloning: Small RNA cloning of total RNA was performed as described in Aravin et al., 2008. All further bioinformatic analysis on mapped sequences was done using Unix-based utilities (Galaxy, Goecks et al., 2010). .
DNA Methylation Profiles of Chimp and Human Sperm: A Look into Epigenetic
- Résumé en Français
- Specific contribution to the publication
- Publication reference
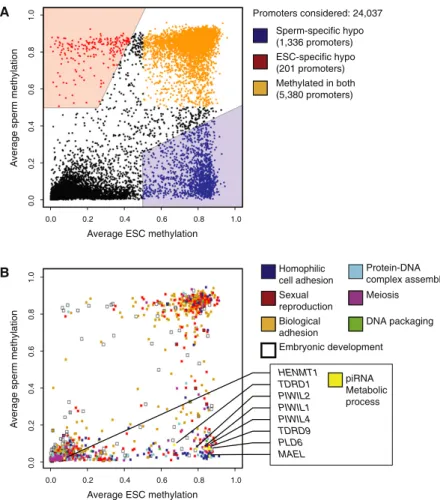
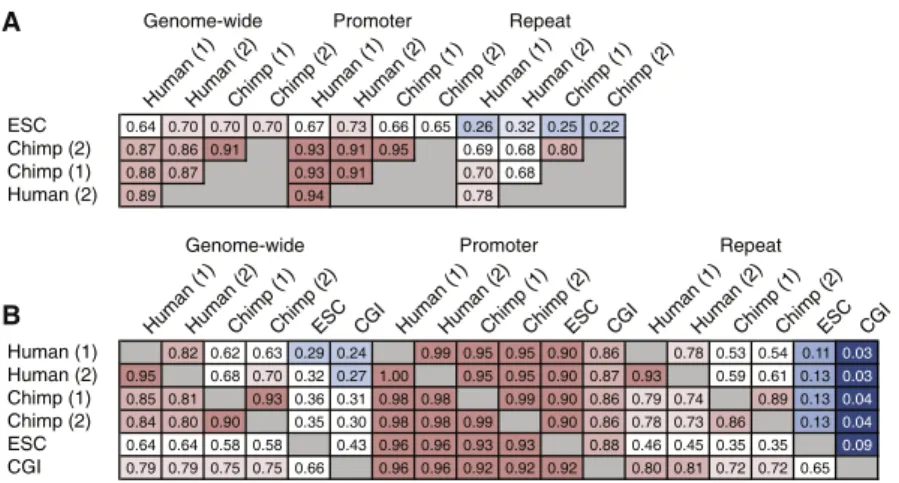
I participated in the preparation and analysis of all data reported here (except the methylome of ES cells previously published by Laurent et al., 2010). We therefore produced DNA methylation profiles at single CpG resolution in human and chimpanzee sperm and compared them with methylation maps from human ESCs (Laurent et al., 2010). The L1PA subfamilies are considered the most active in the human genome (Khan et al., 2006), and the youngest of them (L1HS and L1PA2) were among the few subfamilies enriched for hypomethylation in ESCs relative to sperm.
Among the SVAs, the newest subfamilies, D-F (Wang et al., 2005), showed the highest frequency of hypomethylation in human sperm (Figure 5A). We also annotated 921 SVA elements that appear to represent new insertions occurring after the human-chimpanzee divergence ( Mills et al., 2006 ). Although the full set of HMRs in human sperm and ESCs did not reach this empirical cutoff, they did pass the 0.4 standard used by Weber and colleagues (Figure 6A) (Weber et al., 2007).
Rather, promoter regions appear to be generally identified and characterized in sperm (see Zaidi et al., 2010). The phenomenon we observe is similar to the concept of CpG shores (Doi et al., 2009). Low levels of methylation have also been associated with the ability to bind cohesin complexes (Parelho et al., 2008).
Transduction of SVAs has been implicated in human disease and regeneration (Damert et al., 2009; Ostertag et al., 2003). To eliminate redundancy in sets of Gene Ontology categories identified as enriched, we used the REVIGO software through the web interface (Skunca et al., 2009).^.
Discussion and Perspectives
Towards an understanding of piRNA cluster biology
Using various labeled piRNA clusters as transgenes in both the mouse and drosophila genomes, we were able to show that piRNA clusters can be programmed to produce new piRNAs upon introduction of ectopic sequence, out of their context maternal genomic. These results suggest that the cues that tag a transcript for piRNA processing lie within the cluster sequences themselves. Surprisingly, piRNA meiotic clusters in mouse, rat, and human are positioned within syntenic parts of their genomes, despite the cluster sequences being extremely divergent ( Aravin et al., 2006 ; Girard et al., 2006 ).
This suggests that different selective pressures drive the evolution of piRNA cluster positions and cluster sequences. Clusters of piRNAs targeting transposons have been proposed to act as graveyards, keeping track of previous waves of transposon activity and hunting new ones throughout the animal's lifespan. The ability to ectopically program piRNA clusters could be very useful to control, for example, gene expression during meiosis or to direct site-specific chromatin changes if the target group is expressed during embryogenesis.
Furthermore, our transgenic lines provide the opportunity to study the yet unknown silencing capacity of meiotic piRNAs. To address the silencing function of meiotic piRNAs, both transgenic lines with GFP-tagged cluster were crossed with a reporter mouse expressing a GFP-tagged protein with appropriate expression patterns (in this case, a MILI-GFP-tagged transgenic line used from Aravin et al., 2008). These data may indicate that coexpression of a meiotic piRNA and its cognate target sequence is not sufficient to induce silencing or that meiotic.
To answer this question, one could perform MIIWI-IPs in animals expressing the target GFP reporter and examine whether the presence of a target changes the MIWI loading rate with GFP piRNAs. Finally, one could rule out the implication of the local chromatin environment in both silencing potential and MIWI burden by generating knock-in-labeled piRNA clusters in mouse ES cells.
Transposon de novo methylation in the male germ line: insight into the ecology of
Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during mouse male germ cell development. Complex genome-wide transcriptional dynamics orchestrated by Blimp1 for mouse germ cell lineage specification. Multiple domains are involved in the targeting of mouse DNA methyltransferase to DNA replication foci.
Sp1 sites in the mouse aprt gene promoter are required to prevent CpG island methylation. Role of piRNAs and non-coding RNAs in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Under normal circumstances, commitment is thought to be unidirectional with repressive epigenetic marks stabilizing the loss of plasticity (De Carvalho et al., 2010).
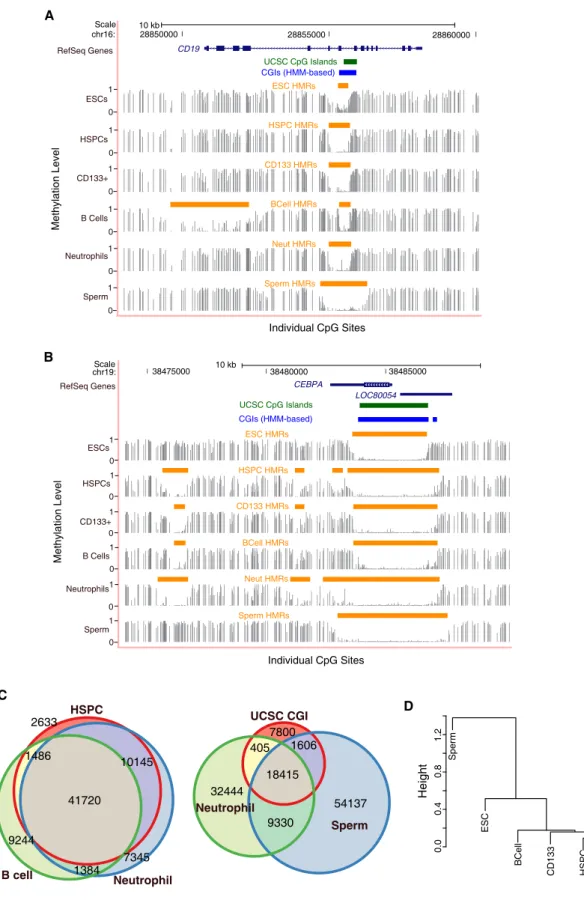
Higher resolution maps of DNA methylation with shotgun bisulfite sequencing have been produced mainly from cultured cells (Laurent et al., 2010; Lister et al., 2009) or mixed cell types (Li et al., 2010). Other genome-wide studies have implicated DNA methylation in the regulation of alternative promoters and even RNA splicing patterns (Maunakea et al., 2010). Although CGIs have been computationally defined (Irizarry et al., 2009b), we developed an algorithm to empirically identify hypomethylated regions (HMRs) in bisulfite sequencing datasets based solely on their methylation state (see Figures 1A and 1B).
UCSC predicted/annotated CpG islands (green lines) and HMM-based CpG islands (blue lines) are also shown (Irizarry et al., 2009b). Therefore, we investigated the existing ChIP-seq data of PU.1 from human HSPCs (Novershtern et al., 2011). Previous studies experimentally identified binding sites for the insulator protein, CTCF, by chromatin immunoprecipitation (Kim et al., 2007).
Sequencing traces of two loci (A, B) with ChIP-seq peaks derived from HSPCs enriched for PU.1 transcription factor (Novershtern et al., Cell.